环肽类化合物的组合物的制作方法
本发明涉及药物纯化和制备领域,尤其涉及一种环肽类化合物的组合物,以及制备该环肽类化合物的组合物的方法。
背景技术:
米卡芬净(Micafungin)是一种新型棘白菌素类抗真菌药物,通过抑制真菌细胞壁的组成成分β-1,3-D-葡聚糖合成酶,破坏真菌细胞结构,使之溶解。米卡芬净广泛用于治疗各种感染,尤其是曲霉菌、念珠菌、隐球菌、毛霉菌、放线菌、组织胞浆菌、皮肤癣菌和镰刀菌等引起的感染。特别是用于治疗化疗、AIDS等免疫力低下患者的真菌感染。临床还与两性霉素和三唑类抗真菌药物进行联合用药。
米卡芬净钠(Micafungin Sodium,又称FK463)是药品Mycamine(米开民)的活性药物成分。米卡芬净钠盐的化学结构如式I所示:
澄清度检查是检查药物中的微量不溶性杂质,出于用药安全性考虑,用于注射剂的原料药,一般应进行此项检查。药物澄清度是注射剂药物的一重要指标,间接地反应出药物中部分不溶性微粒的含量,反应出药物的纯度。不溶性 微粒进入血管能引起血管肉芽肿、静脉炎、血栓及血小板减少,对心肌、肝、肾亦有损害。来自国家药品不良反应检测中心的监测数据显示,抗生素类注射剂的药物不良反应发生率高,其主要原因为药物的澄清度项不符合要求。
米卡芬净钠注射剂作为一种注射剂药物,日用量较高,澄清度是一项重要质量指标。控制米卡芬净钠注射剂的澄清度,势必首先需要控制米卡芬净钠原料药的不溶性微粒含量,即需要控制米卡芬净钠原料药的澄清度。而影响米卡芬净澄清度的主要物质,是其在制备方法中产生的脂类杂质。
美国专利6,107,458和WO2004014879A1以及WO96/11210公开了制备和提纯式I化合物的方法。其中,WO2004014879将米卡芬净DIPEA(二异丙基乙基胺)盐(式VIII化合物)通过过滤和色谱分离提纯后,再使用溶剂沉淀,得到式I化合物。
Atsushi Ohigashi等人在Journal of Synthesit Organic Chemistry(合成有机化学杂志)2006年第64卷第12期上发表的论文“Process Development of Micafungin,a Novel Lipopeptide Antifungal Agent”中介绍,式VIII化合物经过离子交换得到式I化合物的溶液,然后在该溶液中加入有机溶剂形成沉淀,得到式I化合物。
Xellia Pharmaceuticals在专利WO2012136498中公开了式III化合物与式VII化合物一锅缩合得到式VIII化合物,并在专利WO2012143293中公开了由 式VIII化合物一步大孔树脂层析制备得到式I化合物的方法。
上述现有技术在制备式I化合物的方法均是采用发酵得到的式II化合物为起始原料,经酰化酶水解脱除侧链生成式III化合物。式III化合物与式V化合物酰化缩合得到式VIII化合物,即式I化合物DIPEA盐,最后经离子交换得到式I化合物。具体制备方法如下所示:
该方法制备得到的式I化合物常常会含有与式I化合物结构的酰化侧链类似物的微量杂质,包括主原料式II化合物生成的棕榈酸(式IV化合物)和反应过程中残留的式V、VI和VII化合物。而现有技术公开的制备方法中并没有有效去除这些杂质的纯化工艺。
式II化合物经酰化酶水解生成式III化合物的同时,生成另一副产物式IV化合物(棕榈酸)。棕榈酸为长链脂肪酸,在水中几乎不溶解,在式III化合物纯化时,通常有部分的棕榈酸残留在式III化合物中,即使反复使用已知方法进行结晶和层析,也无法降低式III化合物中棕榈酸的含量。而在后续制备和纯化式I化合物的过程中,棕榈酸与式I化合物的侧链以非极性键和氢键相互结合和缠绕,导致棕榈酸仍残留在式I化合物中。
式III化合物与式V化合物酰化缩合制备式VIII化合物的过程中,式V化合物中本身也残留有少量式VI和式VII化合物,且过量的式V化合物会转化为式VI和式VII化合物,最终导致获得的式VIII化合物样品中残留有式VI和式VII化合物。式VI和式VII化合物为长链芳香类化合物,酯和水溶解性均较差,在式VIII化合物和式I化合物纯化时难以去除,即使反复使用已知方法进行结晶和层析,也无法降低式VI和式VII化合物的含量。这是由于式VI和式VII化合物与式VIII和式I化合物的侧链以非极性键和氢键相互结合和缠绕,导致式VI和式VII化合物始终保留在其中。
可见,采用现有技术公开的层析和结晶工艺均难以完全去除棕榈酸、式VI化合物和式VII化合物,尤其是含量较低时,这些物质与式I化合物无法完全分离。
发明人在式I化合物研究过程中发现,上述杂质的残留最终会影响溶液的澄清度,产生“乳光”现象。如果不将上述杂质含量控制在一定限度的范围内,最终获得式I化合物溶液澄清度检查表现显著浑浊,无法达到药品的质量标准。而目前所公开的式I化合物制备方法均无法将这些微量杂质良好去除或控制在合理限度内。
因此,本领域迫切需要获得一种高纯度组合物及其制备方法,以便能够更 好的解决溶液澄清度的问题,确保最终制备的抗真菌药物的安全性。
技术实现要素:
发明人在制备式I化合物的初期,采用本领域公开的方法进行制备得式I化合物,得到的式I化合物经澄清度检查时表现显著浑浊。发明人尝试将式I化合物溶解在水、醇水溶液或醇类溶剂中,然后采用0.22μm膜微滤,再采用已知的方法将式I化合物进行固体析出沉淀,得到的化合物复溶后仍表现出浑浊,出现“乳光”现象,使用分光光度计在410nm波长下测定透光率小于97%。发明人继续进行研究,采用活性炭吸附、有机溶剂萃取、超滤、硅藻土层析、氧化铝层析等方式,都未能解决上述“乳光”现象。
为了解决这一难题,发明人将高浓度的式I化合物溶解在水中,采用膜微滤,将膜上极微量的物质进行质谱分析。发明人惊奇的发现,质谱检测结果显示含有式IV、VI和VII化合物。由此发明人推断,式I化合物出现的“乳光”现象是由制备过程中式IV、VI和VII化合物的残留所引起。
发明人为了进一步明确引起“乳光”现象的原因,采用具有高灵敏度的UV检测器的HPLC检测样品,检测结果显示未发现式IV化合物、式VI化合物和式VII化合物,推测这些物质含量极低,低于UV检测器的最低检测限度。发明人继续使用更高灵敏度的分析仪器LC-MS(3Q),该仪器通常用于临床血药浓度的检测,检测灵敏度极高,可达到ng或pg级别。发明人使用LC-MS检测后,惊奇地发现澄清度良好的样品式IV化合物、式VI化合物、式VII化合物含量低。而澄清度表现“乳光”现象的样品,式IV化合物、式VI化合物和式VII化合物含量较高。由检测结果可知,样品中式IV化合物、式VI化合物和式VII化合物残留量过多时会导致澄清度表现出“乳光”现象。
在上述检查结果的基础上,结合式I化合物的合成路线,发明人认为对前端工序的式VIII化合物进行纯化,将有可能解决这一难题。对此,发明人针对式VIII化合物的纯化工艺进行了研究,采用了多种树脂层析和结晶方法,包括使用C-18、HP20SS等树脂层析、甲醇/乙酸乙酯体系或乙醇/乙酸乙酯或甲醇/乙酸正丙酯等体系结晶,均未解决上述难题。
发明人在进一步研究后惊奇地发现,将式VIII化合物溶解在甲醇、乙醇、正丙醇或异丙醇的水溶液,并控制溶液水分含量在一定的比例范围内,调节溶解液pH值,通过降温或添加难溶性溶剂等方法得到式VIII化合物,经澄清度检查未呈现乳光现象,澄清度检查与水一致,将VIII化合物溶解在水中,浓度控制在100mg/ml以下时,使用分光光度计在410nm波长下测定透光率均大于等于97.5%。然后经用离子交换、HP20SS层析制备得到式I化合物,所得到的样品经澄清度检查未呈现乳光现象,澄清度检查与水一致,透光率也均在97.5%以上。
发明人为了证明上述结晶工艺的有效性,进行了对比实验研究。采用上述方法多次结晶得到的式VIII化合物,该样品澄清度检查与水一致,将样品分别配制浓度为10mg/ml,20mg/ml,50mg/ml,100mg/ml的溶液,在410nm波长下,使用分光光度计测定透光率都达到了97.5%以上。经LC-MS(3Q)方法检测,式IV化合物、式VI化合物和式VII化合物含量分别为8ppm、0.2ppm和0.4ppm。而以该样品为起始原料,在该样品中加入含量分别为100ppm、10ppm和30ppm的式IV化合物、式VI化合物和式VII化合物,然后采用甲醇/水/乙酸乙酯进行结晶(pH 7.5,水分含量控制在一定的比例范围内),过滤得到的样品经LC-MS(3Q)方法检测发现式IV化合物、式VI化合物和式VII化合物含量分别为10ppm、0.4ppm和1.2ppm。由此数据说明,该方法对这些微量杂质去除效果显著。同样地,本发明人按照美国专利7,199,248的方法,将式I化合物进行固体析出,XRD分析固体为无定型固体形式,且检测结果显示该方法去除上述杂质的效果不明显。
本发明人进一步研究利用上述结晶工艺所获得的固体经XRD分析,式VIII化合物为形态优异的晶体。本发明人通过研究惊奇地发现醇与水体积比为10-100,优选11-50,更优选12-25时,且结晶溶液的pH值控制在5.0至8.5时,式VIII化合物在形成晶体的过程中,去除式IV化合物、式VI化合物和式VII化合物的效果明显。发明人主要推断在pH值5.0至8.5范围,式VIII化合物为中性或带负电荷,与杂质式IV化合物、式VI化合物和式VII化合物非极性作用力减弱,相互结合作用减少。另外,在此结晶过程,式VIII化合物分子 排列规整,同样有利于杂质式IV化合物、式VI化合物和式VII化合物的去除,从而去除上述杂质的效果明显。
在将式VIII化合物溶解在甲醇、乙醇、正丙醇或异丙醇的水溶液,并控制溶液水分含量在一定的比例范围内,调节溶解液pH值的条件下,通过分子重排结晶的方式,将IV化合物、式VI化合物和式VII化合物保留在结晶母液中,获得意想不到的纯化效果。同时本发明人按照WO2004014879A1制备式VIII化合物,获得的固体经XRD分析为无定型,对IV化合物、式VI化合物和式VII化合物去除效果不好。
据此,发明人获得了一种含有环肽类化合物式VIII化合物的组合物,含有极其微量的IV化合物、式VI化合物和式VII化合物,有效地解决了式I化合物澄清度的问题;同时获得了所述的组合物的制备方法和检测方法;式VIII化合物的组合物进一步用于制备式I化合物的用途;还获得了一种式I化合物的组合物,其含有极其微量的式IV化合物或其盐,式VI化合物和式VII化合物;以及式I化合物的组合物用于制备治疗真菌感染的药物的用途。
含式VIII化合物的组合物
本发明提供了一种环肽类化合物的组合物,所述组合物包含:结构如式VIII所示化合物,结构如式IV所示化合物或其盐,结构如式VI所示化合物,结构如式VII所示化合物;所述组合物溶于水后形成的溶液无乳光现象。
在一优选例中,所述组合物溶于水后形成的溶液在410nm波长下测定的透光率大于等于97.5%,优选大于等于98.0%,更优选大于等于98.5%。
在另一优选例中,所述组合物溶于水后形成的溶液中VIII所示化合物的浓度为10mg/ml,优选20mg/ml,更优选50mg/ml,最优选100mg/ml。
在一优选例中,所述组合物中式IV所示化合物或其盐的含量为0.01-100ppm,优选0.01-50ppm,更优选0.01-10ppm。
在另一优选例中,所述组合物中式VI所示化合物的含量为0.001-10ppm,优选0.001-1ppm,更优选0.001-0.5ppm。
在另一优选例中,所述组合物中如式VII所示化合物的含量为0.01-30ppm,优选0.01-30ppm,优选0.01-10ppm,更优选0.01-5ppm。
在另一优选例中,所述组合物中式IV所示化合物或其盐的含量为0.01-100ppm,优选0.01-50ppm,更优选0.01-10ppm;式VI所示化合物的含量为0.001-10ppm,优选0.001-1ppm,更优选0.001-0.5ppm;如式VII所示化合物的含量为0.01-30ppm,优选0.01-10ppm,更优选0.01-5ppm。
在另一优选例中,所述组合物中各组分含量用超高效液相和三重四极杆串联质谱联用仪测定。
含式VIII化合物的组合物的制备方法
本发明提供了一种制备含有式VIII化合物的组合物的方法,所述方法包括步骤:
(a)将如式VIII所示化合物粗品溶解在含水的醇溶液中;
(b)调节pH,通过降温和/或添加有机溶剂(i),形成晶体或沉淀,过滤,干燥得到所述环肽类化合物的组合物。
在一优选例中,步骤(a)中所述醇选自:甲醇、乙醇、正丙醇和异丙醇中的一种或其混合。
在另一优选例中,步骤(a)中所述含水的醇溶液中,醇与水体积比为10-100,优选11-50,更优选12-25。
在另一优选例中,步骤(b)中所述pH调节为5.0至8.5。
在另一优选例中,步骤(b)中所述有机溶剂(i)选自:正丙醇、异丙醇、异丁醇、丙酮、乙酸甲酯、乙酸乙酯、乙酸正丙酯、乙酸异丙酯或其混合。
在另一优选例中,步骤(b)中所述的降温的温度为-40至35℃,优选-20至35℃,更优选-10至30℃,最优选-5-20℃。
在另一优选例中,所述步骤(a)-(b)可以重复一次或一次以上。
含有式I化合物的组合物
本发明提供了一种含有式I化合物的组合物,所述组合物包含:结构如式I所示化合物,结构如式IV所示化合物或其盐,结构如式VI所示化合物,结构如式VII所示化合物;所述组合物溶于水后形成的溶液无乳光现象。
在一优选例中,所述组合物为原料药。
在另一优选例中,所述原料药中主成分式I化合物含量大于90%,优选大于95%。
在另一优选例中,所述组合物溶于水后形成的溶液在410nm波长下测定的透光率大于等于97.5%,优选大于等于98.0%,更优选大于等于98.5%。
在另一优选例中,所述组合物溶于水后形成的溶液中I所示化合物的浓度为10mg/ml,优选20mg/ml,更优选50mg/ml,最优选100mg/ml。
在另一优选例中,所述组合物中式IV所示化合物或其盐的含量为0.01-100ppm,优选0.1-50ppm,更优选1-10ppm。
在另一优选例中,所述组合物中式VI所示化合物的含量为0.001-10ppm,优选0.001-1ppm,更优选0.001-0.5ppm。
在另一优选例中,所述组合物中如式VII所示化合物的含量为0.01-30ppm,优选0.01-10ppm,更优选0.01-5ppm。
在另一优选例中,所述组合物中式IV所示化合物或其盐的含量为0.01-100ppm,优选0.1-50ppm,更优选1-10ppm;式VI所示化合物的含量为0.001-10ppm,优选0.001-1ppm,更优选0.001-0.5ppm;如式VII所示化合物的含量为0.01-30ppm,优选0.01-10ppm,更优选0.01-5ppm。
在另一优选例中,所述组合物中各组分含量用超高效液相和三重四极杆串联质谱联用仪测定。
式I化合物组合物的鉴定和性质
“高效液相色谱法”(HPLC)是用于检测化合物纯度的常用方法,是以液体为流动相,采用高压输液系统,将具有不同极性的单一溶剂或不同比例的混 合溶剂、缓冲液等流动相泵入装有固定相的色谱柱,在柱内各成分被分离后,进入检测器进行检测,从而实现对试样的分析。本发明中采用HPLC测定式I化合物纯度,所述的HPLC检测方法如下:
分析柱:YMC-ODS 250×4.6mm,5μm
流动相:乙腈∶磷酸盐缓冲液(pH 3.0)=45∶70
流速:1ml/min
柱温:35℃
稀释液:水的磷酸盐缓冲液
检测波长:210nm
进样量:10μl
“UPLC-三重四级杆联用”是用于检测微量化合物含量的一种有效方法。本发明中采用UPLC-三重四级杆联用方法测定环肽类化合物的组合物中式VI化合物和式VII化合物的含量,所述的检测方法如下:
(1)UPLC色谱条件:
色谱柱:Waters Acquity TM BEH C18柱100×2.1mm,1.7μm
流动相:A为0.1%冰乙酸水,B为乙腈,0→10min,60%→100%(B)
柱温:40℃
流速:0.4mL/min
进样量:5μl
(2)质谱条件:
电喷雾离子化正离子检测方式;气帘气(CUR)20mL/min;碰撞气(CAD)10mL/min;离子喷射电压(IS)5500v;雾化气温度*(TEM)600℃;GAS1 50mL/min;GAS2 50mL/min.
本发明中采用UPLC-三重四级杆联用方法测定环肽类化合物的组合物中式IV化合物的含量,所述的检测方法如下:
(1)色谱条件:
色谱柱Agilent SB-C18柱(2.1mm×50mm,1.8μm);
流速0.4ml/min;柱温35℃;
流动相甲醇∶水=90∶10;
进样2μl
(2)质谱条件:
SIR模式(m/z 255.2);ESI(-);毛细管电压3.5kV;锥孔电压30V
本发明中药物组合物中,环肽类化合物的组合物中式VI化合物和式VII化合物的含量、式IV化合物的含量是通过上述UPLC-三重四级杆联用方法测定,及外标法计算的。
“溶液透光率”指溶液透过光的效率,是透过溶液的光通量与其入射光通量的百分率。其数值大小可以反映溶液的澄清度。
(1)测定方法
仪器:分光光度计(精度±0.5T%)。
步骤:按照规定的样品浓度,称取样品,加水溶解并定容,摇匀,作为试液。用试液冲洗并注入10mm比色皿中,以溶解样品的同批水调节仪器零点,于波长410nm处,测定其透光率。
药物组合物
本发明提供了一种用于治疗真菌感染的药物组合物,所述的药物组合物中含有本发明所获得的式I化合物组合物和药学上可接受的载体。所述组合物溶于水后形成的溶液无乳光现象。
在一优选例中,所述组合物溶于水后形成的溶液在410nm波长下测定的透光率大于等于97.5%,优选大于等于98.0%,更优选大于等于98.5%。
在一优选例中,所述组合物溶于水后形成的溶液中I所示化合物的浓度为10mg/ml,优选20mg/ml,更优选50mg/ml,最优选100mg/ml
在一优选例中,所述药物组合物中,以式I化合物的含量计,式IV所示化合物或其盐的含量为0.01-100ppm,优选0.01-50ppm,更优选0.01-10ppm。
在另一优选例中,所述药物组合物中,以式I化合物的含量计,式VI所示 化合物的含量为0.001-10ppm,优选0.001-1ppm,更优选0.001-0.5ppm。
在另一优选例中,所述药物组合物中,以式I化合物的含量计,式VII所示化合物的含量为0.01-30ppm,优选0.01-10ppm,更优选0.01-5ppm。
相关术语
如本文所用,“‘乳光’现象”又称丁达尔现象,是指胶体分散相或超显微粒子的漫反射效应。在光的传播过程中,光线照射到粒子时,如果粒子大于入射光波长很多倍,则发生光的反射;如果粒子小于入射光波长,则发生光的散射,这时观察到的是光波环绕微粒而向其四周放射的光,称为散射光或乳光。
如本文所用,术语“原料药”,根据ICH Q7A中的规定是指用于药品制造中的任何一种物质或物质的混合物,而且在用于制药时,成为药品的一种活性成分。此种物质在疾病的诊断,治疗,症状缓解,处理或疾病的预防中有药理活性或其他直接作用,或者能影响机体的功能或结构。指用于生产各类制剂的原料药物,是制剂中的有效成份,但病人无法直接服用的物质。所述原料药中主成分含量一般大于90%,优选大于95%,更优选大于98%。
如本文所用,术语“澄清度检查”是指溶液澄清度的检查;澄清度检查方法是指在室温条件下,将用水稀释至一定浓度的供试品溶液与等量的浊度标准液分别置于配对的比浊用玻璃管中,在暗室内照度为1000lx的条件下检查溶液的澄清度或浑浊程度。中国药典也对澄清度进行了规定,“澄清”系指溶液的澄清度相当于所用溶剂,或未超过0.5号浊度标准液。
如本文所用,术语“如式VIII所示化合物粗品”,“式VIII化合物粗品”可以互换使用,是制备本发明所述的含式VIII化合物的组合物的起始原料,含有式VIII化合物以及较高含量的式IV、VI和VII化合物。可采用现有技术进行制备,例如但不限于WO2004014879A所公开的方法。
如本文所用,术语“以式I化合物的含量计,药物组合物中式VI化合物、式VII化合物和式IV化合物的含量”是指本发明中药物组合物中,式VI化合物和式VII化合物的含量、式IV化合物的含量通过UPLC-三重四级杆质谱联用方法测定,外标法计算得到的式VI化合物、式VII化合物和式IV化合物浓度,用上述计算得 到的各化合物浓度除以式I化合物在药物组合物中的浓度,即得到药物组合物中以式I化合物含量计的式VI化合物、式VII化合物和式IV化合物的含量。
如本文所用,术语“药学上可接受的载体”指用于治疗剂给药的载体,包括各种赋形剂和稀释剂。该术语指这样一些药剂载体:它们本身并不是必要的活性成分,且施用后没有过分的毒性。合适的载体是本领域普通技术人员所熟知的。在Remington′s Pharmaceutical Sciences(Mack Pub.Co.,N.J.1991)中可找到关于药学上可接受的赋形剂的充分讨论。在组合物中药学上可接受的载体可包括液体,如水、盐水、甘油和乙醇。另外,这些载体中还可能存在辅助性的物质,如崩解剂、润湿剂、乳化剂、pH缓冲物质等。
附图说明
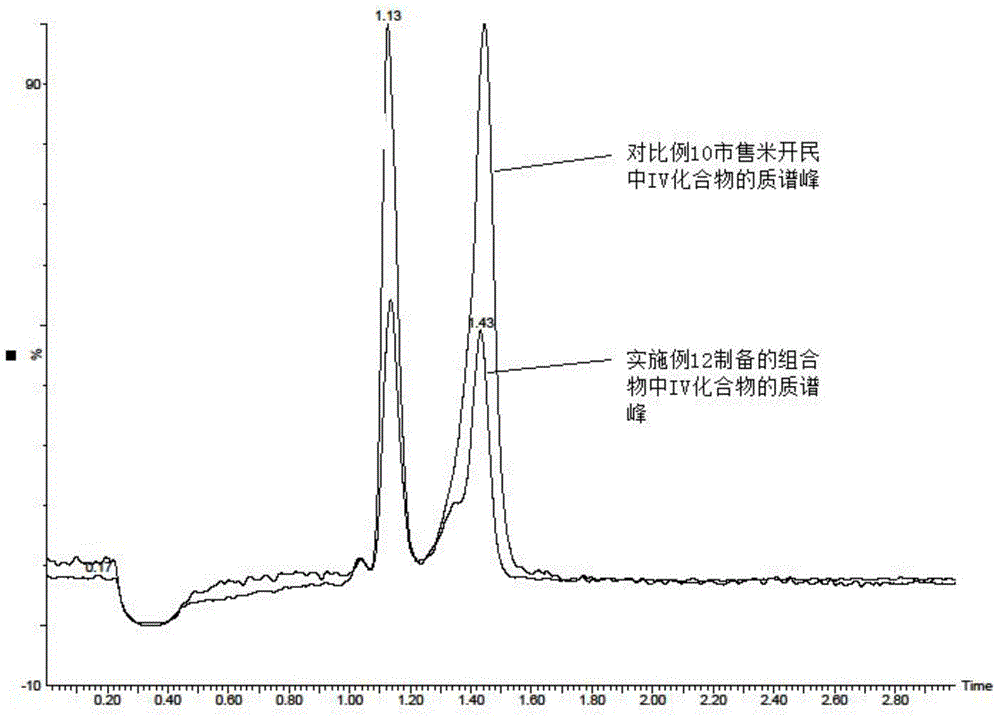
图1显示了本发明实施例12制备得到的含式VIII化合物的组合物中式IV化合物和对比例10中市售米开民中式IV化合物的质谱图。
图2显示了本发明实施例12制备得到的含式VIII化合物的组合物中式VI化合物的质谱图。
图3显示了本发明实施例12制备得到的含式VIII化合物的组合物中式VII化合物的质谱图。
图4显示了本发明对比例10中市售米开民中式VI化合物的质谱图。
图5显示了对比例10中市售米开民中式VII化合物的质谱图。
具体实施方式
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。除非另外说明,否则所有的百分数、比率、比例、或份数按重量计。
本发明中的重量体积百分比中的单位是本领域技术人员所熟知的,例如是指在100毫升的溶液中溶质的重量。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于 本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
实施例1
制备化合物VIII粗品
按照WO2004014879A1实施例5的方法制备得到式VIII化合物的固体,其式IV、VI和VII化合物的含量分别为200ppm、20ppm、60ppm,澄清度检查有明显的“乳光”现象,浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为95.6%。
实施例2
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,25℃下溶解于100ml 95%甲醇水溶液(19∶1)中,调节pH值到7.0。过0.2um膜微滤后,缓慢流加100ml乙酸乙酯,控制温度在20℃,溶液中有固体析出,并保持此温度继续搅拌1h大量固体析出,缓慢流加700ml丙酮/乙酸乙酯混合液(1∶1),过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为20ppm、0.5ppm和8ppm。
实施例3
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,30℃下溶解于80ml乙醇水溶液(乙醇∶水=10∶1)中,调节pH值到6.0。缓慢加入加500ml乙酸乙酯,过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为60ppm、5ppm和17ppm。
实施例4
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,20℃下溶解于90ml正丙醇水溶液(正丙醇∶水=11∶1)中,调节pH值到6.5。缓慢加入500ml乙酸 正丙酯,过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为8ppm、0.4ppm和4ppm。
实施例5
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,40℃下溶解于70ml异丙醇水溶液(异丙醇∶水=20∶1)中,调节pH值到8.5。降温至-10℃搅拌3h,大量固体析出,过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为100ppm、10ppm和30ppm。
实施例6
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,25℃下溶解于100ml乙醇/异丙醇水溶液(乙醇∶异丙醇∶水=5∶5∶1)中,调节pH值到6.5。过0.2um膜微滤后,降温至-5℃搅拌3.5h,大量固体析出,过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为50ppm、1ppm和10ppm。
实施例7
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,20℃下溶解于120ml甲醇/正丙醇水溶液(甲醇∶正丙醇∶水=5∶10∶1)中,调节pH值到6。缓慢加入600ml乙酸甲酯,过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为9ppm、0.3ppm和0.1ppm。
实施例8
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,45℃下溶解于750ml正丙醇/异丙醇水溶液(正丙醇∶异丙醇∶水=50∶50∶1)中,调节pH值到8。降温至35℃,溶液中有固体析出,并保持此温度继续搅拌1h大量固体析出,缓慢 加入3500ml正丙醇/乙酸甲酯混合溶液(4∶3),过滤得到含式VIII化合物的组合物,其中式IV、VI和VII的含量分别为80ppm、8ppm和9ppm。
实施例9
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,25℃下溶解于200ml甲醇/乙醇水溶液(甲醇∶乙醇∶水=20∶30∶1)中,调节pH值到7.5。降温至-20℃,溶液中有固体析出,并保持此温度继续搅拌2.5h大量固体析出,缓慢加入400ml异丙醇/乙酸正丙酯混合溶液(1∶3),过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为5ppm、0.2ppm和5ppm。
实施例10
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,20℃下溶解于120ml乙醇/正丙醇水溶液(乙醇∶正丙醇∶水=20∶5∶1)中,调节pH值到5.5。降温至20℃,溶液中有固体析出,并保持此温度继续搅拌4h大量固体析出,缓慢加入500ml乙酸异丙酯,过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为80ppm、1ppm和10ppm。
实施例11
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,45℃下溶解于90ml甲醇/异丙醇水溶液(甲醇∶异丙醇∶水=10∶10∶1)中,调节pH值到5。降温至30℃,溶液中有固体析出,并保持此温度继续搅拌1.5h大量固体析出,缓慢加入800ml异丁醇/乙酸异丙酯混合溶液(1∶1),过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为30ppm、0.8ppm和6ppm。
实施例12
制备含式VIII化合物的组合物
将实施例2获得的含式VIII化合物的组合物按照实施例2的方法重复一次,得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为1ppm、0.04ppm和0.01ppm。
实施例2至实施例12制备的含式VIII化合物的组合物,澄清度检查全部合格,没有明显的“乳光”现象。浓度分别为10mg/ml、20mg/ml、50mg/ml、100mg/ml的式VIII化合物溶液在410nm下的透光率,如下表所示:
实施例13
制备含式I化合物的组合物
取实施例2、5、8、12制备得到的式VIII化合物的组合物各5g,参照CN102627688A实施例20的方法制备得到含式I化合物的组合物,其中式IV、VI和VII化合物的含量分别如下表所示:
实施例13制备的含式I化合物的组合物,澄清度检查全部合格,没有明显的“乳光”现象。浓度分别为10mg/ml、20mg/ml、50mg/ml、100mg/ml的式I化合物溶液在410nm下的透光率,如下表所示:
对比例1
制备含式VIII化合物的组合物
按照WO2004014879A1实施例5和实施例6中的方法制备得到式VIII化合物,其中式IV、VI和VII化合物的含量分别为900ppm、47ppm和250ppm,澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为93.7%。
对比例2
制备含式VIII化合物的组合物
将对比例1所得到的组合物按照WO2004014879A1实施例6中纯化化合物(5)的方法进行纯化,得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为190ppm、18ppm和57ppm,澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为96.2%。
对比例3
制备含式I化合物的组合物
按照WO2004014879A1实施例6的方法制备得到含式I化合物的组合物,其中式IV、VI和VII化合物的含量分别为146ppm、16ppm和42ppm,澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为96.4%。
对比例4
制备含式I化合物的组合物
将对比例3获得的组合物按照WO2004014879A1实施例6中纯化化合物(6)的方法进行纯化,得到含式I化合物的组合物,其中式IV、VI和VII化合物的含量分别为146ppm、15ppm和41ppm,澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为96.5%。
对比例5
制备含式I化合物的组合物
将对比例3获得的组合物,按照WO03/018615A1实施例1的方法重结晶后得到含式I化合物的组合物,其中式IV、VI和VII化合物的含量分别为141ppm、15ppm和37ppm,澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为96.4%。
对比例6
制备含式VIII化合物的组合物
取按照CN102627688A实施例19的方法制备得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为150ppm、43ppm和49ppm,澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为96.5%。
对比例7
制备含式I化合物的组合物
取按照对比例6方法制备得到的5g样品,按照CN102627688A实施例20的方法制备得到含式I化合物的组合物,其中式IV、VI和VII化合物的含量分别为129ppm、33ppm和39ppm,澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为96.8%。
由对比例1-7的结果可以看出,采用现有技术的纯化及制备方案无法有效去除式IV、VI和VII化合物,而且即使将现有技术的纯化及制备方案进行重复,效果也不明显,无法获得澄清度检查合格的产品。
对比例8
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,25℃下溶解于200ml甲醇/乙醇水溶液(甲醇∶乙醇∶水=60∶50∶1)中,调节pH值到7.5。降温至-20℃,溶液中有固体析出,并保持此温度继续搅拌2.5h大量固体析出,缓慢加入400ml异丙醇/乙酸正丙酯混合溶液(1∶3),过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为181ppm、20ppm和48ppm。澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为96.3%。
对比例9
制备含式VIII化合物的组合物
将由实施例1制备得到的式VIII化合物粗品10g,40℃下溶解于70ml异丙醇水溶液(异丙醇∶水=1∶20)中,调节pH值到8.5。降温至15℃搅拌3h,大量固体析出,过滤得到含式VIII化合物的组合物,其中式IV、VI和VII化合物的含量分别为192ppm、18ppm和59ppm。澄清度检查有明显的“乳光”现象。浓度为10mg/ml的式VIII化合物溶液,在410nm下的透光率为96.2%。
由此可见,水与醇的比例超出本发明保护的范围时无法获得晶体,也无法达到去除杂质的效果。
实施例14
制备药物组合物
按照专利CN100352495C实施例1进行药物组合物的制备,配方(1)如下:
将乳糖在低于50℃加热下溶于纯水(200ml)。冷却至20℃以下后,向乳糖溶液中加入实施例13获得的含式I化合物的组合物,在温和搅拌下避免产生气泡。在加入2%柠檬酸水溶液(0.95ml)后,向溶液中加入0.4%氢氧化钠水溶液(约24ml),以调节pH5.5,然后用纯水稀释,产生给定的体积(250ml)。将所得的溶液分装到100个10ml体积的管形瓶中,每个管形瓶2.5ml。用常规方法,用冻干机将各个管形瓶中的溶液冻干,以获得冻干组合物,组合物中含式I化合物的组合物中,式IV、VI和VII化合物的含量分别如下表所示:
浓度分别为10mg/ml、20mg/ml、50mg/ml、100mg/ml的药物组合物溶液在410nm下的透光率,如下表所示:
实施例15
制备药物组合物
按照实施例14制备各含25mg的米卡芬净钠的冻干组合物(配方2),只是用15g麦芽糖代替乳糖。所得冻干组合物,式IV、VI和VII化合物的含量(以式I化合物的含量计)同实施例14测定值一致。
将蔗糖溶于纯水(100ml)。向蔗糖溶液中加入实施例13获得的含式I化合物的组合物,在温和搅拌下避免产生气泡。在加入2%柠檬酸水溶液(0.48ml)后,向溶液中加入0.4%氢氧化钠水溶液(约12ml),以调节pH5.5,然后用纯水稀释,产生给定的体积(125ml)。将所得的溶液分装到50个10ml体积的管形瓶中,每个管形瓶2.5ml。用常规方法,用冻干机将各个管形瓶中的溶液冻干,以获得冻干组合物。所得冻干组合物,式IV、VI和VII化合物的含量(以式I化合物的含量计)同实施例14测定值一致。
实施例16
制备药物组合物
将海藻糖溶于纯水(100ml)。向海藻糖溶液中加入实施例13获得的含式I化合物的组合物,在温和搅拌下避免产生气泡。在加入2%柠檬酸水溶液(0.48ml)后,向溶液中加入0.4%氢氧化钠水溶液(约12ml),以调节pH5.5,然后用纯水稀释,产生给定的体积(125ml)。将所得的溶液分装到50个10ml体积的管形瓶中,每个管形瓶2.5ml。用常规方法,用冻干机将各个管形瓶中的溶液冻干,以获得冻干组合物。所得冻干组合物,式IV、VI和VII化合物的含量(以式I化合物的含量计)同实施例14测定值一致。
实施例14至实施例16制备的药物组合物,澄清度检查全部合格,没有明显的“乳光”现象。由此可见药物组合物中赋形剂的种类,对其澄清度无影响。
对比例10
与市售制剂产品杂质含量比较
市售制剂产品:商品名称:米开民
通用名:注射用米卡芬净钠
厂家:安斯泰来富山株式会社
批号:022510
其中,市售制剂产品米开民中,按照式I化合物含量计,式IV、VI和VII化合物的含量分别为150ppm,12ppm,32ppm,其澄清度检查中,有乳光现象。浓度为10mg/ml的药物组合物(以式I化合物的含量计)溶液,在410nm下的透光率为97.3%。
以上所述仅为本发明的较佳实施例而已,并非用以限定本发明的实质技术内容范围,本发明的实质技术内容是广义地定义于申请的权利要求范围中,任何他人完成的技术实体或方法,若是与申请的权利要求范围所定义的完全相同,也或是一种等效的变更,均将被视为涵盖于该权利要求范围之中。
- 还没有人留言评论。精彩留言会获得点赞!