新型阳离子聚磷腈化合物、聚磷腈‑药物偶联化合物和其制备方法与流程
本发明涉及高肿瘤选择和生物相容性阳离子直链聚磷腈载体聚合物和其抗癌药物偶联物,和其制备方法。
背景技术:
目前在临床上用于化疗的大多数的抗癌药物为小于1000Da的低分子量的单体化合物。众所周知,由于此类单体低分子量抗癌药物在静脉注射时对肿瘤细胞和组织的无选择性,会引起严重的毒性和副作用,此外,它们在血液循环中的半衰期低于几小时,限制了它们可持续性药效。因此,在新型抗癌药物开发中待攻克的最重要关键技术是将抗癌药物选择性递送到肿瘤部位的肿瘤靶向技术和抗癌药物的活性组分在肿瘤部位的及时释放技术。在过去的几十年中,全世界为克服此限制已经作出了大量的工作,并由此已经发现聚合物药物递送系统是带来突破的最有效和实际的方法之一,由其出现了被称为“聚合物疗法”的新领域(R.Haag,F.Kratz,Angew.Chem.Int.Ed.45(2006)1198-1215)。
在药物递送系统中采用的大多数聚合物都是合成或天然的有机聚合物。许多天然和合成的聚合物都被尝试用作聚合物疗法中的药物递送系统,但是发现只有有限的药物递送系统才是有效的,因为除了前文所述的肿瘤靶向性和释放动力学之外,癌症的聚合物疗法中还需要满足诸如水溶性、生物可降解性、自毒性、与所负载药物的相容性等要求。
发明人在几十年前发现了相对于上述有机聚合物,通过将各种有机基团接枝到由交替的氮原子和磷原子组成的被称作磷腈的无机聚合物主链上而设计出了一类新型有机/无机杂化聚合物(Y.S.Sohn等人,Macromolecules,1995,28,7566),已将其作为癌症治疗的药物递送系统进行了深入开发。在早期,将各种亲水性聚(乙二醇)(PEG)和疏水性寡肽引入到磷腈主链上,而得到了用于热敏性药物递送系统的两亲性聚磷腈类。同时还发现,该两亲性聚磷腈类是自组装成各种纳米结构,如有助于在水溶液中递送药物的热敏性胶束和水凝胶,但是也观察到,由于将一些疏水性寡肽接枝到聚磷腈主链上,使得水溶性下降并产生了一定的毒性。通常已知的是,当缓慢加热此类两亲性聚合物时,其展现出低临界溶解温度(LCST),在该温度下聚合物开始从其水溶液中沉淀出来。因此,两亲性聚合物药物递送系统应具有高于体温的LCST以用于静脉注射,从而避免其在血液循环过程中的沉淀。
关于疏水性抗癌药物,包括紫杉醇和多西紫杉醇的紫杉烷族是最广泛用于包括乳腺癌、卵巢癌和非小细胞肺癌的广谱癌症的有效化疗的一种。由于紫衫烷抗癌剂仅微溶于水(<1μg/ml),因此它们不能被直接注射,而是应该采用表面活性剂(诸如聚山梨酯80或克列莫佛EL和乙醇)配制后进行IV注射。然而,这样配制的紫杉烷抗癌剂展示出包括由抗癌剂本身引起的神经毒性和中性粒细胞减少症和由于溶剂系统引起的超敏反应的若干不良反应,这限制了它们在临床上的广泛使用。
因此,在过去的十年里,各个领域中都作出大量的研究来攻克此类不良反应,在这些研究中,采用各种形貌结构的纳米技术取得了最积极的进展。特别地,由亲水性外壳和疏水性核构成的聚合物胶束可以通过将疏水性抗癌分子装入疏水性胶束核而使疏水性抗癌药物(如紫杉烷)溶解。此外,通过将紫杉烷药物分子化学性连接至亲水性聚(乙二醇)可以将其偶联以溶解,这正处于临床试验阶段。
此类由偶联至聚合物药物递送系统的小分子抗癌药物组成的聚合物前药有望延长其血液循环时间并通过提高的通透性和滞留(EPR)效应(H.Maeda等人,J.Control.Release 65(2000)271-284)连同控制药物释放而提供肿瘤靶向性,以产生最大的药效和最低的毒性。根据最近的报道,为了通过EPR效应实现肿瘤靶向性,聚合物颗粒的尺寸应该在50-200nm的范围内(V.P.Torchilin,J.Control.Release 73(2001)137-172)。另外,基因递送的研究中报道,阳离子聚合物可以容易地渗入阴离子肿瘤细胞(N.P.Gabrielson,D.W.Park,J.Control.Release 136(2009)54-61)。然而,由将偶联至聚(谷氨酸)的紫杉醇构成的聚谷氨酸(Poliglumex)目前在临床III期是强阳离子型的,但已知的是,它在其它器官上的聚积比在肿瘤组织上更多,这延缓了它的商业化(S.Wallace;C.Li,Adv.Drug Deliv.Rev.60(2008)886-898)。
[现有技术文件]
[非专利文件1]
R.Haag,F.Kratz,Angew.Chem.Int.Ed.45(2006)1198-1215)
技术实现要素:
技术问题
因此,本发明的目的是提供一类新型的用于药物递送的阳离子聚磷腈类化合物,和其具有优异的肿瘤选择性和在肿瘤部位的容易的药物释放性的抗癌药物偶联化合物,和它们的制备方法。
本发明的发明人一直在寻找在上述技术背景下具有优异肿瘤选择性和容易的药物释放性的更有效的临床药物递送系统,并且最终发现,接枝了作为增溶基团的亲水性PEG并接枝了选自多官能团氨基酸、包含氨基酸或直链氨基醇的寡肽之一作为间隔基团以与疏水性的抗癌药物偶联的聚磷腈类,由于其阳离子性和较长的血液循环,展现出优异的肿瘤选择性。本发明的发明人还发现,当利用于酸可裂解的连接基团将疏水性抗癌药物偶联至上述聚磷腈载体聚合物时,通过酸降解得到了在肿瘤部位的具有优异肿瘤选择性和可控释放性的智能聚合物抗癌药。
技术方案
为了实现上述目标,本发明提供了由以下化学式1表示的直链聚磷腈类。
[化学式1]
其中n是1至300的整数;OMPEG表示具有350至1000分子量的甲氧基聚(乙二醇);S是选自由赖氨酸、精氨酸、谷氨酰胺、天冬酰胺、酪氨酸、含有赖氨酸的寡肽、含有精氨酸的寡肽、含有谷氨酰胺的寡肽、含有天冬酰胺的寡肽、含有酪氨酸的寡肽、氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇组成的组的间隔基团;l等于0-0.9;m等于0.1-1,且l+m等于1。
本发明还提供了由以下化学式2表示的聚磷腈-抗癌药物偶联物。
[化学式2]
其中n是从1至300的整数;OMPEG表示具有350至1000分子量的甲氧基聚(乙二醇);S是选自由赖氨酸、精氨酸、谷氨酰胺、天冬酰胺、酪氨酸、含有赖氨酸的寡肽、含有精氨酸的寡肽、含有谷氨酰胺的寡肽、含有天冬酰胺的寡肽、含有酪氨酸的寡肽、氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇组成的组的间隔基团;L是连接聚合物的间隔基团和具有羟基或胺基团的药物分子D的连接基团;x和y独立地在0-0.5的范围内;z在0-1的范围内;且x+y+z=1。
此外,本发明提供制备由化学式2表示的聚磷腈-药物偶联化合物的方法,其包括步骤(a)至步骤(d):
(a)通过使起始物料六氯环三磷腈进行热聚合得到直链聚(二氯磷腈)并继而使该直链聚(二氯磷腈)与甲氧基聚(乙二醇)的钠盐反应,而制备出PEG化的聚磷腈中间体,
(b)使PEG化的聚磷腈中间体与选自由赖氨酸酯、含有赖氨酸的寡肽酯、氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇组成的组的间隔基团反应,而制备出亲水性阳离子聚磷腈载体聚合物,
(c)通过使具有OH或NH2官能团的药物分子与合适的连接基团反应,而制备出药物-连接基团前体;
(d)将由步骤(c)得到的药物-连接基团前体连接到由步骤(b)得到的亲水性阳离子聚磷腈载体聚合物的间隔基团上。
可选地,本发明提供了制备由化学式2表示的聚磷腈-药物偶联化合物的方法,其包括步骤(a)至步骤(d):
(a)通过使起始物料六氯环三磷腈进行热聚合得到直链聚(二氯磷腈)并继而使该直链聚(二氯磷腈)与甲氧基聚(乙二醇)的钠盐反应,而制备出PEG化的聚磷腈中间体,
(b)通过使PEG化的聚磷腈中间体与选自由赖氨酸酯、含有赖氨酸的寡肽酯、氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇组成的组的间隔基团反应,而制备出亲水性阳离子聚磷腈载体聚合物,
(c)将连接基团连接到由步骤(b)得到的亲水性阳离子聚磷腈载体聚合物的间隔基团上,
(d)将具有OH或NH2官能团的药物分子连接到由步骤(c)得到的连接至亲水性阳离子聚磷腈载体聚合物的连接基团上。
有益效果
本发明的阳离子直链聚磷腈化合物显示出高肿瘤选择性,并且发现,相较于传统有机聚合物-药物偶联物,本发明的直链聚磷腈-药物偶联化合物在肿瘤组织中比在其它主要器官(如肝脏和肾脏)中有更高选择性地聚积。此外,本发明的直链聚磷腈-药物偶联化合物在中性血液和器官中较为稳定而不会释放偶联的药物,而在酸性肿瘤微环境中其更容易释放,从而得到最高的药效和最小的毒性。因而,本发明的阳离子直链聚磷腈化合物和其抗癌药物偶联化合物是具有高度商业化前景的新材料。
附图说明
图1示出了示例1的阳离子聚磷腈化合物的粒径分布(平均直径=3.0nm)。
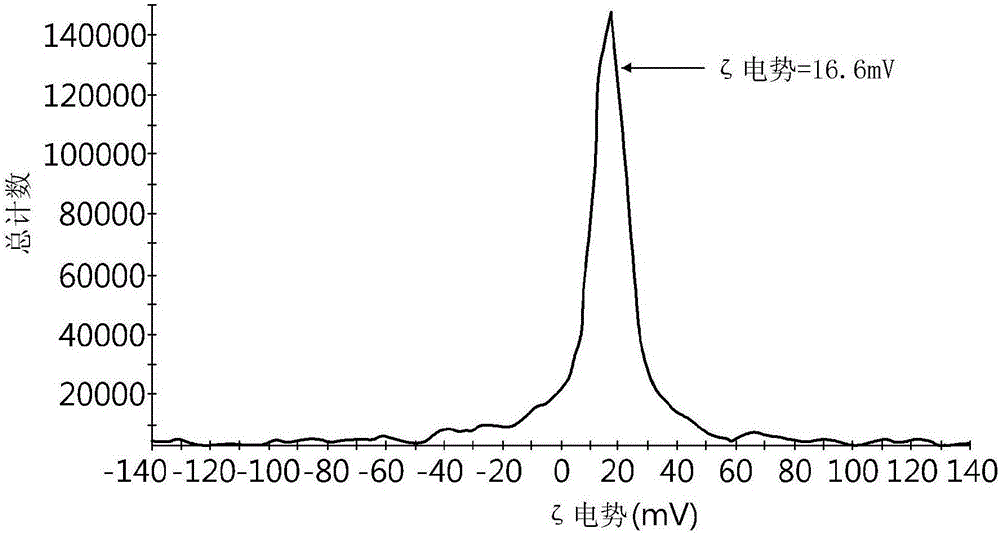
图2示出了示例1的阳离子聚磷腈化合物的所测得的ζ电位。
图3示出了示例12的聚磷腈-多西紫杉醇偶联物的粒径分布(平均直径=60nm)。
图4示出了由荧光芘法测定的示例17的聚磷腈-多西紫杉醇偶联物的临界胶束浓度(CMC)。
图5示出了示例17的聚磷腈-紫杉醇偶联物在酸性pH 5.4和中性pH 7.4的时间依赖性降解。
图6(a)示出了,示例1中接种了非小细胞肺肿瘤细胞A549的小鼠的主要器官中(1:肝;2:肺;3:肾;4:脾;5:肿瘤;6:肌肉)Cy标记的聚磷腈-多西紫杉醇偶联物随时间依赖性(注射12h、24h、48h、72h后)生物分布的体外NIR荧光图像,(b)示出了血液(WB)和血浆(PL)的时间依赖性NIR荧光图像。
图7示出了,示例12中接种了SCC7肿瘤细胞的小鼠在IV注射后主要器官(1:肝;2:肺;3:脾;4:肾;5:肿瘤;6:心脏)中Cy标记的聚磷腈-多西紫杉醇偶联物时间依赖性(注射24h和48h后)的生物分布。
图8示出了与未采用药物处理的小鼠的各个器官相比,在图7中处理过的小鼠的各个器官的时间依赖性荧光强度。
图9示出了采用Sprague-Dawley大鼠,在IV注射后,示例12(■)中聚磷腈-多西紫杉醇偶联物和作为参照的泰索帝(◆)的药物代谢动力学研究结果中多西紫杉醇的平均血浆浓度-时间曲线。
图10示出了,示例12中采用BALB/C裸鼠和MKN-28胃癌细胞系的聚磷腈-多西紫杉醇偶联物的异种移植试验的结果。
图11示出了图10中异种移植试验期间(40天)小鼠体重的变化。
图12示出了,示例14中采用BALB/C裸鼠和非小细胞癌A549细胞系的聚磷腈-多西紫杉醇偶联物的异种移植试验的结果。
具体实施方案
最佳方式
提供本发明的组成和更详细的手段。本发明体现在以下描述中实施方案,但并不限于此。本发明的详细组成和手段在下文中举例说明。为了实现上述目的,提供了由以下化学式1表示的聚磷腈化合物。
[化学式1]
其中n是1至300的整数;OMPEG表示具有350至1000分子量的甲氧基聚(乙二醇);S是选自由赖氨酸、精氨酸、谷氨酰胺、天冬酰胺、酪氨酸、含有赖氨酸的寡肽、含有精氨酸的寡肽、含有谷氨酰胺的寡肽、含有天冬酰胺的寡肽、含有酪氨酸的寡肽、氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇组成的组的间隔基团;l等于0-0.9;m等于0.1-1,且l+m等于1。
上述本发明的聚磷腈化合物包括亲水性和多官能化的赖氨酸或含有赖氨酸的亲水性寡肽作为侧基基团,连同包括亲水性MPEG作为另一侧基基团,显示出高于100℃的较低临界溶解温度(LCST),此LCST远高于体温,而常规两亲性聚磷腈的LCST低得多并接近体温。此外,本发明的聚磷腈化合物显著地延长了血液半周期并展示出较低的全身性毒性,且更令人惊奇的是该聚磷腈化合物本身明显地显示出较高的肿瘤靶向性,这可能是因为尽管其较小的颗粒尺寸(平均直径<6nm),其阳离子性质和长血液循环的原因。亲水性寡肽的代表性例子是甘氨酰赖氨酸。
本发明还提供了由以下化学式2表示的聚磷腈-抗癌药物偶联物。
[化学式2]
其中n是从1至300的整数;OMPEG表示分子量为350至1000的甲氧基聚(乙二醇);S是选自由赖氨酸、精氨酸、谷氨酰胺、天冬酰胺、酪氨酸、含有赖氨酸的寡肽、含有精氨酸的寡肽、含有谷氨酰胺的寡肽、含有天冬酰胺的寡肽、含有酪氨酸的寡肽、氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇组成的组的间隔基团;L是连接聚合物间隔基团和具有羟基或胺基团的药物分子D的偶联基团;x和y独立地在0-0.5的范围内;z在0-1的范围内;且x+y+z=1。
对于本发明的实施方案,S表示赖氨酸或含有赖氨酸的二肽或三肽,但并不仅限于此。本发明的另一个实施方案中,S表示氨基乙醇和氨基丙醇,但不限于此。期望的药物分子D为疏水性抗癌剂,诸如多西紫杉醇、紫杉醇、喜树碱和(反式-(±)-1,2-二氨基环己烷)铂(II),但不限于此。根据上述化学式2,本发明的实施方案可以由下列化学式19至21之一来表示。
[化学式19]
[化学式20]
在化学式19和20中,n是独立地从3至300的整数;OMPEG表示具有350至1000分子量的甲氧基聚(乙二醇);D表示多西紫杉醇、紫杉醇、喜树碱和(反式-(±)-1,2-二氨基环己烷)铂(II);R为C1-6直链、支化或环状的烷基基团或OCH2Bz;x和y独立地在0-0.5的范围内;z大于0且小于1.0;x+y+z=1。
[化学式21]
其中n是从3至300的整数;OMPEG表示具有350至1000分子量的甲氧基聚(乙二醇);D表示多西紫杉醇、紫杉醇或喜树碱;R’表示t-Boc或CBZ;x和y独立地在0-0.5的范围内;z大于0且小于1.0;x+y+z=1。
本发明的构成组分在下文中说明。
[聚磷腈载体化合物]
本发明的药物载体聚磷腈化合物是独特的无机/有机杂化聚合物,其由交替的氮原子和磷原子组成的无机聚合物主链构成,并接枝了两个有机基团,一个是为实现长血液循环的亲水性聚(乙二醇)和另一个是为了与疏水性抗癌药物偶联的亲水性多官能化的赖氨酸、含有赖氨酸的寡肽或直链氨基醇作为间隔基团。本发明的发明人发现,接枝到聚磷腈主链上的赖氨酸基团或含有赖氨酸的肽能根据氨基酸的pKa值提供聚磷腈化合物的阳离子性质,其中氨基酸的pKa值可以通过作为间隔基团的氨基酸来控制。在本发明中使用的聚(乙二醇)是分子量在300~2000范围内的甲氧基聚(乙二醇),且其含量是由它与间隔基团的摩尔比决定的,而该摩尔比是由x和y确定,并且x取决于最终偶联药物的所需性质(诸如溶解性、形貌和生物可降解性)。聚磷腈化合物的分子量可以通过重复单元n,也可以通过PEG的分子量来控制。与支化的直链有机聚合物相比,本发明的具有两个有机侧基基团的直链聚磷腈化合物具有较高的分子量,但较低的水合量,从而产生了更高的原子密度和更好的肿瘤选择性。
[抗癌药物]
与本发明的支化的聚磷腈化合物偶联的抗癌活性药物成分应该至少具有一个羟基(OH-)基团或胺基(NH2)基团的官能团,且更希望的是其为诸如紫杉烷、喜树碱和铂(II)的药物的疏水性抗癌药物,但并不限于此。除了这些小分子抗癌药物,任何带有至少一个羟基或氨基官能团的抗癌药物分子都可以利用本发明中采用的合适的间隔基团和偶基团系统与本发明聚磷腈载体聚合物偶联。本发明的示例将通过展示两种显示出较低空间位阻的抗癌药物来说明。
通过将疏水性紫杉烷药物分子化学性偶联至亲水性聚磷腈载体聚合物而制备的聚磷腈-紫杉烷偶联化合物是一类用于静脉注射的新型聚合物前药,它们的分子量控制为3000至300,000Da,但我们发现,30,000至100,000Da的级分对于偶联药物的生物相容性和功效是最优的。本发明的亲水性聚磷腈载体聚合物不能在水介质中形成聚合物胶束,但是却发现其与疏水性紫衫烷分子化学连接的偶联物能自组装形成平均直径在20-100nm范围内的强聚合物胶束,如上所述它们通过EPR效应表现出优异的肿瘤靶向性连同由于胶束的PEG外壳而导致了长血液循环。最后,为了实现聚磷腈-紫杉烷偶联物的最大药效和最低全身性毒性,采用酸(pH=4-7)可裂解的连接基团乌头酸酐将抗癌药物成分紫杉烷分子连接至载体聚磷腈,以使得强细胞毒性的紫杉烷药物分子在中性血液系统(pH=7.2)的循环中不会从聚磷腈-紫杉烷偶联物中显著地释放出来,但在酸性肿瘤微环境的肿瘤部位却容易地释放出来。
如化学式3所示,多西紫杉醇药物分子在其2、7、10和2’位置处具有四个羟基基团,但2’位的羟基基团是已知最活跃的,因此,通过酯化反应将该药物分子连接至连接基团乌头酸酐的羧酸基团而形成了前体,之后使该前体反应形成了与聚磷腈载体聚合物的间隔基团氨基酸或氨基乙醇的具有胺基团的酰胺键。在紫杉醇的情况下,可以采用相同的反应方案。此处应当指出采用多功能化的乌头酸酐作为用于将紫杉烷药物分子偶联至本发明的聚磷腈化合物的连接基团的两个重要原因。众所周知,当将空间位阻的药物分子引入以通过酯化反应偶联至聚合物药物载体,其产率不仅非常低,而且可能产生立体异构体。从初步的研究发现,当将多西紫杉醇直接引入到连接了聚磷腈载体聚合物的乌头酸酐上时,不仅需要超过24小时长的反应时间,而且导致产生了一些立体异构体。然而,本发明的发明人发现,当多西紫杉醇先与连接基团乌头酸酐反应制备前体时,将其与本发明的聚磷腈载体聚合物反应,可以得到高产率和高纯度的最终的聚磷腈-多西紫杉醇偶联物。
[化学式3]
本发明涉及喜树碱系列的下文示例性化合物中,但并不限于此,其包括所有药学活性的衍生物,特别是伊立替康、托泊替坎和贝洛替康。
[间隔基(S)]
在本发明中,将多功能化间隔基团引入到聚磷腈载体聚合物以将它们化学地连接到连接基团乌头酸酐上,实现与抗癌药物的偶联。间隔基团应包含伯胺基团来连接上述连接基团和另一个伯胺或醇基基团来将其接枝到聚磷腈主链,并且,该间隔基团是选自赖氨酸、精氨酸、谷氨酰胺、天冬酰胺、酪氨酸的多官能化的氨基酸基团和包含这些氨基酸的寡肽或直链氨基醇类的一种。上述氨基酸被归类为基础氨基酸类,且当将此类基础氨基酸引入到聚磷腈主链,根据氨基酸的pKa值,在特定的pH范围内,可赋予聚磷腈化合物阳离子特性。间隔基团不仅可以是单个氨基酸,也可以是多于两个氨基酸的组合。例如,可以采用甘氨酰赖氨酸(Gly-Lys)、丙氨酰赖氨酸(Ala-Lys)或三肽。此外,对于某些多官能化的氨基酸或肽,可以使羧酸酯基团水解以便与药物分子的直接偶联而无需连接基团。此代表性示例是赖氨酸,这将在实施例中进行说明。
[连接基团]
众所周知,聚合物药物偶联物的药效严重依赖于聚合物载体中活性药物分子的释放动力学。在本发明中,根据药物和载体系统的分子结构,通过适当选择间隔基团和连接基团乌头酸酐,利用酰胺键(-CONH-)或酯键(-COO-)将活性药物分子偶联至聚磷腈载体聚合物,这与仅对带有伯胺基团的药物分子有用的常规方法不同。因此,本发明提供了用于为药物偶联引入酸可裂解连接基团的新型、升级的间隔基-连接基团系统。
前述偶联反应可以在包括二氯甲烷、氯仿、乙腈、1,4-二恶烷、二甲基甲酰胺和四氢呋喃的对偶联反应为惰性的任何有机溶剂中,在通常的偶联反应条件下进行。但是,由于在引入连接基团的过程中形成的反应中间体的不稳定性,整个反应在干燥的溶剂中并在干燥的氩气气氛和低温下(-10-0℃)进行,以提高反应效率。特别是,已知顺式乌头酸酐连接基团会在碱性溶液中进行酸酐开环反应,从而产生异构体,并因此,在中性条件下采用DPTS(二甲基氨基吡啶/对-甲苯磺酸)作为催化剂进行反应,来代替碱性催化剂DMAP(二甲基氨基吡啶)。适于制备聚磷腈偶联化合物的代表性药物是寡肽、多肽、单体药物、抗体、核苷酸,脂质类和其它带有-OH或NH2官能团的材料。
在本发明中可用的连接基团列于以下化学式4至8中,而化学式4是最优选的。
[化学式4]
[化学式5]
[化学式6]
[化学式7]
[化学式8]
在采用上述化学式7的酸可裂解(pH=5.5)的乌头酸酐作为连接基团的情况下,聚合物偶联药物可以根据下述反应方案3来制备。
[反应方案3]
然而,在该反应方案中形成了不能释放药物部分的非活性异构体,且根据偶联位点的不同,药物释放速率和药效都难以控制。特别是,当间隔基团/连接基团以及连接基团/药物的化学键都是酰胺键时,可能会形成通过水解难以释放药物分子的异构体(反应方案1非活性),这导致了低的药效。在反应方案4间隔基团/连接基团以及连接基团/药物的化学键都是酯键的情况下,尽管通过酶水解可能可行,但是依然会导致非活性体的形成。此外,当采用药物分子的OH基团进行酸酐开环反应时,特别对于空间位阻的药物应该采用大量过量的亲核剂。在本发明的紫杉烷药物的情况下,大约需要20倍过量的量才能完成反应。在此情况下,发现最终聚磷腈-药物偶联物的产率低于5%,这在商业上是不可行的。
[反应方案4]
在本发明中发现,以下反应方案5现实,代替乌头酸酐的开环反应,使其羧基基团与药物分子(HOR’)的羟基基团反应以形成酯键,从而得到药物前体。随后通过采用载体聚合物的间隔胺基团进行酸酐开环反应而将该前体偶联至药物载体聚合物(RNH2)。乌头酸酐的开环反应也可以采用上述反应方案中所示的过量的NHS(N-羟基琥珀酰亚胺)来进行。此合成路线与上述方法相比提供了重要的优势。首先,可以避免上述反应方案1和2中所示的非活性异构体,从而得到更高产率的最终偶联药物,且该偶联药物的纯度以及药效都可以得到合理的控制。
[反应方案5]
本发明还提供了直链聚磷腈化合物和聚磷腈-药物偶联化合物的详细合成方法。在这方面,会对药物、间隔基团和连接基团的具体示例进行说明,但本发明并不限于这些示例。
本发明的直链聚磷腈化合物是通过将亲水性的聚(乙二醇)(PEG)和带有两个伯胺和一个羧基基团的多官能化的赖氨酸、含有赖氨酸的寡肽或氨基乙醇接枝到二氯磷腈主链(-N=PCl2-)上而合成的。由此制备聚磷腈化合物的多官能化的赖氨酸、含有赖氨酸的寡肽或直链氨基醇可以作为间隔基团,以采用如乌头酸酐的连接基团与如紫杉烷、铂(II)络合物或喜树碱偶联,从而制备出新型的酸可裂解的直链聚磷腈-药物偶联物。
本发明的直链聚磷腈-药物偶联化合物在肿瘤组织中通过选择性聚积显示出了优异的肿瘤选择性,并且发现,通过控制上述化学式1中的x、y和z值,可以制备高水溶性、长血液循环和优异的肿瘤靶向抗癌药物。
在本发明中,期望的选自上述赖氨酸、精氨酸、谷氨酰胺、天冬酰胺或含有酪氨酸的寡肽的间隔基团是赖氨酸和含有甘氨酸的寡肽,例如,二肽和三肽,且更期望的是甘氨酰赖氨酸,但并不限于此。在上述寡肽中,期望赖氨酸位于末端位置,以与紫杉烷抗癌药物偶联。期望的选自直链氨基醇类的间隔基团是氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇,且更能期望的是氨基乙醇和氨基丙醇。
在本发明中,将紫杉烷抗癌药物(D)的分子通过赖氨酸的氨基或羧基基团偶联至聚磷腈载体聚合物,并且为了防止将羧基用于偶联,应利用如t-Boc、FMOC或CBZ的保护基团对胺基团进行保护。在上述化学式1中,L是连接抗癌药物(D)和载体聚合物间隔基团(S)的连接基团并且是顺式乌头酸酐、琥珀酸酐或马来酸酐,并且期望地是顺式乌头酸酐。在本发明中举例说明的抗癌药物组分(D)是紫杉烷族、喜树碱族和包括多西紫杉醇、紫杉醇、喜树碱、托泊替坎、伊立替康、贝洛替康和奥沙利铂的铂络合物,但并不限于这些药物。
本发明的聚磷腈-药物偶联物具有在3,000-300,000范围内的窄分子量分布,并且期望地是在30,000-100,000的范围内,且根据药物分子(D)的疏水性,本发明的聚磷腈-药物偶联物在水中通过自组装成平均直径在20-200nm范围内的聚合物胶束而高度可溶。如上所述,本发明中由聚磷腈-药物偶联物组装而得的此类聚合物胶束由于其EPR效应而显示出优异的肿瘤选择性。
聚磷腈和聚磷腈-紫杉烷偶联化合物可以通过以下四个步骤的合成反应来制备。
(a)通过热聚合,将起始物料六氯环三磷腈转变为直链聚(二氯磷腈)((N=PCl2)n),继而使其与聚(乙二醇)的钠盐反应,而得到中间体聚磷腈,[N=PCl(MPEG)]n;
(b)使上述聚磷腈中间体与选自由赖氨酸酯、含有赖氨酸的寡肽、氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇组成的组中的其中之一反应,而制备出亲水性阳离子聚磷腈载体聚合物;
(c)使带有羟基(OH)或伯胺(NH2)基团的抗癌药物与连接基团(例如乌头酸酐)反应,而制备出前体;以及
(d)最后,使步骤(c)的前体与步骤(b)的聚磷腈载体聚合物反应,而得到化学式2的最终聚磷腈偶联化合物。
[化学式2]
其中n是从1至300的整数;OMPEG表示分子量为350至1000的甲氧基聚(乙二醇);S是选自由赖氨酸、精氨酸、谷氨酰胺、天冬酰胺、酪氨酸、含有赖氨酸的寡肽、含有精氨酸的寡肽、含有谷氨酰胺的寡肽、含有天冬酰胺的寡肽、含有酪氨酸的寡肽、氨基乙醇、氨基丙醇、氨基丁醇、氨基戊醇和氨基己醇组成的组的间隔基团;L是连接聚合物的间隔基团与具有羟基或胺基团的药物分子D的连接基团;x和y独立地在0-0.5的范围内;z在0-1的范围内;且x+y+z=1。
在本发明的另一实施方案中,带有羟基(OH)或伯胺(NH2)基团的抗癌药物可以是选自多西紫杉醇、紫杉醇、喜树碱和(反式-(±)-1,2-二氨基环己烷)铂(II)的组的一种,但不限于这些药物。
另外,可以使制备聚磷腈-药物偶联物的上述四步反应进行取决于待偶联的药物分子的结构的轻微改性。例如,在铂(II)络合物的情况下,在步骤(c)中使连接基团与聚合物载体反应并继而在步骤(D)中将药物连接到与载体聚合物相连的连接基团上,而不是在步骤(c)中通过将药物与连接基团反应而制备前体并继而在步骤(d)中使前体与聚合物载体反应。
所有下述的合成反应过程都应该在小心控制的惰性气氛下,采用干燥的氩气或氮气以及完全干燥的溶剂来进行。详细的合成方法在下文中进行描述。
步骤(a)
六氯环三磷腈((N=PCl2)3)如化学式9所示,将其根据文献方法(Youn Soo Sohn等人,Macromolecules,1995,28,7566)进行热聚合,以得到平均分子量为104-105的聚(二氯磷腈)(N=PCl2)n,如化学式10所示。
[化学式9]
[化学式10]
其中n表示范围为3至300的聚合度。
将化学式11中的单甲氧基聚(乙二醇)用甲苯和水的共沸混合物干燥,然后与金属钠反应,以转化为化学式12中的钠盐,随后使其在三乙胺存在下与化学式10中的聚(二氯磷腈)反应,以完成化学式15中的聚磷腈的PEG化。
[化学式11]
[化学式12]
在上述化学式11和12中,a是PEG的聚合度且为7至22。
在下文中对PEG化的聚磷腈的制备进行更详细地说明。
将化学式9的六氯环三磷腈(N=PCl2)3与3至10%的无水氯化铝在手套箱中混合均匀,随之在真空下被密封在Pyrex安瓿瓶中。在加热室中在转动条件下在3-5h内使该安瓿瓶加热至230–250℃,以得到聚(二氯磷腈)的透明粘性液体。同时,使化学式11的单甲氧基聚(乙二醇)与1.2-1.5当量的金属钠在非反应性无水有机溶剂如四氢呋喃(THF)、苯、甲苯中反应,以得到化学式12的钠盐。向一摩尔的化学式10的聚(二氯磷腈)溶解在相同溶剂的溶液加入上述制备的0.5-1.8当量的化学式12的钠盐。反应溶剂可以是任何非反应性溶剂,但期望的是四氢呋喃、苯、甲苯和氯仿。应将化学式12的钠盐溶液缓慢地在2-8小时内加入冷却至低于0℃的聚(二氯磷腈)溶液中,然后使反应混合物在环境温度下进一步反应6–24h,以得到化学式13的PEG化的聚磷腈中间体。
[化学式13]
其中n是聚磷腈的聚合度,在3至300的范围内,a是甲氧基聚(乙二醇)的聚合度,在7至22的范围内,b是甲氧基聚(乙二醇)的取代摩尔分数,在0.5至1.8的范围内。
步骤(b)
为了用间隔基团取代化学式13的PEG化的聚磷腈的其余的(2-b)氯原子以用于药物偶联,将1.5-1.8当量的赖氨酸酯或含赖氨酸的寡肽酯与6当量的三乙胺一起溶解于任何不参与反应的有机溶剂中,理想的是将四氢呋喃、氯仿或二氯甲烷缓慢地加入到上述化学式13的PEG化的聚磷腈中间体溶液中,然后在40-60℃下回流12h至3天。可以将化学式14的赖氨酸衍生物用作赖氨酸酯,并将化学式15的衍生物用作包含赖氨酸的寡肽酯。在该化学式中,可以用亮氨酸、异亮氨酸、苯丙氨酸和缬氨酸来取代甘氨酸。
[化学式14]
其中R是直链、支化或环状的C1-6烷基基团或OCH2Bz,且R’是胺保护基团,t-Boc(叔丁氧羰基)、Fmoc(芴甲氧羰基)或CBZ(苄氧羰基)。
[化学式15]
其中R是直链、支化或环状的C1-6烷基基团或OCH2Bz,且R’是胺保护基团,t-Boc(叔丁氧羰基)、Fmoc(芴甲氧羰基)或CBZ(苄氧羰基)。
在本发明中,R可以是甲基、乙基、正丙基、正丁基或叔丁基,但不限于此。
将上述反应溶液离心或过滤以去除副产物沉淀(Et3NHCl或NaCl),并对滤液进行浓缩。将乙醇加入到该浓缩物中,随后进行真空浓缩以完全去除有机溶剂。将油状产物溶解于少量的乙醇(100ml)中,然后加入大量的水(900ml),以在低温下进行重结晶,然后用不同孔径的膜进行膜超滤,以将聚合物分级为50,000至100,000的分子量。对分级的聚磷腈溶液进行冷冻干燥,以得到约40-50%产率的化学式16和化学式17的聚磷腈化合物。
[化学式16]
[化学式17]
在化学式16和17中,n是聚磷腈的聚合度,其范围为3至300;MPEG表示分子量为350至1000的甲氧基聚(乙二醇);b的值为0.5-1.8;R是直链、支化或环状的C1-6烷基基团或OCH2Bz;R’是胺保护基团,t-Boc(叔丁氧羰基)、Fmoc(芴甲氧羰基)或CBZ(苄氧羰基)。
步骤(c)
不难将具有官能团的药物分子偶联至在步骤(b)中制备的具有多官能间隔基团的聚磷腈化合物,但将具有多官能团的药物分子(如紫杉烷)以临床应用中需要的键合方式偶联至多官能的聚磷腈载体聚合物并不容易。换言之,除了令人满意的物理化学性质外,抗癌药物-聚磷腈偶联物还需要表现出强的肿瘤靶向性和药物释放动力学。特别地,已知的是聚合物偶联物的药物释放动力学对于药效至关重要,因此,本发明中连接基团的作用是非常重要的。更具体地,连接基团应该不仅能够容易地将药物分子的官能团(OH,NH2)连接至聚磷腈载体聚合物的官能团(COOH,NH2),而且能够使偶联药物在被靶向的肿瘤部位能容易地释放出药物分子。
本发明中已经发现,化学式18的顺式乌头酸酐是在合成和药物释放动力学方面将紫杉烷偶联至聚磷腈载体聚合物的最佳连接基团,特别是在首先将连接基团与紫杉烷反应制备前体,然后将其连接到聚磷腈类的间隔基团上的情况下。紫杉烷药物前体的制备如下。
在氩气气氛下将多西紫杉醇(1.0mmol,803mg)和顺式乌头酸酐(2.0mmol,312mg)混合并溶解在THF或二氯甲烷中。将混合溶液冷却至凝固点,然后加入DIC(N,N'-二异丙基碳二亚胺)(2.0mmol,0.252g)和DPTS(4-(N,N'-二甲基氨基)吡啶鎓-4-甲苯磺酸盐)(2.0mmol,0.58g),使连接基团乌头酸的羧酸基团与多西紫杉醇的2'-羟基基团之间进行12小时的酯化反应。通过TLC确认反应完成后,加入过量的NHS(N-羟基琥珀酰亚胺)和DIPEA(二异丙基乙胺)以使未反应的乌头酸的酸酐环开环。已知乌头酸酐环容易在碱性溶液中通过羟基和胺基开环。搅拌12小时后,将反应混合物冷却至0℃保温3小时,然后真空过滤去除沉淀物。将滤液进行真空蒸馏,除去所有的有机溶剂,以得到油状混合物,将其溶解于乙醇(50ml)中。向该乙醇溶液中加入大量过量的水(500ml),在冰箱中重结晶3小时。去除上清液以得到油状产物,将得到的油状产物完全溶解在300ml二氯甲烷中。将该二氯甲烷溶液用饱和盐溶液洗涤三次,随后将所收集的有机层用无水NaHCO3干燥,然后进行真空蒸发,以得到95%产率的前体。
[化学式18]
步骤(d)
将疏水抗癌药物如紫杉烷偶联至上述在步骤(b)中制备的聚磷腈载体聚合物,以得到由化学式2表示的新型聚磷腈-紫杉烷偶联物。将紫杉烷分子偶联至聚磷腈载体聚合物的方法有两种:一种方法是将紫杉烷分子通过乌头酸以酰胺键合的方式偶联至聚磷腈载体的赖氨酸胺上,另一种方法是聚磷腈载体的赖氨酸羧基基团与紫杉烷分子的2'-羟基基团的酯化反应。因此,步骤(d)以两种不同的方法进行。
在酰胺键合方法中,应该通过将上述的化学式16或化学式17的聚磷腈载体聚合物的保护基团(t-Boc)与三氟乙酸和二氯甲烷(2:1)的混合物反应6h,然后进行中和、水洗并随之冻干,而将该保护基团除去。将脱保护的聚磷腈载体聚合物与药物前体(通过使连接基团乌头酸酐与药物如紫杉烷反应,随后加入过量的NHS(N-羟基琥珀酰亚胺)和DIPEA(二异丙基乙胺)而制备的乌头酸紫衫烷-NHS(N-羟基琥珀酰亚胺基))反应12h。将所得的反应混合物真空蒸发浓缩12h,然后溶解于乙醇中。对最终的溶液进行真空蒸发(37℃,5mm巴),去除所有痕量溶剂和DIPEA。将残留的聚合物偶联产物溶解于50ml乙醇中,然后在该乙醇溶液中加入950ml蒸馏水,以在冰箱中重结晶。3h后,对该溶液混合物进行真空过滤,以收集最终的聚合物偶联产物,用30%的乙醇溶液洗涤数次、然后使用超滤膜用蒸馏水超滤,直至通过UV光谱法检测到未反应的紫杉烷小于0.1%为止。将纯化的聚磷腈-紫杉烷偶联物溶液进行冻干,以得到化学式19或20的最终产物。
[化学式19]
[化学式20]
在化学式19和20中,n是独立地从3至300的整数;OMPEG表示具有350至1000分子量的甲氧基聚(乙二醇);D表示多西紫杉醇、紫杉醇、喜树碱和(反式-(±)-1,2-二氨基环己烷)铂(II);R为C1-6直链、支化或环状的烷基基团或OCH2Bz;x和y独立地在0-0.5的范围内;z大于0且小于1.0;x+y+z=1。
在酯键合的方法中,可以用碱将聚磷腈载体聚合物的间隔基团的末端赖氨酸酯水解成羧酸形式,然后与紫杉烷的2'-羟基基团进行酯化反应。例如,将化学式19的聚磷腈载体聚合物溶解在甲醇中,然后向其中加入过量的KOH或NaOH(150~200%),以得到载体聚合物的赖氨酸的钾盐或钠盐。通过真空蒸发去除甲醇溶剂,将所得金属盐溶解于蒸馏水(100ml)中。将二氯甲烷或氯仿(300ml)加入到该溶液中,通过缓慢加入有机酸将其酸化。当水层的pH降低到4-3时,通过振荡该两层溶剂层,将酸形式的聚磷腈载体聚合物萃取到有机层中。将该萃取程序重复三次,用无水碳酸氢钠干燥所收集的有机层溶液。对干燥的溶液进行过滤和真空蒸发,以得到具有末端被水解的赖氨酸基团的聚磷腈载体聚合物。将所得的具有被水解的赖氨酸的聚磷腈载体聚合物和等量的多西紫杉醇溶解于四氢呋喃中,然后加入N,N'-二环己基碳二亚胺和三乙胺进行酯化反应。通过TLC利用溶剂混合物(CHCl3:MeOH3=10:1)确认反应完成,然后将反应混合物进行真空蒸馏以浓缩反应混合物,随后通过超滤膜分级出分子量为30至100kDa的化学式21的最终聚磷腈-多西紫杉醇偶联物。
[化学式21]
其中n是从3至300的整数;OMPEG表示具有350至1000分子量的甲氧基聚(乙二醇);D表示多西紫杉醇、紫杉醇或喜树碱;R’表示t-Boc或CBZ;x和y独立地在0至0.5的范围内;z大于0且小于1.0;x+y+z=1。
在下文中,将参考以下示例和示范更详细地描述本发明的构成和作用,但是本发明不限于此。在以下的示例中,采用Perkin-Elmer C、H和N分析仪对本发明化合物进行碳、氢和氮元素分析。采用Varian Gemini-500NMR谱仪测量氢和氮的核磁共振谱。采用Malvern Zeta-sizer(Nano-ZS)测量聚磷腈载体聚合物及其药物偶联物的粒径分布。
发明的方式
聚磷腈化合物(药物递送系统)的合成
示例1、[NP(MPEG550)1.5(赖氨酸乙基)0.5]n的合成
将六氯环三磷腈([NPCl2]3,11.54g,100mmol)和无水氯化铝(AlCl3,7.5重量%)在手套箱中均匀混合,然后根据文献方法(Sohn YS等人,Macromolecules 1995,28,7566)在250℃下进行热聚合5h,得到聚(二氯膦腈)([NPCl2]n)。同时,在氩气气氛下,在干燥的甲苯溶剂中将平均分子量为550的甲氧基聚(乙二醇)(MPEG550)(82.5g,150mmol)与金属钠(4.9g,200.4mmol)在120℃下反应6h得到MPEG550的钠盐。在玻璃容器中,在冰浴(0℃)上向上述溶解于四氢呋喃的聚(二氯膦腈)(100ml)中在60min内缓慢加入所制备的MPEG550的钠盐溶液。1h后,移除冰浴,然后将反应混合物在环境温度下进一步反应12h,以得到PEG化的聚磷腈中间体溶液。用在干燥的氯仿(200ml)中干燥的三甲胺去中和Boc-赖氨酸乙酯,在单独的容器中,将经过中和的Boc-赖氨酸乙酯(N-Boc-赖氨酸乙基,20.5g,75.0mmol)缓慢地加入到上述PEG化的聚磷腈中间体溶液中,将该反应混合物在室温下进一步地反应24h。过滤该反应混合物,去除所得的副产物沉淀(NEt3·HCl或NaCl),并将滤液进行真空蒸发以浓缩该溶液(将该溶液溶解于乙醇中,随后进行真空蒸发)。最后,将残余物溶解在蒸馏水中,并过滤去除任何不溶物,并使用超滤膜(CE,MWCO=3000)对滤液进行透析,去除低于3000Da的较低分子量的级分。将分级的聚磷腈聚合物用蒸馏水洗涤多于5次,然后进行冻干。将得到的聚磷腈聚合物溶解在含有30%(体积/体积)三氟乙酸的二氯甲烷溶液(200ml)中,搅拌6h,以从聚合物中去除保护基团(t-Boc),得到产率65%(3.37g)的脱保护的聚磷腈聚合物[NP(MPEG550)1.5(赖氨酸乙基)0.5]n。
通过质子NMR确定出脱保护的赖氨酸胺后,采用三乙胺中和该聚合物溶液,然后通过真空蒸馏去除有机溶剂。向所得的聚合物中加入碳酸氢钠水溶液以完全溶解聚合物,将其脱盐,然后使用分子量截留值为3k Da、30k Da、100k Da的超滤膜进行分级。总回收率为90%。
组成:C83H170N4O41P2
元素分析数据(%):C(51.66);H(8.59);N(2.83)。理论值:C(51.33);H(8.82);N(2.88)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,1.5H,赖氨酸-OCH2CH3),2.49(br,1.00H,琥珀酰基-CH2),2.90(br,1.00H,赖氨酸-ε-CH2),3.38(s,4.50H,MPEG550-OCH3),3.65(br,66.0H,MPEG550-OCH2CH2),4.4(s,1H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例2、[NP(MPEG750)1.5(赖氨酸乙基)0.5]n的合成
根据与示例1中所述的相同的方法,使用六氯环三磷腈([NPCl2]3,11.54g,100mmol)、平均分子量为750的甲氧基聚(乙二醇)(MPEG750,112.5g,150mmol)、三甲胺(80.0ml,600mmol)和Boc-赖氨酸乙基(20.5g,75mmol)制备出产率为74%的标题聚磷腈化合物的期望产物。
组成:C107H218N4O53P2
元素分析数据(%):C(51.30);H(8.99);N(2.36)。理论值:C(52.01);H(8.89);N(2.27)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,1.50H,赖氨酸-OCH2CH3),1.39-1.98(br,3.00H,赖氨酸-CH2),2.90(br,1H,赖氨酸-ε-CH2),3.38(s,4.50H,MPEG750-OCH3),3.65(br,98.0H,MPEG750-OCH2CH2),4.01(bs,6H,MPEG750-CH2),4.45(m,0.5H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例3、[NP(MPEG1000)1.5(赖氨酸乙基)0.5]n的合成
根据与示例1中所述的相同的方法,使用六氯环三磷腈([NPCl2]3,11.54g,100mmol)、平均分子量为1000的甲氧基聚(乙二醇)(MPEG1000,150g,150mmol)、三甲胺(80.0ml,600mmol)和Boc-赖氨酸乙基(20.5g,75mmol)制备出产率为74%的标题聚磷腈化合物的期望产物。
组成:C143H290N4O71P2
元素分析数据(%):C(53.01);H(8.70);N(2.36)。理论值:C(52.62);H(8.96);N(1.72)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,1.50H,赖氨酸-OCH2CH3),1.39-1.98(br,3.00H,赖氨酸-CH2),2.90(br,1H,赖氨酸-ε-CH2),3.38(s,4.50H,MPEG1000-OCH3),3.65(br,130.0H,MPEG1000-OCH2CH2),4.45(m,0.5H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例4、[NP(MPEG550)1.25(赖氨酸乙基)0.75]n的合成
根据与示例1中所述的相同的方法,使用六氯环三磷腈([NPCl2]3,11.54g,100mmol)、平均分子量为550的甲氧基聚(乙二醇)(MPEG550,69.0g,126mmol)、三甲胺(80.0ml,600mmol)和Boc-赖氨酸乙基(27.4g,100mmol)制备出产率为74%的标题聚磷腈化合物的期望产物。
组成:C74.5H153N5O35.5P2
元素分析数据(%):C(50.87);H(9.02);N(4.31)。理论值:C(51.14);H(8.82);N(4.14)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,2.25H,赖氨酸-OCH2CH3),1.39-1.98(br,4.50H,赖氨酸-CH2),2.90(br,1.5H,赖氨酸-ε-CH2),3.38(s,3.75H,MPEG550-OCH3),3.65(br,82.5H,MPEG550-OCH2CH2),4.45(m,0.7H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例5、[NP(MPEG550)1.0(赖氨酸乙基)1.0]n的合成
根据与示例1中所述的相同的方法,使用六氯环三磷腈([NPCl2]3,11.54g,100mmol)、平均分子量为550的甲氧基聚(乙二醇)(MPEG550,55.0g,100mmol)、三甲胺(80.0ml,600mmol)和Boc-赖氨酸乙基(35.6g,130mmol)制备出产率为74%的标题聚磷腈化合物的期望产物。
组成:C66H136N6O30P2
元素分析数据(%):C(50.29);H(8.95);N(5.51)。理论值:C(50.95);H(8.81);N(5.40)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,3.00H,赖氨酸-OCH2CH3),1.39-1.98(br,6.00H,赖氨酸-CH2),2.90(br,2.0H,赖氨酸-ε-CH2),3.38(s,3.0H,MPEG550-OCH3),3.65(br,66.0H,MPEG550-OCH2CH2),4.45(m,1.0H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例6、[NP(MPEG550)1.5(赖氨酸乙基)0.5]n的合成
根据与示例1中所述的相同的方法,使用六氯环三磷腈([NPCl2]3,11.54g,100mmol)、平均分子量为550的甲氧基聚(乙二醇)(MPEG550,82.5g,150mmol)、三甲胺(80.0ml,600mmol)和Gly(Nε-Boc赖氨酸乙基(24.8g,90mmol)制备出产率为74%的标题聚磷腈化合物的期望产物。
组成:C85H173N5O42P2
元素分析数据(%):C(50.75);H(8.82);N(3.61)。理论值:C(51.06);H(8.72);N(3.50)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,3.00H,赖氨酸-OCH2CH3),1.39-1.98(br,6.00H,赖氨酸-CH2),2.90(br,2.0H,赖氨酸-ε-CH2),3.38(s,3.0H,MPEG750-OCH3),3.65(br,66.0H,MPEG500-OCH2CH2),3.92(bs,2H,甘氨酸-CH2),4.45(m,1.0H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例7、[NP(MPEG550)1.5(Nα-Boc赖氨酸)0.5]n的合成
根据与示例1中所述的相同的方法,使用六氯环三磷腈([NPCl2]3,11.54g,100mmol)、平均分子量为550的甲氧基聚(乙二醇)(MPEG550,82.5g,150mmol)、三甲胺(80.0ml,600mmol)和Nε-Boc赖氨酸乙基(20.5g,75mmol)制备出标题聚磷腈化合物产物的酯形式[NP(MPEG550)1.5(Nα-Boc赖氨酸乙基)0.5]n(产率75%)。将该合成的衍生物(10g,10mmol)和NaOH(0.4g,10mmol)溶解在甲醇中,然后在环境温度下水解4小时。通过使用质子NMR确认水解完全,然后将溶液进行真空蒸发,得到固态聚合物,将该固态聚合物溶解在蒸馏水中,并通过有机酸酸化至pH=3。将该酸化的聚合物溶液用氯仿或二氯甲烷萃取三次。有机层用无水碳酸氢钠干燥,然后进行真空蒸发,得到该标题化合物(产率95%)。
组成:C86H174N4O43P2
元素分析数据(%):C(51.02);H(8.94);N(2.91)。理论值:C(51.28);H(8.71);N(2.78)。
1H-NMR谱(CDCl3)(δ,ppm):1.32(s,4.5H,Boc-CH3),1.39-1.98(br,6.00H,赖氨酸-CH2),2.90(br,2.0H,赖氨酸-ε-CH2),3.38(s,3.0H,MPEG550-OCH3),3.65(br,66.0H,MPEG550-OCH2CH2),4.45(m,1.0H,赖氨酸-CH)。
示例8、[NP(MPEG550)(AE)]n的合成
根据与示例1中所述的相同的方法,使用六氯环三磷腈([NPCl2]3,2.0g,5.72mmol)、催化氯化铝(AlCl3,7.0重量%)、平均分子量为550的甲氧基聚(乙二醇)(MPEG550,9.48g,17.2mmol)和金属钠(0.59g,25.7mmol)制备出PEG化的聚磷腈中间体。同时,在干燥的四氢呋喃(50ml)中,将2-氨基乙醇(AE,1.30g,21.3mmol)和氢化钠(0.61g,25.4mmol)在环境温度下反应5h,得到黄色的2-氨基乙醇的钠盐沉淀物,用乙醚充分洗涤后,将其溶解在二甲基亚砜(50ml)中。将所得溶液加入到上述PEG化的聚磷腈中间体中,并将反应混合物在50℃下进一步反应24小时。将反应混合物过滤以去除副产物氯化钠,并使用纤维素膜(MWCO:3.5kDa)对滤液进行透析,在冻干后,得到70%产率的新型聚磷腈载体聚合物。
组成:C27H57N2O14P·H2O
元素分析数据(%):C(47.38);H(8.61);N(3.95)。理论值:C(47.45);H(8.64);N(4.10)。
1H-NMR谱(CDCl3)(δ,ppm):3.26(s,3H,MPEG的OCH3),3.50-3.52(m,4H,氨基乙醇的(CH2)2),3.54-3.81(brm,46H,MPEG的CH2),3.83-4.10(brm,2H,MPEG的-P-O-CH2-)。
31P-NMR谱:(CDCl3,ppm):δ-2.66(O-P-O)。
示例9、[NP(MPEG750)(AE)]n的合成
根据与示例8中所述的相同的方法,使用六氯环三磷腈([NPCl2]3,2.0g,5.72mmol)、催化氯化铝(AlCl3,7.0重量%)、平均分子量为750的甲氧基聚(乙二醇)(MPEG750,12.9g,17.2mmol)、金属钠(0.59g,25.7mmol)、2-氨基乙醇(1.30g,21.3mmol)和氢化钠(0.61g,25.4mmol)制备出产率为78%的标题聚磷腈化合物的期望产物。
组成:C35H73N2O18P·H2O
元素分析数据(%):C(48.02);H(8.96);N(3.55)。理论值:C(48.89);H(8.73);N(3.26)。
1H-NMR谱(CDCl3)(δ,ppm):3.41(s,3H,MPEG的OCH3),3.49-3.53(m,4H,氨基乙醇的(CH2)2),3.57-3.83(brm,62H,MPEG的CH2),3.83-4.05(brm,2H,MPEG的-P-O-CH2-)。
31P-NMR谱:(CDCl3,ppm):δ-3.95(O-P-O)。
抗癌药物-连接基团前体的合成
示例10、2'-乌头酸多西紫杉醇NHS酯的合成
将乌头酸酐(3.12g,20mmol)、多西紫杉醇(8.03g,10mmol)和催化剂DPTS(二甲基氨基吡啶/对甲苯磺酸)(6.0g,20mmol)真空干燥4h,并冷却至-10℃,随后将其完全溶解在干燥的四氢呋喃(100ml)中。向该溶液中缓慢地加入也溶解在干燥的四氢呋喃中的DIC(N,N'-二异丙基碳二亚胺)(2.5g,20mmol)。将混合反应溶液在-10℃下反应6h,然后在0℃下反应6至12h。反应的进程通过使用二氯甲烷:甲醇(95:5)的溶剂混合物的TLC监测,直到检测不到游离多西紫杉醇为止。在确认反应完成后,向上述反应溶液中加入过量的NHS(N-羟基琥珀酰亚胺)(11.5g,100mmol)和大量过量的碱性DIPEA(二异丙基乙胺),并使反应混合物进一步反应12h。最后将反应混合物进行真空蒸发,得到固体聚合物产物,将其溶解于少量乙醇(50ml)中,然后加入大量的水(950ml),在0℃下重结晶3h。除去上清液水层,得到棕色油状产物,将其溶解在氯仿或二氯甲烷中,依次用盐溶液(pH=7)、柠檬酸溶液(pH=2)、碳酸氢钠溶液(pH=9)、最后用盐溶液洗涤。最终的有机溶液用无水硫酸镁干燥,然后经过真空干燥,最终获得由2'-乌头酸-多西紫杉醇-NHS酯构成的前体。
组成:C53H60N2O21
元素分析数据(%):C(60.07);H(5.86);N(2.71)。理论值:C(59.99);H(5.70);N(2.64)。
1H-NMR谱(CDCl3)(δ,ppm):1.13ppm(s,3H,C17-CH3),1.24ppm(s,3H,C16-CH3),1.34ppm(s,9H,C60-叔丁基),1.75ppm(s,3H,C19-CH3),1.96ppm(s,3H,C18-CH3),2.18ppm(d,2H,C14-CH2),2.43ppm(s,3H,C22-CH3),2.64(t,4H,NHS-CH2CH2),2.92(s,2H,乌头酸-CH2),4.21ppm(d,1H,C20-CHa),4.24ppm(m,1H,C7-CH),4.32ppm(d,1H,C20-CHb),4.95ppm(dd,1H,C5CH),5.23ppm(d,1H,C10-CH),5.40ppm(d,1H,C30-CH),5.69ppm(d,1H,C2-CH),6.40-6.68(m,1H,乌头酸-CH),7.51ppm(m,2H,C33,C27-CH),7.53(m,6H,C32,C34-CH;C31,C35-CH;C26,C28-CH),8.12(d,2H,C25,C29-CH)。
示例11、2'-乌头酸紫杉醇NHS酯的合成
根据与示例10中所述相同的方法,使用乌头酸酐(3.12g,20mmol)、紫杉醇(8.53g,10mmol)、DPTS(5.88g,20mmol)、NHS(11.5g,100mmol),DIC(2.52g,20mmol)和DIPEA(10ml)制备出由2'-乌头酸紫杉醇NHS酯构成的前体的期望产物。
组成:C55H58N2O19
元素分析数据(%):C(61.90);H(5.75);N(2.80)。理论值:C(62.85);H(5.56);N(2.67)。
1H-NMR谱(CDCl3)(δ,ppm):1.13ppm(s,3H,C17-CH3),1.24ppm(s,3H,C16-CH3),1.34ppm(s,9H,C60-叔丁基),1.75ppm(s,3H,C19-CH3),1.96ppm(s,3H,C18-CH3),2.18ppm(d,2H,C14-CH2),2.43ppm(s,3H,C22-CH3),2.64(t,4H,NHS-CH2CH2),2.92(s,2H,乌头酸-CH2),4.21ppm(d,1H,C20-CHa),4.24ppm(m,1H,C7-CH),4.32ppm(d,1H,C20-CHb),4.95ppm(dd,1H,C5CH),5.23ppm(d,1H,C10-CH),5.40ppm(d,1H,C30-CH),5.69ppm(d,1H,C2-CH),6.40-6.68(m,1H,乌头酸-CH),7.51ppm(m,2H,C33,C27-CH),7.53(m,6H,C32,C34-CH;C31,C35-CH;C26,C28-CH),8.12(d,2H,C25,C29-CH)。
示例12、2'-乌头酸喜树碱NHS酯的合成
根据与示例10中所述相同的程序,使用乌头酸酐(3.12g,20mmol)、喜树碱(3.48g,10mmol)、DPTS(5.88g,20mmol)、NHS(11.5g,100mmol),DIC(2.52g,20mmol)和DIPEA(10ml)制备出由2'-乌头酸喜树碱NHS酯构成的前体的期望产物。
组成:C30H23N3O11
元素分析数据(%):C(60.29);H(3.98);N(6.57)。理论值:C(59.90);H(3.85);N(6.99)。
1H-NMR谱(CDCl3)(δ,ppm):0.9(t,3H,C18-CH3),2.0(m,2H,C19-CH2),2.64(t,4H,NHS-CH2CH2),2.92(s,2H,乌头酸-CH2),4.20(d,2H,C5-CH2),4.76(m,2H,C22-CH2),6.40-6.68(m,1H,乌头酸-CH),6.70(s,1H,C14-CH),7.59(s,1H,C11-CH),7.80(m,2H,C12-CH;C7-CH),8.0(m,2H,C9-CH;C12-CH)。
示例13、2'-乌头酸甘氨酸喜树碱的合成
将t-Boc-甘氨酸(0.5g,2.85mmol)和喜树碱(CPT)(0.5g,1.43mmol)溶解于无水二氯甲烷(20ml)中,并向其中加入DIPC(0.36ml,2.85mmol)和DMAP(0.31g,2.53mmol)。在室温下将混合物溶液搅拌反应16h。通过振荡,用稀盐酸水溶液(pH=2)萃取所得混合物。分离水层,并用无水硫酸镁干燥,然后进行真空干燥,得到t-Boc-甘氨酸喜树碱。在二氯甲烷和三氟乙酸的混合物(10ml/10ml)中将该中间体反应1h,除去t-Boc基团,在0℃下将得到的喜树碱-甘氨酸-NH2和顺式乌头酸酐(0.57g,3.63mmol)在二甲基甲酰胺溶剂(2ml)中反应16h。向反应混合物中加入过量的乙醚以沉淀喜树碱前体,将其进行过滤并真空干燥,得到80%产率的2'-乌头酸-甘氨酸喜树碱。
组成:C28H23N3O10
元素分析数据(%):C(59.72);H(4.01);N(7.39)。理论值:C(59.84);H(4.09);N(7.48)。
1H-NMR谱(CDCl3)(δ,ppm):0.88-0.93(brm,3H,CPT-C18的-CH3),2.12-2.16(m,2H,CPT-C-19的-CH2),2.86(s,2H,顺式乌头酸盐的-CH2),3.92-4.42(brm,2H,甘氨酸的-CH2),5.27(brs,2H,CPT-C5的-CH2),5.49(brs,2H,CPT-C22的-CH2),5.97(s,1H,顺式乌头酸盐的=CH),7.18-7.21(m,1H,CPT-C14的=CH),7.69-7.72(m,1H,CPT-C11的=CH),7.84-7.89(m,1H,CPT-C10的=CH),8.10-8.21(m,2H,CPT-C12和C9的=CH),8.68(brs,1H,CPT-C7的=CH),12.3-12.8(brs,顺式乌头酸的-COOH)。
聚磷腈-抗癌药物偶联物的合成
示例14、[NP(MPEG550)1.5(赖氨酸乙基-2'-乌头酸-多西紫杉醇)0.5]n的合成
在反应烧瓶中,将示例1中得到的聚磷腈化合物(4.85g,5.0mmol)溶解在二氯甲烷中,用冰浴冷却。将示例8中得到的2'-乌头酸-多西紫杉醇-NHS酯也溶解在二氯甲烷中,并加入到该反应烧瓶中,反应混合物在低温(0-5℃)下反应12h。反应12h后,将反应混合物进行真空蒸发,将残余物溶解在少量乙醇中,然后加入大量过量的水进行重结晶,重复两次。使用膜过滤器去除剩余的不溶性杂质,将滤液用30%乙醇水溶液洗涤5次,然后用纯水洗涤5次,随后使用超滤膜分级并冻干,得到产率为90%的最终聚磷腈-多西紫杉醇偶联物,[NP(MPEG550)1.5(赖氨酸乙基-2'-乌头酸-多西紫杉醇)0.5]n。
组成:C132H225N5O59P2
元素分析数据(%):C(53.62);H(7.92);N(2.67)。理论值:C(54.89);H(7.85);N(2.42)。
1H-NMR谱(CDCl3)(δ,ppm):1.13ppm(s,1.5H,C17-CH3),1.24ppm(s,1.5H,C16-CH3),1.34ppm(bs,4.1H,C60-叔丁基),1.75ppm(s,1.5H,C19-CH3),1.96ppm(s,1.5H,C18-CH3),2.18ppm(d,1.0H,C14-CH2),2.43ppm(s,1.5H,C22-CH3),4.21ppm(d,0.5H,C20-CHa),4.24ppm(m,0.5H,C7-CH),4.32ppm(d,0.5H,C20-CHb),4.95ppm(dd,0.5H,C5-CH),5.23ppm(d,0.5H,C10-CH),5.40ppm(d,0.5H,C30-CH),5.69ppm(d,0.5H,C2-CH),7.51ppm(m,1.0H,C33,C27-CH),7.53(m,3.0H,C32,C34-CH;C31,C35-CH;C26,C28-CH),8.12(d,0.6H,C25,C29-CH),1.24(s,1.5H,赖氨酸-OCH2CH3),1.29(bs,1H,赖氨酸-CH2),1.55ppm(bs,1H,赖氨酸-CH2),1.80(bs,1H,赖氨酸-CH2),2.90(br,1H,赖氨酸-e-CH2),3.38ppm(s,4.50H,CH3O-,PEG),和3.63ppm(m,66.0H,-CH2CH2-O-),4.4(s,0.51H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例15、[NP(MPEG550)1.5(赖氨酸乙基)0.2(赖氨酸乙基-2'-乌头酸-多西紫杉醇)0.3]n的合成
根据与示例14中所述相同的方法,使用示例1的聚磷腈化合物(9.7g,10mmol)、前体(示例11的2'-乌头酸多西紫杉醇NHS酯(3.18g,3.0mmol))和DIPEA(5ml)得到产率为90%的期望的标题产物。
组成:C112.4H203N4.6O51.8P2
元素分析数据(%):C(53.48);H(8.31);N(2.64)。理论值:C(53.79);H(8.15);N(2.57)。
1H-NMR谱(CDCl3)(δ,ppm):1.13ppm(s,0.9H,C17-CH3),1.24ppm(s,0.9H,C16-CH3),1.34ppm(bs,3.31H,C60-叔丁基),1.75ppm(s,0.9H,C19-CH3),1.96ppm(s,0.9H,C18-CH3),2.18ppm(d,0.6H,C14-CH2),2.43ppm(s,0.9H,C22-CH3),4.21ppm(d,0.3H,C20-CHa),4.24ppm(m,0.3H,C7-CH),4.32ppm(d,0.3H,C20-CHb),4.95ppm(dd,0.31H,C5-CH),5.23ppm(d,0.3H,C10-CH),5.40ppm(d,0.3H,C30-CH),5.69ppm(d,0.3H,C2-CH),7.51ppm(m,0.6H,C33,C27-CH),7.53(m,1.8H,C32,C34-CH;C31,C35-CH;C26,C28-CH),8.12(d,0.6H,C25,C29-CH),1.24(s,1.5H,赖氨酸-OCH2CH3),1.29(bs,1H,赖氨酸-CH2),1.55ppm(bs,1H,赖氨酸-CH2),1.80(bs,1H,赖氨酸-CH2),2.90(br,1H,赖氨酸-e-CH2),3.38ppm(s,4.50H,CH3O-,PEG),和3.63ppm(m,66.0H,-CH2CH2-O-),4.4(s,0.51H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例16、[NP(MPEG750)1.5(赖氨酸乙基)0.2(赖氨酸乙基-2'-乌头酸-多西紫杉醇)0.3]n
根据与示例14中所述相同的方法,使用示例2的聚磷腈化合物(12.3g,10mmol)、前体(示例8的2'-乌头酸多西紫杉醇NHS酯(5.3g,5.0mmol))和DIPEA(10ml)得到产率为89%的期望的标题产物。
组成:C136.4H251N4.6O63.8P2
元素分析数据(%):C(53.27);H(8.45);N(2.31)。理论值:C(53.92);H(8.33);N(2.12)。
1H-NMR谱(CDCl3)(δ,ppm):1.13ppm(s,0.9H,C17-CH3),1.24ppm(s,0.9H,C16-CH3),1.34ppm(bs,3.31H,C60-叔丁基),1.75ppm(s,0.9H,C19-CH3),1.96ppm(s,0.9H,C18-CH3),2.18ppm(d,0.6H,C14-CH2),2.43ppm(s,0.9H,C22-CH3),4.21ppm(d,0.3H,C20-CHa),4.24ppm(m,0.3H,C7-CH),4.32ppm(d,0.3H,C20-CHb),4.95ppm(dd,0.31H,C5-CH),5.23ppm(d,0.3H,C10-CH),5.40ppm(d,0.3H,C30-CH),5.69ppm(d,0.3H,C2-CH),7.51ppm(m,0.6H,C33,C27-CH),7.53(m,1.8H,C32,C34-CH;C31,C35-CH;C26,C28-CH),8.12(d,0.6H,C25,C29-CH),1.24(s,1.5H,赖氨酸-OCH2CH3),1.29(bs,1H,赖氨酸-CH2),1.55ppm(bs,1H,赖氨酸-CH2),1.80(bs,1H,赖氨酸-CH2),2.90(br,1H,赖氨酸-ε-CH2),3.38ppm(s,4.50H,CH3O-,PEG),和3.63ppm(m,98.0H,-CH2CH2O-),4.0(bs,4H,MPEG750-CH2),4.51(s,0.51H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例17、[NP(MPEG1000)1.5(赖氨酸乙基)0.2(赖氨酸乙基-2'-乌头酸-多西紫杉醇)0.3]n
根据与示例14中所述相同的方法,使用示例3的聚磷腈化合物(15.6g,10mmol)、前体(示例11的2'-乌头酸多西紫杉醇NHS酯(5.3g,5.0mmol))和DIPEA(10ml)得到产率为89%的期望的标题产物。
组成:C172.4H323N4.6O48.2P2
元素分析数据(%):C(53.71);H(8.74);N(2.01)。理论值:C(54.04);H(8.50);N(1.68)。
1H-NMR谱(CDCl3)(δ,ppm):1.13ppm(s,0.9H,C17-CH3),1.24ppm(s,0.9H,C16-CH3),1.34ppm(bs,3.31H,C60-叔丁基),1.75ppm(s,0.9H,C19-CH3),1.96ppm(s,0.9H,C18-CH3),2.18ppm(d,0.6H,C14-CH2),2.43ppm(s,0.9H,C22-CH3),4.21ppm(d,0.3H,C20-CHa),4.24ppm(m,0.3H,C7-CH),4.32ppm(d,0.3H,C20-CHb),4.95ppm(dd,0.31H,C5-CH),5.23ppm(d,0.3H,C10-CH),5.40ppm(d,0.3H,C30-CH),5.69ppm(d,0.3H,C2-CH),7.51ppm(m,0.6H,C33,C27-CH),7.53(m,1.8H,C32,C34-CH;C31,C35-CH;C26,C28-CH),8.12(d,0.6H,C25,C29-CH),1.24(s,1.5H,赖氨酸-OCH2CH3),1.29(bs,1H,赖氨酸-CH2),1.55ppm(bs,1H,赖氨酸-CH2),1.80(bs,1H,赖氨酸-CH2),2.90(br,1H,赖氨酸-ε-CH2),3.38ppm(s,4.50H,MPEG-CH3O-),和3.63ppm(m,128H,MPEG-CH2CH2O),4.0(bs,4H,MPEG1000-CH2),4.51(s,0.51H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例18、[NP(MPEG550)1.5(赖氨酸乙基)0.2(赖氨酸乙基-2'-乌头酸-多西紫杉醇)0.5]n的合成
根据与示例14中所述相同的方法,使用示例5的聚磷腈化合物(7.7g,10mmol)、前体(示例8的2'-乌头酸多西紫杉醇NHS酯(6.36g,6.0mmol))和DIPEA(10ml)得到产率为89%的期望的标题产物。
组成:C115H191N7O48P2
元素分析数据(%):C(54.92);H(8.01);N(3.99)。理论值:C(55.21);H(7.70);N(3.92)。
1H-NMR谱(CDCl3)(δ,ppm):1.13ppm(s,0.9H,C17-CH3),1.24ppm(s,0.9H,C16-CH3),1.34ppm(bs,3.31H,C60-叔丁基),1.75ppm(s,0.9H,C19-CH3),1.96ppm(s,0.9H,C18-CH3),2.18ppm(d,0.6H,C14-CH2),2.43ppm(s,0.9H,C22-CH3),4.21ppm(d,0.3H,C20-CHa),4.24ppm(m,0.3H,C7-CH),4.32ppm(d,0.3H,C20-CHb),4.95ppm(dd,0.31H,C5-CH),5.23ppm(d,0.3H,C10-CH),5.40ppm(d,0.3H,C30-CH),5.69ppm(d,0.3H,C2-CH),7.51ppm(m,0.6H,C33,C27-CH),7.53(m,1.8H,C32,C34-CH;C31,C35-CH;C26,C28-CH),8.12(d,0.6H,C25,C29-CH),1.24(s,3.0H,赖氨酸-OCH2CH3),1.29(bs,2H,赖氨酸-CH2),1.55ppm(bs,2H,赖氨酸--CH2),1.80(bs,2H,赖氨酸-CH2),2.90(br,2H,赖氨酸-e-CH2),3.38ppm(s,3.00H,CH3O-,PEG),和3.63ppm(m,44.0H,-CH2CH2-O-),4.4(s,0.51H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例19、[NP(MPEG550)1.5(甘氨酸赖氨酸乙基)0.2(甘氨酸赖氨酸乙基-2'-乌头酸-多西紫杉醇)0.3]n的合成
根据与示例14中所述相同的方法,使用示例6的聚磷腈化合物(10.4g,10mmol)、前体(示例10的2'-乌头酸多西紫杉醇NHS酯(5.3g,5.0mmol))和DIPEA(10ml)得到产率为89%的期望的标题产物。
组成:C114.4H206N5.6O52.8P2
元素分析数据(%):C(52.98);H(8.23);N(3.19)。理论值:C(53.53);H(8.09);N(3.06)。
1H-NMR谱(CDCl3)(δ,ppm):1.13ppm(s,0.9H,C17-CH3),1.24ppm(s,0.9H,C16-CH3),1.34ppm(bs,3.31H,C60-叔丁基),1.75ppm(s,0.9H,C19-CH3),1.96ppm(s,0.9H,C18-CH3),2.18ppm(d,0.6H,C14-CH2),2.43ppm(s,0.9H,C22-CH3),4.21ppm(d,0.3H,C20-CHa),4.24ppm(m,0.3H,C7-CH),4.32ppm(d,0.3H,C20-CHb),4.95ppm(dd,0.31H,C5-CH),5.23ppm(d,0.3H,C10-CH),5.40ppm(d,0.3H,C30-CH),5.69ppm(d,0.3H,C2-CH),7.51ppm(m,0.6H,C33,C27-CH),7.53(m,1.8H,C32,C34-CH;C31,C35-CH;C26,C28-CH),8.12(d,0.6H,C25,C29-CH),1.24(s,1.5H,赖氨酸-OCH2CH3),1.29(bs,1H,赖氨酸-CH2),1.55ppm(bs,1H,赖氨酸-CH2),1.80(bs,1H,赖氨酸-CH2),2.90(br,1H,赖氨酸-e-CH2),3.38ppm(s,4.50H,CH3O-,PEG),和3.63ppm(m,66.0H,-CH2CH2-O-),3.98(bs,2H,甘氨酸-CH2),4.4(s,0.51H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例20、[NP(MPEG550)1.50(赖氨酸乙基)0.2(赖氨酸乙基-2'-琥珀酰基紫杉醇)0.3]n的合成
通过使用DCL(2.54g,20mmol)和DIPEA(10ml),将示例1的聚磷腈化合物(9.7g,10mmol)与通过文献方法(C.-M.Huang等,Chem.Biol.2000,7,453-461)制备的2'-琥珀酰基紫杉醇(5.26g,5.0mmol)进行酯化反应,得到产率为90%的期望的标题产物。
组成:C113.6H202N4.6O51.8P2
元素分析数据(%):C(54.15);H(8.26);N(2.61)。理论值:C(54.49);H(8.12);N(2.57)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,1.5H,赖氨酸-OCH2CH3),2.49(br,1.00H,琥珀酰基-CH2),2.90(br,1.00H,赖氨酸-ε-CH2),3.38(s,4.50H,MPEG550-OCH3),3.65(br,66.0H,MPEG550-OCH2CH2),1.13(s,12H),1.25(s,12H),1.35(s,36H),1.68(m,8H),1.75(s,12H),1.86(m,8H),1.96(s,12H),2.36(m,20H),2.60(m,4H),3.98(s,8H),4.06(d,8H),4.30(m,12H),4.33(m,8H),4.97(d,4H),5.22(m,4H),5.36(s,4H),5.60(m,4H),5.69(m,8H),6.20(t,4H),7.33(m,8H),7.41(m,8H),7.52(m,8H),7.61(m,4H),7.33(m,4H),8.12(d,8H)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例21、[NP(MPEG550)1.50(赖氨酸乙基)0.2(赖氨酸乙基-2'-琥珀酰基紫杉醇)0.50]n的合成
根据与示例20中所述相同的方法,通过使用DCL(2.54g,20mmol)和DIPEA(10ml),将示例6的聚磷腈化合物(10.4g,10mmol)与通过文献方法(C.-M.Huang等人,Chem.Biol.2000,7,453-461)制备的2'-琥珀酰基紫杉醇(7.36g,7.0mmol)进行酯化反应,得到产率为80%的期望的标题产物。
组成:C136H226N6O58P2
元素分析数据(%):C(55.36);H(7.99);N(2.93)。理论值:C(55.67);H(7.76);N(2.86)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,1.5H,赖氨酸-OCH2CH3),2.49(br,1.00H,琥珀酰基-CH2),2.90(br,1.00H,赖氨酸-ε-CH2),3.38(s,4.50H,MPEG550-OCH3),3.65(br,66.0H,MPEG550-OCH2CH2),1.13(s,12H),1.25(s,12H),1.35(s,36H),1.68(m,8H),1.75(s,12H),1.86(m,8H),1.96(s,12H),2.36(m,20H),2.60(m,4H),3.98(s,8H),4.06(d,8H),4.30(m,12H),4.33(m.8H),4.97(d,4H),5.22(m,4H),5.36(s,4H),5.60(m,4H),5.69(m,8H),6.20(t,4H),7.33(m,8H),7.41(m,38H),7.52(m,8H),7.61(m,4H),7.33(m,4H),8.12(d,8H)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例22、[NP(MPEG550)1.50(Nα-Boc赖氨酸)0.2(Nα-Boc赖氨酸-多西紫杉醇)03]n的合成
将示例7的聚磷腈化合物(9.57g,10.0mmol)和多西紫杉醇(10.6g,10.0mmol)真空干燥,然后在反应容器中将其溶解于四氢呋喃、二氯甲烷或氯仿的干燥溶剂中,进行冰浴冷却后向其中加入溶解在相同溶剂中的催化剂DCl(2.54g,20mmol)和DIPEA(10ml)。将反应混合物在冰温下反应24h后,将反应溶液减压过滤并将滤液真空干燥。将所得产物用示例14中相同的方法纯化,得到产率为60%的标题化合物。
组成:C113.8H204.2N4.6O50.8P2
元素分析数据(%):C(55.06);H(7.98);N(2.60)。理论值:C(54.42);H(8.19);N(2.57)。
1H-NMR谱(CDCl3)(δ,ppm):1.25(s,1.5H,赖氨酸-OCH2CH3),2.49(br,1.00H,琥珀酰基-CH2),2.90(br,1.00H,赖氨酸-ε-CH2),3.38(s,4.50H,MPEG550-OCH3),3.65(br,66.0H,MPEG550-OCH2CH2),1.13(s,12H),1.25(s,12H),1.35(s,36H),1.68(m,8H),1.75(s,12H),1.86(m,8H),1.96(s,12H),2.36(m,20H),2.60(m,4H),3.98(s,8H),4.06(d,8H),4.30(m,12H),4.33(m.8H),4.97(d,4H),5.22(m,4H),5.36(s,4H),5.60(m,4H),5.69(m,8H),6.20(t,4H),7.33(m,8H),7.41(m,8H),7.52(m,8H),7.61(m,4H),7.33(m,4H),8.12(d,8H)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例23、[NP(MPEG550)1.5(赖氨酸乙基)0.2(赖氨酸乙基-2'-乌头酸-喜树碱)0.3]n的合成
根据与示例14的相同方法,将示例1的聚磷腈化合物(9.7g,10mmol)和示例12的2'-乌头酸-喜树碱-NHS酯(2.5g,5.03mmol)反应,得到产率为75%的所需的标题化合物。
组成:C98.6H180.8N5.2O45.8P2
元素分析数据(%):C(52.61);H(8.42);N(3.34)。理论值:C(53.01);H(8.16);N(3.26)。
1H-NMR谱(CDCl3)(δ,ppm):0.9(t,3H,C18-CH3),2.0(m,2H,C19-CH2,2.64(t,4H,NHS-CH2CH2),2.92(s,2H,乌头酸-CH2),4.20(d,2H,C5-CH2),4.76(m,2H,C22-CH2),6.40-6.68(m,1H,乌头酸-CH),6.70(s,1H,C14-CH),7.59(s,1H,C11-CH),7.80(m,2H,C12-CH;C7-CH),8.0(m,2H,C9-CH;C12-CH),1.24(s,1.5H,赖氨酸-OCH2CH3),1.29(bs,1H,赖氨酸-CH2),1.55ppm(bs,1H,赖氨酸-CH2),1.80(bs,39 1H,赖氨酸-CH2),2.90(br,1H,赖氨酸-e-CH2),3.38ppm(s,4.50H,CH3O-,PEG),和3.63ppm(m,66.0H,-CH2CH2-O-),4.4(s,0.51H,赖氨酸-CH)。
31P-NMR谱:(CDCl3,ppm):δ-0.014(s),δ-5.551(s)。
示例24、[NP(MPEG550)(赖氨酸乙基)0.2(乌头酸-甘氨酰喜树碱)]n的合成
根据与示例14中所述相同的方法,将示例5的聚磷腈化合物(0.5g,0.25mmol)和前体(示例13的2'-乌头酸-甘氨酸喜树碱(0.17g,0.25mmol))和DIPEA(10ml)制备出产率为85%的期望的标题化合物。
组成:C111H191N7O50P2
1H-NMR谱(CDCl3)(δ,ppm):0.89-0.92(brm,3H,CPT-C18的-CH3),1.13-1.58(brm,6H,赖氨酸的-CH2),2.12-2.17(brm,2H,CPT-C-19的-CH2),3.01(s,2H,顺式乌头酸盐的-CH2),3.21(s,9H,MPEG的-OCH3),3.34-3.54(brm,144H,MPEG的-CH2-CH2),3.94-4.41(brm,5H,甘氨酸的-CH2,赖氨酸的P-NH-CH2和顺式乌头酸盐的=CH),5.29(brs,2H,CPT-C5的-CH2),5.47(brs,2H,CPT-C22的-CH2),7.15-7.17(m,1H,CPT-C14的=CH),7.69-7.72(m,1H,CPT-C11的=CH),7.84-7.94(m,1H,CPT-C10的=CH),8.10-8.21(m,2H,CPT-C12和CPT-C9的=CH),8.68(brs,1H,CPT-C7的=CH)。
31P-NMR(DMSO,ppm):δ-5.19(O-P-O),0.85(O-P-N)。
示例25、[NP(MPEG550)(AE)(ACA)Pt(dach)]n的合成
将示例8的聚磷腈载体聚合物[NP(MPEG550)(AE)]n(1g,1.5mmol)溶解于碳酸氢钠水溶液(pH=9.0)中,并加入连接基团顺式乌头酸酐(ACA)(2.35g,15mmol),在4℃下进一步反应5h。使用纤维素膜(MWCO:3.5kDa)对反应混合物进行透析,得到具有连接基团的新型聚磷腈中间体[NP(MPEG550)(AE)(ACA)]n。将氢氧化钡(1.33mmol)的甲醇溶液(20ml)加入到经过透析的溶液中,并将反应混合物进一步搅拌5h,以将酸性连接基团转化为聚合物的钡盐。将反应溶液真空蒸发直至干燥。将固体聚合物溶解于蒸馏水(10ml)中,向该聚合物溶液中缓慢加入(dach)Pt(SO4)(dach:反式±1,2-二氨基环己烷)(0.49g,1.21mmol)的水溶液(10ml),并将反应混合物进一步搅拌3h。过滤掉硫酸钡沉淀物后,将滤液用纤维素膜(MWCO:3.5kDa)透析、冻干,得到产率为74%的新型聚磷腈-奥沙利铂偶联物药物。
组成:C39H73N4O19PPt·H2O
元素分析数据(%):C(40.54);H(6.34);N(4.62)。理论值:C(40.83);H(6.54);N(4.88)。
1H-NMR谱(CDCl3)(δ,ppm):1.04-1.20(brm,4H,dach的C-4和C-5),1.44(brs,2H,dach的C-3),1.82-1.93(brm,2H,dach的C-6),2.02-2.52(brm,2H,C-1,dach的C-2),3.26(s,3H,MPEG的OCH3),3.41-3.45(m,6H,顺式乌头酸盐和氨基乙醇的CH2),3.47-3.81(brm,46H,MPEG的-O-CH2),3.95-4.21(brm,2H,MPEG的-P-O-CH2-),4.69(s,1H,顺式乌头酸盐的-C=CH)。
31P-NMR(DMSO,ppm):-4.53(O-P-O)。
示例26、[NP(MPEG750)(AE)(ACA)Pt(dach)]n的合成
根据示例25中所述相同的方法,使用示例9的聚磷腈化合物(1.0g,11.91mmol)、顺式乌头酸酐(1.86g,1.191mmol)、Ba(OH)2·8H2O(0.35g,1.11mmol)和(dach)PtSO4(0.4g,0.99mmol)制备得到产率为72%的所期望的标题化合物。
组成:C47H89N4O23PPt·H2O
元素分析数据(%):C(42.32);H(7.64);N(3.88)。理论值:C(42.65);H(6.88);N(4.23)。
1H-NMR谱(CDCl3)(δ,ppm):1.05-1.21(brm,4H,dach的C-4和C-5),1.47(brs,2H,dach的C-3),1.80-1.93(brm,2H,dach的C-6),2.01-2.45(brm,2H,dach的C-1和C-2),3.27(s,3H,MPEG的-OCH3),3.41-3.45(m,6H,顺式乌头酸盐和氨基乙醇的-CH2),3.50-3.72(brm,62H,MPEG的-CH2),3.99-4.13(brm,2H,MPEG的-P-O-CH2),4.70(s,1H,顺式乌头酸盐的-C=CH)。
31P-NMR(DMSO,ppm):-4.52(O-P-O)。
示例27、[NP(MPEG550)(赖氨酸乙醇)(ACA)Pt(dach)]n的合成
根据示例25中所述相同的方法,使用示例5的聚磷腈化合物(1.0g,1.28mmol)、顺式乌头酸酐(2g,1.191mmol)、Ba(OH)2·8H2O(0.44g,1.39mol)和(dach)PtSO4(0.52,1.28mmol)制备得到产率为79%的所期望的标题化合物。
组成:C45H84N5O20PPt·H2O
元素分析数据(%):C(42.63);H(6.61);N(5.42)。理论值:C(42.88);H(6.83);N(5.56)。
1H-NMR谱(CDCl3)(δ,ppm):1.07-1.21(brm,7H,赖氨酸的-CH3和(CH2)2),1.43-1.48(brm,6H,dach的-C-3、-C-4和C-5),1.80-1.93(brm,8H,赖氨酸的-CH2和dach的-C-6),2.02-2.26(brm,2H,dach的C-1和C-2),2.82(brs,2H,顺式乌头酸盐的CH2),3.26(s,3H,MPEG的-OCH3),3.44-3.72(brm,46H,MPEG的-CH2-CH2),3.96-4.03(brm,3H,乙酯的CH2和赖氨酸的-N-CH),4.62(s,1H,顺式乌头酸盐的=CH)。
物理化学性质和药效的测量
实验1、粒径和胶束形成
将示例1的聚磷腈载体化合物和示例11的聚磷腈-多西紫杉醇偶联物分别溶解于蒸馏水(0.2%)中,并通过DLS(动态光散射)方法测量其粒径分布和ζ电位,结果示于图1、2和3。
图1示出了聚磷腈载体聚合物的粒径分布,其平均直径为约3~4nm,对应于未组装的聚合物的流体动力学体积的粒径,这可能是由于聚合物的赖氨酸胺基团的阳离子性质,如图2所示。然而,如图3所示,聚磷腈-多西紫杉醇偶联物的粒径增加到60nm,这清楚地表明,由于通过将引入疏水多西紫杉醇分子引入到载体聚合物而导致偶联分子产生两亲性,偶联药物分子自组装形成了较大的胶束纳米颗粒。进一步证实在5~70℃的温度范围内胶束尺寸没有显着变化。
实验2、聚磷腈-紫杉醇偶联物的临界胶束浓度(CMC)的测量。
如上所述,本发明的聚磷腈-多西紫杉醇偶联物在水溶液中形成聚合胶束。为了将这种聚合物-药物偶联物在临床上用于IV注射,由聚合物-药物偶联物自组装的胶束的溶液稳定性是关键的。这种胶束稳定性用“临界胶束浓度(CMC)”表示,其通过多种方法测量,但最广泛使用的方法是“芘荧光法”(K.Kalyanasundoram等人,J.Am.Chem.Soc.1988,99,2039)。根据该方法,本发明的聚磷腈-多西紫杉醇偶联物的CMC值的测量如下:
制备芘水溶液(6×10-7M),并使用该溶液制备一系列浓度范围为5.0~0.0005%(重量/重量)的示例17的聚磷腈-紫杉醇偶联物的样品溶液。在339nm(Iex)和390nm(Iem)处测量荧光光谱,然后,如图4所示,根据谱带I和谱带III的荧光强度的比率,测定CMC的值。
由此获得的聚磷腈-紫杉醇偶联物的CMC值为41mg/L,其非常低,并且当静脉注射时,在血液系统中聚合物胶束将会是稳定的。
实验3、聚磷腈-药物偶联物的生物可降解性。
仪器:Yonglin GPC系统
柱:Waters水凝胶HR柱(1×保护,1×线性,2×HR2)
流出物:水/乙腈(8:2)(0.5%NaNO3)
流速:1ml/min
通过将示例17的聚磷腈-紫杉醇偶联物(250mg)溶解在pH=5.4的缓冲溶液(5ml)和另一种pH=7.4的缓冲溶液(5ml)中,制备出两种样品溶液。在37℃下将两种样品溶液在水浴中缓慢振荡,并且在预定的时间(培养后0.5、1、2、4、6、8、16天)取500μl样品溶液并冻干。将干燥的样品溶解在含有0.2%叔丁基溴化铵的四氢呋喃中,并经过凝胶渗透色谱分析(GPC)。根据GPC数据,在图5中示出了聚磷腈-紫杉醇偶联药物的降解模式。
从图中可以看出,聚磷腈偶联药物的平均分子量首先在几天内快速下降,但是特别在中性介质下其降解降低。预计聚磷腈主链在血液系统中的半衰期为约16天,但最令人鼓舞的是,聚磷腈主链在酸性介质中是连续可降解的,这类似于与药物释放动力学相关的肿瘤微环境。在本发明中,将所有最终聚合物产物分级成不同的分子量,研究它们的生物降解性、排泄性和药物释放动力学。
实验4、通过成像研究聚磷腈化合物的肿瘤靶向性
仪器:Kodak成像站4000mm电子成像系统(Kodak,New Haven,CT)
激发和发射过滤器:Omega Optical,Battlebor,VT(激发:560nm,发射:700nm)
购买十只八周龄的CH3/HeN裸鼠(东京医科大学医学研究所),并在用非小细胞肺癌A549(1×106)接种适应一周后。当肿瘤尺寸生长至约300mm3时,将小鼠分为两组,一组注射用荧光染料Cy5.5标记的示例1的聚磷腈载体化合物,另一组不作处理作为参照组。在预定时间(IV注射后12h,24h,48h,72h),处死小鼠,分离全部主要器官(肝、肺、肾、脾、肿瘤、肌肉),利用CCD相机进行NIR荧光成像(Kodak成像站4000MM)研究,结果示于图6中。
从图中器官的荧光强度,与其它器官相比,示例1的聚磷腈化合物清楚地显示出其主要聚积在肿瘤组织中,尽管其具有3-4nm的小粒径而不能产生EPR效应。因此,可以推测出,该聚磷腈载体聚合物的肿瘤选择性是由于聚合物的赖氨酸游离胺导致其所具有的阳离子性质和由于其PEG化结构而引起的长血液循环。
实验5、聚磷腈-多西紫杉醇偶联药物的肿瘤靶向性
示例12的聚磷腈-多西紫杉醇偶联物用Cy5.5标记,并且以与实验4(图7)相同的方式比较其器官分布。通过将用Cy5.5标记的偶联药物处理的小鼠的每个器官的荧光强度与未处理的小鼠的每个器官的荧光强度进行比较,获得偶联药物的定量生物分布数据,结果示于图8中。
图7显示出偶联药物主要在肿瘤中聚积,并且特别地,该聚积在注射后48h后达到最大值,这可能是由于偶联物的较大粒度(60nm)的EPR效应(提高的渗透性和滞留效应)和长血液循环。图8所示的聚磷腈-多西紫杉醇偶联药物的生物分布的定量数据清楚地显示,与之前的聚磷腈载体聚合物由于其阳离子性造成的肿瘤选择性相比,该偶联药物的肿瘤选择性要高得多,这是由于其大粒径的EPR效应。
实验6、聚磷腈-多西紫杉醇偶联物中多西紫杉醇含量的分析。
聚磷腈-药物偶联物中药物组分的含量可以通过1H NMR光谱、UV光谱或HPLC测定。在质子NMR光谱法中,偶联药物中的多西紫杉醇含量可以从接枝到聚合物主链的PEG基团在3.4ppm(3H-CH3)处的甲氧基质子与被偶联的多西紫杉醇在7.33ppm(C10,2H)处的C10质子的积分面积之比估算而得。然而,甲氧基质子和多西紫杉醇C10质子的化学位移都与相邻的质子峰轻微重叠,这影响了积分比的准确性。在HPLC方法中,通过酸性分解从偶联药物中释放出的游离多西紫杉醇的总量可以通过HPLC来测量,但是不能得到可重复的结果。
另一方面,使用UV光谱法可以获得可重复的结果,因为多西紫杉醇分子在230nm处显示出强的UV吸收,而聚磷腈载体聚合物在约230nm处几乎没有吸收。因此,使用水和乙腈(1:1)的溶剂混合物(多西紫杉醇在其中可完全溶解)测量的校准曲线。通过使用10.0mg多西紫杉醇/10ml溶剂混合物的标准溶液及其稀释溶液(1mg/ml,1/2mg/ml等)在230nm处测量的UV吸收而得到校准曲线。
实验7、从聚磷腈-多西紫杉醇偶联物的体外药物释放
仪器:配备DAD检测器(230nm)的Agilent 1100系列
柱:Agilent Zobax Eclipse Plus C18柱(直径=4.6mm;长度=150mm,粒径=3.5μm)
流速:1.p ml/min
洗脱液组成:A:含0.1%TFA的H2O;B:乙腈(等度法)
使用HPLC方法进行聚磷腈-多西紫杉醇偶联物的体外药物释放实验。使用与实验6相同的方式得到的校准曲线测定从偶联物释放的药物多西紫杉醇的量。
实验8、聚磷腈-紫杉醇偶联物的细胞毒性的体外试验。
众所周知,由于抗癌剂紫杉醇是在临床上对许多不同的癌症非常有效,选择乳腺癌(MCF-7)、卵巢癌(SK-OV3)、非小细胞肺癌(A549)和胃癌(SNU638)癌细胞系,根据文献方法(SRB方法)(Rita Song等人,J.Control.Release 105(2005)142-150)测试其体外细胞毒性。
结果示于下表1中。从表中可以看出,示例20和21的聚磷腈-紫杉醇偶联物的IC50值比游离紫杉醇高得多,因为在聚磷腈-紫杉醇偶联物降解后,疏水性紫杉醇组分非常难以从聚合物胶束核中释放出来。
表1聚磷腈-紫杉醇偶联物的体外细胞毒性
实验9、聚磷腈-多西紫杉醇偶联物的药代动力学研究。
为了检测示例12的聚磷腈-多西紫杉醇偶联物相比于目前临床使用的非偶联而配制的“泰索帝”的药代动力学,采用Sprague-Dawley大鼠根据文献方法(Jun等人,Int.J.Pharm.422(2012)374-380)对偶联物进行药代动力学研究。时间依赖性血浆浓度示于图9,药代动力学参数示于表2中。
表2示例14和作为参照的泰索帝的药代动力学参数
实验10、聚磷腈-多西紫杉醇偶联物的裸鼠异种移植试验。
为了评价示例12相比于作为参照的泰索帝体外效应,根据文献方法(Y.J.Jun等人,Int J.Pharm.422(2012)374-380),使用BALB/C裸鼠进行针对胃癌细胞系MKN-28的裸鼠异种移植试验。由于泰索帝的已知最佳剂量为10mg/kg,基于聚磷腈-多西紫杉醇偶联物中多西紫杉醇的含量,将示例14的一次注射剂量确定为10mg/kg和20mg/kg,给药三次(第1、5、9天)。从第一次注射开始至40天,测量每个的肿瘤尺寸和体重。图10示出对MKN-28胃癌肿瘤细胞的抗肿瘤活性,图11示出了40天内的体重变化。从图10中可以看出,聚磷腈-多西紫杉醇偶联物显示出与泰索帝相当的强抗肿瘤活性,更重要的结果可以从图11看出,在药物注射期间用泰索帝处理的小鼠的平均体重显着降低(>10%),而对于偶联药物却没有观察到显着的体重变化,这意味着本发明的偶联药物显示较低的全身毒性。对非小细胞肺癌细胞系A549进行的异种移植试验的结果示于图12,其显示处本发明的偶联药物表现出甚至比参照泰索帝更好的抗肿瘤活性。
- 还没有人留言评论。精彩留言会获得点赞!