抗ErbB2抗体-药物偶联物及其组合物、制备方法和应用与流程
本发明属于生物医药
技术领域:
。具体而言,本发明涉及抗ErbB2抗体-药物偶联物、包含其的组合物、其制备方法和应用。
背景技术:
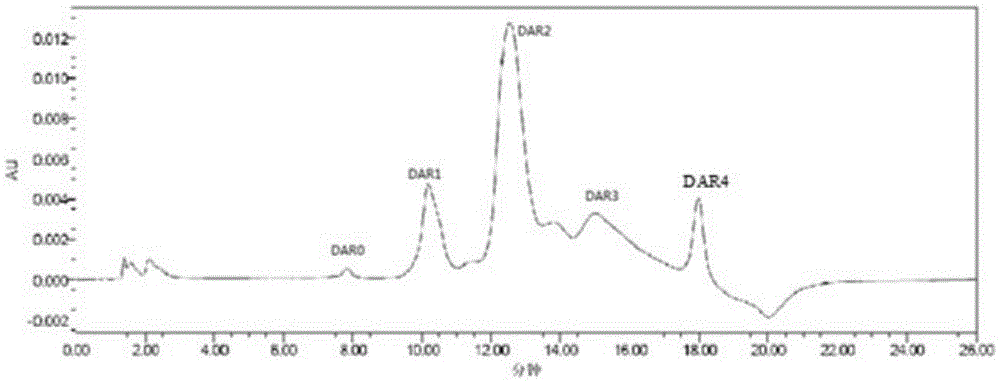
:作为新型的靶向治疗药物,抗体-药物偶联物(antibody-drugconjugate,ADC)开创了肿瘤治疗方法的新纪元。在美国西雅图基因公司(SeattleGenetics,Inc.)和免疫原公司(ImmunoGen,Inc.)的引领下,很多跨国制药企业和初创公司都开展了这一领域的研发。根据MarketResearch的报道,全球共有45个此类候选药处于临床研究中。ADC一般包括采用一定方式连接的三个部分:抗体、连接基和药物。抗体是良好的药物靶向性载体。利用药物分子中的特定官能团如羟基、巯基或氨基,可将药物与抗体连接,组成化学免疫偶联物。抗体的靶向性能将与之相连的药物“精确”地运送到靶细胞,从而有效提高病灶局部的药物浓度,同时极大地降低体内其他组织、器官的药物浓度,从而实现增效减毒。用于这些策略的多克隆抗体和单克隆抗体均已有报导(Rowland等人,1986,CancerImmunol.Immunother.,21:183-87)。目前临床上所用的ADC中的抗体大多是人源化抗体,例如,PSMAADC(抗PSMA抗体-MMAE偶联物)、SGN-75(抗CD70抗体-MMAF偶联物)、T-DM1(曲妥珠单抗-DM1偶联物)中所用均为人源化抗体。目前FDA批准的ADC药物包括(T-DM1)和(brentuximabvedotin)。ADC中作为“弹头”的药物通常为细胞毒性药物,它们主要通过抑制细胞DNA或蛋白质合成、抑制细胞有丝分裂等方式来杀伤肿瘤细胞。由于细胞毒性药物对正常细胞同样具有较大杀伤力,因而其应用和发展受到极大限制。早期的ADC利用常规抗肿瘤药物,但这些ADC临床活性大多低于化学药单体的体外活性。目前的ADC所用的细胞毒性药物主要包括:美登素类(Maytansinoids,参见例如EP0425235、US5208020、US5416064、US7276497、US7473796、US7851432、US2007/0269447、US2011/0158991、WO2004/103272、WO2012/061590)、耳抑素肽类(Auristatins,参见例如US6884869、US7498298)、卡奇霉素类(Calicheamicins,参见例如US5606040、US5770710)、阿霉素类(Doxorubicins,参见例如Dubowchik等人,2002,BioconjugateChem.,13:855-869)、苯并二吡咯类抗生素(duocarmycins和CC-1065,参见例如US7129261)、伊立替康代谢产物(Irinotecanmetabolites,参见例如WO2015/012904)、吡咯并苯二氮卓类(Pyrrolobenzodiazepines,参见例如Biotechnol.Healthc.2012Winter,9(4):28-31)以及吡咯并苯二氮卓二聚体类(PBDdimmers,参见例如WO2005/040170)等。这些细胞毒性药物具有很强的非选择性毒性,会对正常细胞造成伤害,因而本身不能成药。ADC中的连接基需满足以下要求:在细胞外具有足够的稳定性,保证小分子药物不与抗体脱离;进入细胞后,可断裂的连接基在适当条件下断裂,释放出活性小分子药物,而不可断裂的连接基则与小分子药物和来自抗体酶解的氨基酸残基一起形成活性部分。在目前进入临床试验的ADC中,细胞毒性药物通常籍由连接基连接在抗体表面的赖氨酸残基上,或者连接在抗体铰链区的半胱氨酸残基(由链间二硫键部分还原得到)上。通常的药物/抗体比值(drugantibodyratio,DAR)为2-4。当药物连接在抗体表面的赖氨酸残基上时,由于抗体表面存在大量(超过80个)赖氨酸残基并且偶联反应是非选择性的,因而偶联数目和位点具有不确定性,导致生成的ADC的不均一性。例如,T-DM1的DAR值分布为0-8,平均DAR值为3.5(Lazar等人,2005,RapidCommun.MassSpectrom.,19:1806-1814)。当药物连接在抗体铰链区的半胱氨酸残基上时,由于抗体铰链区的链间二硫键只有四对,为了达到平均DAR值为2-4的要求,需要部分还原链间二硫键(Sun等人,2005,BioconjugateChem.,16:1282-1290),然而现有的还原剂如二硫苏糖醇(DTT)和磷酸三氯乙酯(TCEP)等无法选择性地还原链间二硫键,因此所得抗体-药物偶联物也不是均一产物,而是由多种组分组成的混合物,其中主要组分的DAR值为0、2、4、6、8,而且对应每一DAR值的组分又存在由于连接位点不同而形成的异构体。ADC产品的不均一性对于其临床应用是不利的,因为产品中各组分间药代动力学性质、效价和/或毒性不均一(例如,具有较高DAR值的组分在体内被清除得更快,并导致更高的毒性,参见例如Boswell等人,2011,BioconjugateChem.,22:1994-2004),使得其稳定性难以令人满意。ErbB族受体酪氨酸激酶是细胞生长、分化和存活的重要介质。该家族包括四个成员:上皮生长因子受体(EGFR或ErbB1)、Her2(ErbB2)、Her3(ErbB3)和Her4(ErbB4)。临床上用抗ErbB2抗体曲妥珠单抗治疗ErbB2高表达的乳腺癌。在临床试验中,15%的具有免疫组织化学(IHC)2+以上水平的乳腺癌患者对曲妥珠单抗产生临床反应,反应持续中值为9.1个月(参见例如Cobleigh等人,1996,临床肿瘤学杂志,14:737-744)。曲妥珠单抗(商品名Herceptin)在1998年9月25日被美国食品与药品管理局(FDA)批准用于治疗过量表达ErbB2蛋白的乳腺癌患者。曲妥珠单抗虽然挽救了一些乳腺癌患者或延长了患者的生存期,但其仅对ErbB2高表达的患者有效,且临床反应率仅有15%。因此,还需要对更多患者具有更佳疗效的药物。为了提高治疗指数,已将曲妥珠单抗与美登木素(DM1)偶联,制成trastuzumabemtansine(T-DM1,Kadcyla),用于使用曲妥珠单抗、其它抗Her2治疗药物及常用乳腺癌治疗一线化疗药物紫杉烷类治疗无效的患者。T-DM1将药物输送至肿瘤部位,从而缩小肿瘤,减慢疾病进展并延长生存期。在临床试验中,T-DM1的安全性和有效性得到了评价。根据FDA批准Kadcyla的说明书第19页,991名患者参与的临床试验研究结果显示,Kadcyla治疗组的中位无进展生存期为9.6个月,而拉帕替尼+卡培他滨治疗组的中位无进展生存期为6.4个月;Kadcyla治疗组的中位总生存期为30.9个月,而拉帕替尼+卡培他滨治疗组的中位总生存期为25.1个月。T-DM1现已成为FDA批准的第四个以Her2蛋白为靶点的药物。但是,T-DM1在批准时有一项黑框警告,提醒患者与卫生保健专业人员该药物可导致肝毒性、心脏毒性和死亡。这主要是脱落的DM1以及T-DM1进入体内降解释放出的含DM1小分子引起的。T-DM1的制备是利用曲妥珠单抗上的赖氨酸侧链氨基与磺基琥珀酰亚氨基-4-[N-顺丁烯二酰亚氨基甲基]环己烷-1-羧酸酯(磺基-SMCC)偶联,然后再与DM1偶联。T-DM1中,每分子曲妥珠单抗上偶联的DM1分子平均为3.5个。由于曲妥珠单抗上具有88个赖氨酸位点,因而T-DM1实际为曲妥珠单抗偶联了不同数目DM1和具有多种偶联位点的多种抗体-药物偶联物的组合。然而,每种抗体-药物偶联物的药效、药代动力学性质和/或毒性不同。一般而言,载药量高的抗体-药物偶联物具有更强的体外活性,但高载药量也会带来诸多问题,如因抗体-药物偶联物之间聚合而生成的多聚体成分增加、稳定性下降、毒性增加、免疫原性提高、体内清除速度过快、半衰期过短而实际治疗指数不高。本发明旨在解决现有技术中的上述问题。本发明提供抗Her2抗体-药物偶联物,其抑制哺乳动物肿瘤生长,可用于治疗多种癌症。所述偶联物具有更好的生物学活性、稳定性和均一性,并具有降低的毒副作用。技术实现要素:在第一方面,本发明提供通式(I)的抗体-药物偶联物,其药学上可接受的盐或立体异构体,或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物,其中:A为抗ErbB2抗体或者其活性片段或变体;X和Y各自独立地为N或CR1,且每个R1独立地为H或C1-C10烷基;L为二价连接基;D为细胞毒性药物基团;并且a为选自2-10的整数。在第二方面,本发明提供本发明第一方面的抗体-药物偶联物的制备方法,所述方法包括以下步骤:(1).制备通式(I-A)的化合物:其中D、L、X和Y如上文关于通式(I)所定义;(2).活化步骤(1)中得到的通式(I-A)的化合物,得到通式(I-A-G)的化合物其中G选自-F、-Cl、-Br、-I、-N3、-OR、-SR、-ONRR’、RC(O)O-、-OP(O)RR’、RSO2-O-和其中R和R’每次出现时各自独立地是C1-C10烷基、C6-C14芳基、含5-10个环原子的杂环基或苯氧基,所述烷基、芳基、杂环基和苯氧基是未取代的或者各自独立地被一个或多个选自卤素、羟基、C1-C4烷基、C1-C4烷氧基、C3-C8环烷基、含5-8个环原子的杂环基、C6-C10芳基或含5-10个环原子的杂芳基的取代基取代;(3).将步骤(2)中得到的通式(I-A-G)的化合物偶联于所述抗ErbB2抗体或者其活性片段或变体,得到具有不同a值的多种抗体-药物偶联物的混合物;和(4).纯化步骤(3)中得到的混合物,得到抗体-药物偶联物。在第三方面,本发明提供药物组合物,其包含本发明第一方面的抗体-药物偶联物或者其药学上可接受的盐或立体异构体或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物,以及药学上可接受的载体。在第四方面,本发明提供本发明第一方面的抗体-药物偶联物或者其药学上可接受的盐或立体异构体或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物在制备用于预防或治疗癌症的药物中的用途。在第五方面,本发明提供药物制剂,其包含本发明第一方面的抗体-药物偶联物或者其药学上可接受的盐或立体异构体或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物。在第六方面,本发明提供预防或治疗癌症的方法,所述方法包括向有此需要的患者给药本发明第一方面的抗体-药物偶联物,其药学上可接受的盐或立体异构体,或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物,或者向有此需要的患者给药本发明第三方面的药物组合物或本发明第五方面的药物制剂。附图说明图1是抗体-药物偶联物I-1粗产物的HIC-HPLC谱图。图2是抗体-药物偶联物I-1的HIC-HPLC谱图。图3是抗体-药物偶联物I-1与曲妥珠单抗裸抗还原的重叠反相色谱图。图4是将抗体-药物偶联物I-1和曲妥珠裸抗在相同条件下进行酶切所得的肽图谱。图5是抗体-药物偶联物I-2粗产物的HIC-HPLC谱图。图6是抗体-药物偶联物I-2的HIC-HPLC谱图。图7是抗体-药物偶联物I-2与曲妥珠单抗裸抗还原的重叠反相色谱图。图8是抗体-药物偶联物I-3粗产物的HIC-HPLC谱图。图9是抗体-药物偶联物I-3的HIC-HPLC谱图。图10是抗体-药物偶联物I-3与曲妥珠单抗裸抗还原的重叠反相色谱图。具体实施方式除非另有定义,本文使用的所有术语具有与本领域普通技术人员通常所理解相同的含义。相关的定义及术语可参见例如CurrentProtocolsinMolecularBiology(Ausubel)。本文中所述的数值范围应理解为涵盖其中包含的任何和所有子范围。例如,范围“1至10”应理解为不仅包括明确记载的1至10的值,而且还包括1至10范围内的任何单个值(例如2、3、4、5、6、7、8和9)和子范围(例如1至2、1.5至2.5、1至3、1.5至3.5、2.5至4、3至4.5等等)。该原则亦适用于仅用一个数值作为最小值或最大值的范围。本文中提及的文献均以其整体援引加入本文中。在第一方面,本发明提供通式(I)的抗体-药物偶联物,其药学上可接受的盐或立体异构体,或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物,其中:A为抗ErbB2抗体或者其活性片段或变体;X和Y各自独立地为N或CR1(优选Y为CR1,X为N),且每个R1独立地为H或C1-C10烷基,优选为H或C1-C6烷基(例如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、戊基或己基);L为二价连接基;D为细胞毒性药物基团;并且a为选自2-10的整数,例如2、3或4,特别优选2。抗体本发明中所用的抗体为抗ErbB2抗体或者其活性片段或变体,包括双特异性抗体和抗体功能性衍生物。在本文中,ErbB2和Her2可互换使用,二者均表示天然序列的人Her2蛋白(Genebank登录号:X03363,参见例如Semba等人,1985,PNAS,82:6497-6501;和Yamamoto等人,1986,Nature,319:230-234)及其功能性衍生物,例如氨基酸序列变体。erbB2表示编码人Her2的基因,neu表示编码大鼠p185neu的基因。本发明中优选的Her2是天然序列的人Her2。可用于本发明的抗Her2的抗体的实例包括但不限于:MAbs4D5(ATCCCRL10463)、2C4(ATCCHB-12697)、7F3(ATCCHB-12216)和7C2(ATCCHB12215),这些抗体记载于例如US5,772,997、WO98/77797和US5,840,525中。人源化抗Her2抗体包括US5,821,337表3中所列的huMAb4D5、huMAb4D5-1、huMAb4D5-2、huMAb4D5-3、huMAb4D5-4、huMAb4D5-5、huMAb4D5-6、huMAb4D5-7和huMAb4D5-8;CN1370082A图1B中所示的7C2、7F3、2C4、7D3、3E8和2C4。可用于本发明的抗Her2抗体的实例还可以包括:WO2009/123894中记载的F243L,R292P和Y300L;F243L,R292P和V305I;F243L,R292P和P396L;以及R292P,V305I和P396;WO2012/079093中记载的MM-111;CN104418952A权利要求2至10中记载的TPs、TPL、PTs或PTL抗体。WO2010/108127图33A和图33B中记载的Dl、Dl.5、Dl.5-100、DLIl、DLlIb或DLlIf抗体;以及WO93/21319中记载的人源化520C9。本发明中的天然序列的Her2可以从自然界分离得到,也可以通过重组DNA技术、化学合成法或它们的组合制备得到。术语“功能性衍生物”包括氨基酸序列变体以及天然多肽的共价衍生物(例如通过翻译后修饰、焦谷氨酸化等获得的衍生物),条件是它们保留与天然多肽相当或更高的亲和力和生物活性。氨基酸序列变体与天然多肽氨基酸序列的差异一般在于后者中的一个或多个氨基酸的取代、缺失和/或插入。缺失变体包括天然多肽的片段和N端和/或C端截短变体。通常,氨基酸序列变体与天然多肽应具有至少70%或至少80%或至少90%的同源性。天然多肽的共价衍生物可以是通过改变抗体的翻译后加工,例如改变糖基化位点的数目或位置获得的衍生物。术语“同源性”可与“一致性”、“同一性”或“相似性”互换使用,是指在最佳比对(最优比对)后所获得的待比较的两种核酸序列或者氨基酸序列之间相同核苷酸或相同氨基酸残基的百分数,该百分数是纯粹统计学的并且两种序列间的差异随机分布并覆盖其全长。两种核酸序列或者氨基酸序列之间的比对通常是在以最优方式使它们匹配后比较序列而进行,所述比较可通过区段或者通过“比较窗”来实施。除了手工实施外,最优比对还可以通过记载于文献中的其他方法进行,例如Smith和Waterman,1981,Ad.App.Math.,2:482中记载的局部同源性算法,Neddleman和Wunsch,1970,J.MoI.Biol.,48:443中记载的局部同源性算法,Pearson和Lipman,1988,Proc.Natl.Acad.Sci.USA,85:2444中记载的相似性搜索方法,并且可以通过使用这些算法的计算机软件(GAP、BESTFIT、FASTA和TFASTAintheWisconsinGeneticsSoftwarePackage,GeneticsComputerGroup,575ScienceDr.,Madison,WI)实施,或者通过BLASTN或BLASTP比较软件实施。在本文中,术语“抗体”取其最广义的解释,包括完整的单克隆抗体、多克隆抗体以及由至少两个完整抗体形成的多特异性抗体(例如双特异性抗体),只要它们具有所需的生物学活性。在本文中,术语“单克隆抗体”指抗体来自一群基本均一的抗体,即构成该集群的各抗体完全相同,除了可能存在的少量天然突变。单克隆抗体具有针对抗原的一个决定簇(表位)的高特异性,而与其相对的多克隆抗体则包含针对不同决定簇(表位)的不同抗体。除了特异性之外,单克隆抗体的优点还在于合成时可以不受其他抗体的污染。此处修饰语“单克隆”表示该抗体的特征在于来自一个基本均一的抗体群,而不应理解成需由特殊方法制得。在本发明中,单克隆抗体还特别包括嵌合抗体,即重链和/或轻链的一部分与某种、某类或某亚类抗体相同或同源,其余部分则与另一种、另一类或另一亚类抗体相同或同源,只要它们具有所需的生物学活性(参见例如US4,816,567;和Morrison等人,1984,PNAS,81:6851-6855)。可用于本发明的嵌合抗体包括灵长类化(primatized)抗体,其包含来自非人灵长类(例如古猴、猩猩等)的可变区抗原结合序列和人恒定区序列。术语“抗体片段”是指抗体的一部分,优选是抗原结合区或可变区。抗体片段的实例包括Fab、Fab′、F(ab′)2和Fv片段;二抗体(diabody);线性抗体;和单链抗体分子。术语“双特异性抗体”与“双功能抗体偶联物”可互换使用,指由第一抗体(片段)和第二抗体(片段)通过偶联臂所形成的偶联物,该偶联物保留了各自抗体的活性,故具有双功能和双特异性。术语“多特异性抗体”包括例如三特异性抗体和四特异性抗体,前者是具有三种不同抗原结合特异性的抗体,而后者是具有四种不同抗原结合特异性的抗体。术语“完整抗体”指包含抗原结合可变区和轻链恒定区(CL)、重链恒定区(CH1、CH2和CH3)的抗体。恒定区可以是天然序列(例如人天然恒定区序列)或其氨基酸序列变体。完整抗体优选是具有一种或多种效应功能的完整抗体。非人(例如鼠)抗体的“人源化”形式指包含最少量非人免疫球蛋白序列的嵌合抗体。大多数人源化抗体是人接受者免疫球蛋白的超变区残基被置换成具有所需特异性、亲和力和功能的非人(例如小鼠、大鼠、兔或非人灵长类)超变区残基(供者抗体)。在一些实施方案中,人免疫球蛋白的框架区(FR)残基也被置换成非人残基。而且,人源化抗体还可以包含受者抗体或供者抗体中没有的残基。这些修饰是为了进一步优化抗体的性能。人源化抗体一般包含至少一个,通常是两个可变区,其中所有或几乎所有超变环(hypervanableloops)与非人免疫球蛋白的相对应,而FR则完全或几乎完全是人免疫球蛋白的序列。人源化抗体还可以包含免疫球蛋白恒定区(Fc,通常是人免疫球蛋白Fc)的至少一部分。有关细节参见例如Jones等人,1986,Nature,321:522-525;Riechmann等人,1988,Nature,332:323-329;和Presta,1992,CurrOpStructBwl2:593-596。完整抗体可根据重链恒定区的氨基酸序列分为不同的“类”。主要的五类是IgA、IgD、IgE、IgG和IgM,其中几类还可以分为不同的“亚类”(同种型),例如IgG1、IgG2、IgG3、IgG4、IgA1和IgA2。抗体不同类的重链恒定区分别称为α、β、ε、γ和μ。免疫球蛋白不同类的亚基结构和三维构型是本领域中公知的。在本发明中,尽管大多数情况下抗体中的氨基酸取代是被L-氨基酸取代,但也不限于此。在一些实施方案中,抗体肽链中可以包括一个或多个D-氨基酸。认为包含D-氨基酸的肽在口腔、肠道或血浆中比仅包含L-氨基酸的肽更加稳定而不易降解。本发明所用的单克隆抗体可以由许多方法生产。例如,用于本发明的单克隆抗体可以通过杂交瘤方法,使用许多物种(包括小鼠、仓鼠、大鼠和人的细胞)获得(参见例如Kohler等人,1975,Nature,256:495),或者通过重组DNA技术制得(参见例如US4,816,567),或者从噬菌体抗体库中分离得到(参见例如Clackson等人,1991,Nature,352:624-628;和Marks等人,1991,JournalofMolecularBiology,222:581-597)。本发明中所用的抗体优选为抗人ErbB2抗体,并且优选地,所述抗人ErbB2抗体中的CDR1、CDR2和/或CDR3分别为曲妥珠单抗的CDR1、CDR2和/或CDR3。所述抗人ErbB2抗体可以为人源化抗体或全人源抗体。本发明中所用的抗体更优选为US5,821,337图1中所示的鼠源抗人Her2抗体4D5的人源化抗体。本发明中所用的抗体特别优选为曲妥珠单抗。曲妥珠单抗重链序列为:EVQLVESGGGLVQPGGSLRLSCAASGFNIKDTYIHWVRQAPGKGLEWVARIYPTNGYTRYADSVKGRFTISADTSKNTAYLQMNSLRAEDTAVYYCSRWGGDGFYAMDYWGQGTLVTVSSASTKGPSVFPLAPSSKSTSGGTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSSLGTQTYICNVNHKPSNTKVDKKVEPPKSCDKTHTCPPCPAPELLGGPSVFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVKFNWYVDGVEVHNAKTKPREEQYNSTYRVVSVLTVLHQDWLNGKEYKCKVSNKALPAPIEKTISKAKGQPREPQVYTLPPSRDELTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPVLDSDGSFFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK曲妥珠单抗轻链序列为:DIQMTQSPSSLSASVGDRVTITCRASQDVNTAVAWYQQKPGKAPKLLIYSASFLYSGVPSRFSGSRSGTDFTLTISSLQPEDFATYYCQQHYTTPPTFGQGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC抗体片段可由多种方法方法生产,例如通过完整抗体的蛋白水解来生产(参见,例如Morimoto等人,1992,JournalofBiochemicalandBiophysicalMethods,24:107-117;和Brennan等人,1985,Science,229:81),或者由重组宿主细胞和以上讨论的抗体噬菌体文库直接生产,或者直接从大肠杆菌回收Fab'-SH片段并进行化学偶联以形成F(ab')片段(Carter等人,Biotechnology(NY),1992Feb,10(2):163-167。此外,可以直接从重组宿主细胞培养物中分离F(ab')片段。其它生产抗体片段的方法是本领域技术人员公知的。在一些实施方案中,本发明所用的抗体片段是单链Fv片段(scFv)(参见例如WO93/16185、US5,571,894以及US5,587,458)。在一些实施方案中,抗体片段是线性抗体片段(参见例如US5,641,870),其可以是单特异性的或双特异性的。具有针对至少两个不同表位的结合特异性的双特异性抗体(参见例如Millstein等人,1983,Nature,305:537-539)可以结合ErbB2蛋白的两个不同表位。其它双特异性抗体可以将ErbB2结合部位与EGFR、ErbB3和/或ErbB4结合部位组合。或者,抗ErbB2臂可以与结合白细胞上的激发分子如T细胞受体分子(例如CD2或CD3)的臂组合,或着与IgG的Fc受体(FcγR)如FcγRI(CD64)、FcγRII(CD32)和FcγRIII(CD16)的臂组合,以将细胞的防御机制集中于表达ErbB2的细胞。双特异性抗体也可用于将细胞毒性试剂定位于表达ErbB2的细胞(参见例如WO96/16673、US5,837,234、WO98/02463和US5,821,337)。已有文献披露了双特异性抗体的纯化方法(参见例如WO93/08829;Traunecker等人,1991,EMBOJ.,10:3655-3659;WO94/04690;Suresh等人,1986,MethodsinEnzymology,121:210;US5,731,168)。可以利用亮氨酸拉链(参见例如Kostelny等人,1992,J.Immunol.,148(5):1547-1553)和单链Fv(sFv)二聚体(参见例如Gruber等人,1994,J.Immunol.152:5368)生产双特异性抗体。已有文献描述了从抗体片段生产双特异性抗体的技术,例如利用完整抗体在其位置经蛋白水解裂解产生F(ab')片段的化学键(Brennan等人,1985,Science,229:81)。可以从大肠杆菌回收Fab'-SH片段并化学偶联以形成双特异性抗体(参见Shalaby等人,1992,J.Exp.Med.,175:217-225)。“双抗体(diabodies)”技术提供了制备双特异性抗体片段的替代方法(参见Hollinger等人,1993,Proc.Natl.Acad.Sci.USA,90:6444-6448)。可以制备超过二价的抗体。可以通过编码抗体多肽链的核酸的重组表达容易地生产具有三个或更多个抗原结合部位和两个或更多个可变区的多价“章鱼”样抗体(参见例如US2002/0004586和WO01/77342)。例如,可以制备三特异性抗体(参见例如Tutt等人,1991,J.Immunol.,147:60)。通过“结-入-穴”技术,可改变三或四特异性抗体(重链中或经修饰的重链中)的CH3域,“结-入-穴”技术及数个实例详细记载于例如WO96/027011,Ridgway,J.B.等人,1996,ProteinEng.,9:617-621;及Merchant,A.M.等人,1998,Nat.Biotechnol.,16:677-681中。在这种方法中,改变两种CH3域的相互作用界面以提高含有这两种CH3域的两种重链的异二聚化。(两种重链的)两种CH3域的每一个均可以是“结”,而另一是“穴”。引入二硫桥进一步稳定异二聚体(参见例如Merchant,A.M.等人,1998,NatureBiotech.,16:677-681;和Atwell,S.等人,1997,J.Mol.Biol.,270:26-35)及提高产量。通常通过改变基本的核酸序列来改变氨基酸序列。编码抗体氨基酸序列变体的核酸分子可通过本领域已知的多种方法制备。这些方法包括但不限于从天然来源分离(在天然发生的氨基酸序列变体的情况下)或者通过寡核苷酸介导的(或定点)诱变、PCR诱变以及对抗体的早先制备的变体或非变异型式所进行的盒式诱变。最感兴趣的取代诱变部位包括高变区,但也可预期FR改变。如CN103319599A实施例2利用常规分子生物学技术对全基因合成编码曲妥珠单抗重链的DNA片段进行定点突变,将曲妥珠单抗突变体重链K30R克隆至抗体重链表达载体上,酶切和连接的操作按商业提供的试剂盒说明书进行。药物本发明中所用的药物为细胞毒性药物。本文中术语“细胞毒性药物”是指抑制或阻止细胞功能和/或引起细胞破坏的物质。在一些实施方案中,所述细胞毒性药物是耳抑素肽类,如耳抑素E(auristatinE,亦称为海兔毒素(dolastatin)-10衍生物)或其衍生物(例如由耳抑素E和酮酸形成的酯)。举例来说,耳抑素E可与对乙酰基苯甲酸或苯甲酰基戊酸反应,分别产生AEB(耳抑素EB)及AEVB(5-苯甲酰基戊酸耳抑素E酯)。其它典型耳抑素肽类包括AFP(耳抑素F苯二胺)、MMAF(一甲基耳抑素F)及MMAE(一甲基耳抑素E)。示例性耳抑素肽类的合成和结构描述于US6,884,869、US7,098,308、US7,256,257、US7,423,116、US7,498,298和US7,745,394中。其他新的耳抑素肽类的合成和结构描述于WO2013/173393中。这些文献整体援引加入本文。在一些实施方案中,所述细胞毒性药物是美登素类。美登素(maytansine)首先由Kupchan等人从东非灌木齿叶美登木(Maytenusserrata)中分离得到,它比传统化疗剂如甲氨碟呤、柔红霉素和长春新碱的细胞毒性强100至1000倍(参见US3,896,111)。随后,发现一些微生物也产生美登素类,如美登醇(maytansinol)和美登醇的C-3酯(参见US4,151,042)。也已报道了合成的美登醇C-3酯及美登醇类似物(参见Kupchan等人,1978,J.Med.Chem.,21:31-37;Higashide等人,1977,Nature,270:721-722;Kawai等人,1984,Chem.Pharm.Bull.,32:3441-3451)。美登醇C-3酯由美登醇制备得来。美登醇类似物的实例包括:在芳环上有修饰(例如,脱氯)的美登醇,在C-9、C-14处有修饰(例如,羟化的甲基)的美登醇,在C-15、C-18、C-20和/或C-4、C-5处有修饰的美登醇。在一些实施方案中,所述细胞毒性药物例如是但不限于以下化合物:其中R是H、C1-C8烷基或C3-C8环烷基,所述烷基和环烷基任选地被一个或多个(如1、2、3、4或5个)选自卤素(如F、Cl、Br或I)的取代基取代;X是-S-、-CH2-、-CH(OH)-、-CH(OR)-、-CH(ONH2)-或-C(O)-;且Ar是C6-C14芳基(如苯基或萘基)。在优选的实施方案中,所述细胞毒性药物基团来自通式(D1)或(D2)的化合物或它们的立体异构体:其中:R2选自-CH2N3、-CONHSO2(环丙基)、噻唑-2-基、-CH3和-COOH;R3选自H和-OH;并且R4选自H、-NH2、Cl、Br、I、-OS(O)2R6,其中R6是H、C1-C8烷基、C3-C8环烷基或C6-C14芳基,所述烷基、环烷基和芳基各自任选地被一个或多个(如1、2、3、4或5个)选自卤素的取代基如F取代;其中R5选自-CH(CH3)N(CH3)C(O)CH2CH2SH和-CH(CH3)N(CH3)C(O)CH2C(CH3)2SH。在通式(I)中,所述细胞毒性药物基团可以是通式(D1)或(D2)的药物脱除R4或R5或者脱除R4或R5中的氢或R6而得到的基团。在优选的实施方案中,所述细胞毒性药物选自:连接基在本发明的抗体-药物偶联物中,细胞毒性药物基团经二价连接基与抗体分子连接。所述连接基与抗体的连接可经由多种方式完成,例如经由抗体表面的赖氨酸残基连接,或者还原偶合至抗体的经氧化的烃基。所述连接可以是基于腙、二硫键或肽结构的连接。这些连接方式是本领域技术人员公知的。本发明可用的连接基包括可断裂及不可断裂的连接基。可断裂的连接基在细胞内的条件下通常易断裂。适当的可断裂的连接基包括例如可被细胞内蛋白酶(如溶酶体蛋白酶(lysosomalprotease)或核内体蛋白酶(endosomalprotease))切割的肽连接基。在示例性实施方案中,所述可断裂的连接基可为二肽连接基,如缬氨酸-瓜氨酸(val-cit)连接基、苯丙氨酸-赖氨酸(phe-lys)连接基或顺丁烯二酰亚氨基己酰基-缬氨酸-瓜氨酸-对氨基苯甲氧基羰基(mc-Val-Cit-PAB)连接基。其它适当的可断裂的连接基包括可在特定pH或pH范围内被水解的连接基(如腙连接基),以及二硫化物连接基。不可断裂的连接基可以是磺基-SMCC。磺基-SMCC与蛋白质偶联经由顺丁烯二酰亚氨基发生,其磺基-NHS酯可与伯胺(如蛋白质或肽中的赖氨酸ε-氨基及N端的α-氨基)反应。另一不可断裂的连接基是顺丁烯二酰亚氨基己酰基(MC)。连接基可与抗体共价连接,并且所述共价连接的程度应使所述抗体必须在细胞内被降解才能释放药物。连接基包括用于与抗体连接的基团,例如氨基、羟基或羧基。连接基可来自例如顺丁烯二酰亚胺、卤代乙酰胺(例如碘乙酰胺、溴乙酰胺或氯乙酰胺)、卤代酯(例如碘代酯、溴代酯或氯代酯)、卤代甲基酮(例如碘甲基酮、溴甲基酮或氯甲基酮)、卤化苄(例如碘化苄、溴化苄或氯化苄)、乙烯砜(vinylsulfone)及吡啶基硫(pyridylthio)(参见例如Wong,1991,ChemistryofProteinConjugationandCross-linking,CRCPress,Inc.,BocaRaton)。在一些实施方案中,所述连接基至少包括Val-Cit、Val-Cit-PAB、Val-Ala-PAB、Val-Lys(Ac)-PAB、Phe-Lys-PAB、Phe-Lys(Ac)-PAB、D-Val-Leu-Lys、Gly-Gly-Arg、Ala-Ala-Asn-PAB、Ala-PAB或PAB。在一些实施方案中,所述连接基至少包括肽、寡糖、-(CH2)n-、-(CH2CH2O)n-。在一些实施方案中,所述连接基至少包括-C(O)-、-NH-C(O)-、-C(O)-O-、-NH-C(O)-NH-或-NH-C(O)-O-。在一些实施方案中,通式(I)中的连接基L可以选自:其中m、n每次出现时各自为选自1-10的整数,优选1、2、3、4、5或6。抗体-药物偶联物本发明的抗体-药物偶联物由上文中定义的通式(I)表示。最优选的通式(I)的抗体-药物偶联物选自I-1、I-2和I-3:其中A为曲妥珠单抗。本发明的抗体-药物偶联物可以为药学可接受的盐的形式,或者为立体异构体的形式,或者为溶剂合物的形式,并且所述盐或立体异构体也可为溶剂合物的形式。术语“药学上可接受的盐”是指保留化合物的生物有效性和性质的盐,它们对于用做药物在生物学上或在其它方面是符合需要的。在许多情况下,本发明的抗体-药物偶联物凭借其中存在的氨基和/或羧基或类似基团来形成酸加成盐和/或碱加成盐。药学上可接受的酸加成盐可以是与无机酸或有机酸形成的盐。所述无机酸包括,例如,盐酸、氢溴酸、硫酸、硝酸和磷酸等。所述有机酸包括,例如,乙酸、丙酸、羟基乙酸、丙酮酸、草酸、马来酸、丙二酸、琥珀酸、富马酸、酒石酸、柠檬酸、苯甲酸、肉桂酸、扁桃酸、甲磺酸、乙磺酸、对甲苯磺酸和水杨酸等。药学上可接受的碱加成盐可以是与无机碱或有机碱形成的盐。所述与无机碱形成的盐包括,例如,钠盐、钾盐、锂盐、铵盐、钙盐、镁盐、铁盐、锌盐、铜盐、锰盐和铝盐等,特别优选铵盐、钾盐、钠盐、钙盐和镁盐。所述有机碱包括,例如,伯胺、仲胺和叔胺,取代的胺(包括天然存在的取代的胺),环胺,碱性离子交换树脂等。有机碱的具体实例为异丙胺、三甲胺、二乙胺、N-乙基乙胺、三丙胺和乙醇胺。术语“立体异构体”表示由于至少一个不对称中心形成的异构体。在具有一个或多个不对称中心的化合物中,其可产生外消旋体、外消旋混合物、单一对映异构体、非对映异构体混合物和单独的非对映异构体。特定个别分子也可以几何异构体(顺式/反式)存在。除非另外说明,当所公开的化合物的命名或结构中没有明确说明其立体化学并且具有一个或多个不对称中心时,应该理解代表所述化合物的所有可能的立体异构体。术语“溶剂合物”表示由一个或多个溶剂分子与任一通式I的抗体-药物偶联物或其药学可接受的盐或异构体缔合形成的溶剂合物。术语溶剂合物包括水合物(例如半水合物、一水合物、二水合物、三水合物、四水合物以及类似的水合物)。本发明的抗体-药物偶联物可以选择性地将有效量的细胞毒性药物递送到肿瘤组织,由此获得更佳的治疗选择性,并以更低的剂量实现期望的治疗功效。在第二方面,本发明提供本发明第一方面的抗体-药物偶联物的制备方法,所述方法包括以下步骤:(1).制备通式(I-A)的化合物:其中D、L、X和Y如上文关于通式(I)所定义;(2).活化步骤(1)中得到的通式(I-A)的化合物,得到通式(I-A-G)的化合物其中G选自-F、-Cl、-Br、-I、-N3、-OR、SR、-ONRR’、RC(O)O-、-OP(O)RR’、RSO2-O-和其中R和R’每次出现时各自独立地是C1-C10烷基、C6-C14芳基、含5-10个环原子的杂环基或苯氧基,所述烷基、芳基、杂环基和苯氧基是未取代的或者各自独立地被一个或多个选自卤素、羟基、C1-C4烷基、C1-C4烷氧基、C3-C8环烷基、含5-8个环原子的杂环基、C6-C10芳基或含5-10个环原子的杂芳基的取代基取代;(3).将步骤(2)中得到的通式(I-A-G)的化合物偶联于所述抗ErbB2抗体或者其活性片段或变体,得到具有不同a值的多种抗体-药物偶联物的混合物;和(4).纯化步骤(3)中得到的混合物,得到抗体-药物偶联物。优选地,步骤(2)中的通式(I-A-G)的化合物中的G选自-ONRR’和-OP(O)RR’,其中R和R’每次出现时各自独立地是苯氧基。更优选地,步骤(2)中的通式(I-A-G)的化合物为通过使通式(I-A)的化合物与五氟苯酚反应而形成的通式(I-B)的化合物:其中D、L、X和Y如上文关于通式(I)所定义,并且所述反应优选使用EDCI、NHS和/或DCM完成。优选地,步骤(4)的纯化通过HPLC完成。在第三方面,本发明提供药物组合物,其包含本发明第一方面的抗体-药物偶联物或者其药学上可接受的盐或立体异构体或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物,以及药学上可接受的载体。本文中所用的术语“药物组合物”指组合在一起以实现特定目的的至少一种活性成分及药学上可接受的载体和/或辅料的组合。在一些实施方案中,药物组合物是各组分的混合物的形式,而在另一些实施方案中,药物组合物的各组分可以是在时间和/或空间上分开的,只要其能够共同作用以实现本发明的目的。当药物组合物中含有两种或更多种活性成分时,所述活性成分可以作为混合物同时施用于个体,也可以分开施用于个体。当分开施用时,各活性成分可以同时或依次施用。药学上可接受的载体的选择取决于药物组合物的剂型(例如用于口服、经鼻、皮内、皮下、肌内或静脉施用的剂型),例如,可以包括水(如注射用水)、缓冲液、等渗盐溶液如PBS(磷酸盐缓冲液)、葡萄糖、甘露醇、右旋葡萄糖、乳糖、淀粉、硬脂酸镁、纤维素、碳酸镁、0.3%甘油、透明质酸、抗坏血酸、乳酸、乙醇、聚亚烷基二醇如聚乙二醇(例如聚乙二醇4000)或聚丙二醇、甘油三酯等。此外,本发明的药物组合物还可根据需要包含各种添加剂,如润湿剂、乳化剂或缓冲剂等等。在第四方面,本发明提供本发明第一方面的抗体-药物偶联物或者其药学上可接受的盐或立体异构体或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物在制备用于预防或治疗癌症的药物中的用途。所述癌症包括但不限于乳腺癌、胃癌、卵巢癌、非小细胞肺癌和肝癌,特别是乳腺癌,如ErbB2高表达的乳腺癌。在第五方面,本发明提供药物制剂,其包含本发明第一方面的抗体-药物偶联物或者其药学上可接受的盐或立体异构体或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物。优选地,所述药物制剂为固体制剂、半固体制剂、液体制剂或气体制剂的形式。所述药物制剂特别优选为冻干粉针剂,其具有辅料较少,稳定性好,临床使用安全性较高的优点。在第六方面,本发明提供预防或治疗癌症的方法,所述方法包括向有此需要的患者给药本发明第一方面的抗体-药物偶联物,其药学上可接受的盐或立体异构体,或者所述抗体-药物偶联物、盐或立体异构体的溶剂合物,或者向有此需要的患者给药本发明第三方面的药物组合物或本发明第五方面的药物制剂。在以下的实施例中将进一步举例说明本发明。这些实施例仅用于说明本发明,但不以任何方式限制本发明。实施例各实施例中使用的缩写的含义如下表所示:DMF二甲基甲酰胺DIC二异丙基碳二亚胺HOAt1-羟基-7-氮杂苯并三唑EtOAc乙酸乙酯HPLC高压液相色谱DIEA二异丙基乙胺HATU2-(1H-7-氮杂苯并三唑-1-基)-1,1,3,3-四甲基脲鎓六氟磷酸盐DCM二氯甲烷EDCI1-乙基-3-(3-二甲基氨基丙基)碳二亚胺NHSN-羟基琥珀酰亚胺DMAN,N-二甲基乙酰胺HIC疏水作用色谱HPLC高效液相色谱THF四氢呋喃EtOAc乙酸乙酯实施例1.制备抗体-药物偶联物I-1步骤a.中间体D-1的制备在室温下,将化合物D-0(1mmol,1当量)溶于DMF(50mL)中,依次加入DIC(1.1当量)、HOAt(1.1当量)和4-(3-叠氮-2-氨基丙基)苯胺(1.5当量)。将反应混合物在室温下搅拌8小时,然后加入水(600mL)和EtOAc(200mL*3),萃取后收集有机相,浓缩并经HPLC纯化,得到中间体D-1。MSm/z(ESI):773[M+H]+。步骤b.中间体I-A-1的制备在室温下,将化合物S-2(0.1mmol,1当量)溶于DMF(50mL)中,依次加入DIEA(2当量)、HATU(1.05当量)和化合物D-1(2.0当量)。在室温下反应12小时,然后加入水(300mL)和EtOAc(100mL*3),萃取后收集有机相,浓缩并经HPLC纯化,得到中间体D-A-1。在室温下,将化合物D-A-1(0.1mmol,1当量)溶于DMF(5mL)中,依次加入DIC(1.1当量)、HOAt(1.1当量)和哌啶4-羧酸(1.2当量)。将反应混合物在室温下搅拌6小时,然后加入水(60mL)和EtOAc(20mL*3),萃取后收集有机相,浓缩并经HPLC纯化,得到中间体I-A-1。MSm/z(ESI):1240[M+H]+。步骤c.中间体I-B-1的制备在室温下,将化合物I-A-1(0.1mmol,1当量)溶于DCM(50mL)中,依次加入EDCI(1.5当量)、NHS(1.5当量)和五氟苯酚(2.0当量)。在室温下反应18小时。向反应混合物中依次加入水(30mL)、10%(w/v)柠檬酸水溶液(20mL)和饱和氯化钠水溶液(20mL)进行洗涤,收集有机相,浓缩并经HPLC纯化,得到中间体I-B-1。MSm/z(ESI):1406[M+H]+。步骤d.抗体-药物偶联物I-1粗产物的合成向1ml在pH7.4的PBS缓冲液中配制的浓度为10~20mg/ml的曲妥珠单抗溶液中加入4~6倍物质的量的溶解在DMA中的化合物I-B-1。在2~40℃和温和搅拌下反应2~6小时,通过HIC-HPLC监测反应,得到抗体-药物偶联物I-1粗产物,HIC-HPLC谱图如图1所示。HIC-HPLC条件:色谱柱:TosohTSKgelButyl-NPR,4.6×100mm流动相A:1.5M硫酸铵水溶液流动相B:25mM磷酸钠水溶液,pH=7.0,25%(v/v)异丙醇水溶液流速:0.5ml/min梯度:0~2min:17%流动相B+83%流动相A2~15min:17%~40%流动相B+83%~60%流动相A15~15.1min:40%~70%流动相B+60%~30%流动相A15.1~17min,70%流动相B+30%流动相A从图1可以看出,抗体-药物偶联物I-1粗产物为包含I-1-1(图1中的DAR1,n=1)、I-1-2(图1中的DAR2,n=2)、I-1-3(图1中的DAR3,n=3)和I-1-4(图1中的DAR4,n=4)的混合物。步骤e.抗体-药物偶联物I-1粗产物的纯化对步骤d中得到的抗体-药物偶联物I-1粗产物进行HIC,经脱盐换液(即更换缓冲液)和超滤浓缩,获得抗体-药物偶联物I-1。HIC条件:填料:GE填料(PheynlHP)流动相A:1.5M硫酸铵水溶液,25mM磷酸氢二钠水溶液,pH7.0流动相B:25mM磷酸氢二钠水溶液,pH7.0,10%异丙醇溶液流速:1.0ml/min洗脱条件:0%~40%流动相B洗脱20CV;40%~100%流动相B洗脱30CV分管收集通过HIC对所得抗体-药物偶联物I-1进行DAR分析,HIC谱图如图2所示。从图2可以看出,抗体-药物偶联物I-1的DAR为2,表明该抗体-药物偶联物中n=2,是一分子抗体上偶联两分子药物的抗体-药物偶联物,并且产物纯净。进一步对抗体-药物偶联物I-1进行轻重链还原液相色谱分析,得到重叠反相色谱图,如图3所示。在图3中,深色曲线是曲妥珠单抗裸抗的谱图,浅色曲线是抗体-药物偶联物I-1的谱图。在两谱图中,较高的峰指示抗体重链,较低的峰指示抗体轻链。从图3可以看出,与未偶联药物的曲妥珠单抗裸抗相比,抗体-药物偶联物I-1中的抗体轻链因偶联药物分子而疏水性增强,因此保留时间后移。这说明,抗体-药物偶联物I-1中药物分子全部偶联在抗体轻链上。进一步将抗体-药物偶联物I-1和曲妥珠裸抗在相同条件下进行酶切,得到肽图谱,如图4所示。在图4的肽图谱中,上面的图为裸抗在214nm的肽图谱,下面的图为抗体-药物偶联物I-1在214nm的肽图谱。经比较,抗体-药物偶联物I-1中只在保留时间65.5min处增加了一个肽段。结合图3的轻重链还原色谱图可知,本实施例制备得到的抗体-药物偶联物I-1中,毒素分子(D)全部偶联在轻链同一肽段的赖氨酸上,实现了定点偶联,因而获得优异的产品质量均一性、质量可控性和重现性。实施例2.制备抗体-药物偶联物I-2步骤a.中间体I-A-2的制备在室温下,将化合物D-2(购于南京联宁生物制药公司,1mmol,1当量)溶于DMF(50mL)中,加入碳酸钾(2.5当量)和2-氯乙酸甲酯(1.5当量)。将反应混合物升温至40~45℃,反应4小时。反应结束后,过滤除去碳酸钾固体,浓缩滤液。将浓缩所得物质溶于甲醇(30mL)中,加入1M的氢氧化锂水溶液将pH调节至13~14。将反应混合物升温至55℃,搅拌16小时,然后加入10%(w/v)的柠檬酸水溶液,浓缩并经HPLC纯化,得到中间体D-A-2。在室温下,将化合物D-A-2(0.1mmol,1当量)溶于DMF(5mL)中,依次加入DIC(1.1当量)、HOAt(1.1当量)和哌啶4-羧酸(1.2当量)。将反应混合物在室温下搅拌6小时,然后加入水(60mL)和EtOAc(20mL*3),萃取后收集有机相,浓缩并经HPLC纯化,得到中间体I-A-2。MSm/z(ESI):907[M+H]+。步骤b.中间体I-B-2的制备在室温下,将化合物I-A-2(0.1mmol,1当量)溶于DCM(50mL)中,依次加入EDCI(1.5当量)、NHS(1.5当量)和五氟苯酚(2.0当量)。在室温下反应18小时。向反应混合物中依次加入水(30mL)、饱和柠檬酸水溶液(20mL)和饱和氯化钠水溶液(20mL)进行洗涤,萃取后收集有机相,浓缩并经HPLC纯化,得到中间体I-B-2。MSm/z(ESI):1073[M+H]+。步骤c.抗体-药物偶联物I-2粗产物的合成向1ml在pH7.4的PBS缓冲液中配制的浓度为10mg/ml的曲妥珠单抗溶液中加入8倍物质的量的溶解在DMA中的化合物I-B-2。在室温和温和搅拌下反应16小时,通过HIC-HPLC(HIC-HPLC条件与实施例1步骤d中所列相同)监测反应,得到抗体-药物偶联物I-2粗产物,HIC-HPLC谱图如图5所示。从图5可以看出,抗体-药物偶联物I-2粗产物为包含I-2-1(图5中的DAR1,n=1)、I-2-2(图5中的DAR2,n=2)和I-2-3(图5中的DAR3,n=3)的混合物。步骤d.抗体-药物偶联物I-2粗产物的纯化对步骤c中得到的抗体-药物偶联物I-2粗产物进行HIC(HIC条件与实施例1步骤e中所列相同),经脱盐换液和超滤浓缩,获得抗体-药物偶联物I-2。通过HIC对所得抗体-药物偶联物I-2进行DAR分析,HIC谱图如图6所示。从图6可以看出,抗体-药物偶联物I-2的DAR为2,表明该抗体-药物偶联物中n=2,是一分子抗体上偶联两分子药物的抗体-药物偶联物,并且产物纯净。进一步对抗体-药物偶联物I-2进行轻重链还原液相色谱分析,得到重叠色谱图,如图7所示。在图7中,深色曲线是曲妥珠单抗裸抗的谱图,浅色曲线是抗体-药物偶联物I-2的谱图。在两谱图中,较高的峰指示抗体重链,较低的峰指示抗体轻链。从图7可以看出,与未偶联药物的曲妥珠单抗裸抗相比,抗体-药物偶联物I-2中的抗体轻链因偶联药物分子而疏水性增强,因此保留时间后移。这说明,抗体-药物偶联物I-2中药物分子全部偶联在抗体轻链上。由于实现了定点偶联,可获得优异的产品质量均一性、质量可控性和重现性。实施例3.制备抗体-药物偶联物I-3步骤a.中间体I-A-3的制备在室温下,将化合物D-3(根据CN104662000A第65页实施例V-3合成,1mmol,1当量)溶于THF(60mL)和DMF(30mL)的混合溶液中,依次加入二(对硝基苯基)碳酸酯(3当量)和DIEA(2当量)。将反应混合物在室温下搅拌12小时,然后加入水(600mL)和EtOAc(200mL*3),萃取后收集有机相,浓缩得到中间体D-A-3粗品,不经纯化而直接用于下一步反应。在室温下,将哌啶4-羧酸(5当量)溶解在饱和NaHCO3水溶液(5mL)中,加入化合物D-A-3粗品(1当量)。将反应混合物在室温下搅拌8小时。加入10%(w/v)柠檬酸水溶液以将pH调节至4~5,然后用EtOAc(150mL*2)萃取。将有机相干燥并浓缩,得到中间体I-A-3粗品。MSm/z(ESI):1020[M+H]+。步骤b.中间体I-B-3的制备在室温下,将化合物I-A-3(0.1mmol,1当量)溶于DCM(50mL)中,依次加入EDCI(1.5当量)、NHS(1.5当量)和五氟苯酚(2.0当量)。在室温下反应18小时。向反应混合物中依次加入水(30mL)、10%(w/v)柠檬酸水溶液(20mL)和饱和氯化钠水溶液(20mL)进行洗涤,萃取后收集有机相,浓缩并经HPLC纯化,得到中间体I-B-3。MSm/z(ESI):1186[M+H]+。1HNMR(400MHz,DMSO-d6)δ:7.02-7.09(m,4H),4.92(m,1H),4.52(m,1H),3.89(d,1H),3.77(m,1H),3.68-3.55(m,3H),3.46(m,2H),3.24(m,6H),3.05(d,2H),2.92-2.90(m,4H),2.68(m,1H),2.33-2.27(m,11H),1.97-1.93(m,4H),1.73-1.72(m,5H),1.29-1.24(m,5H),1.06-1.01(m,15H),0.96(m,3H),0.74(m,4H),3.34(m,4H)。步骤c.抗体-药物偶联物I-3粗产物的合成向1ml在pH7.8的PBS缓冲液中配制的浓度为10mg/ml的曲妥珠单抗溶液中加入8倍物质的量的溶解在DMA中的化合物D-0913-3。在室温和温和搅拌下反应4小时,通过HIC-HPLC(HIC-HPLC条件与实施例1步骤d中所列相同)监测反应,得到抗体-药物偶联物I-3粗产物,HIC-HPLC谱图如图8所示。从图8可以看出,抗体-药物偶联物I-3粗产物为包含I-3-1(图8中的DAR1,n=1)、I-3-2(图8中的DAR2,n=2)和I-3-3(图8中的DAR3,n=3)的混合物。步骤d.抗体-药物偶联物I-3粗产物的纯化对步骤c中得到的抗体-药物偶联物I-3粗产物进行HIC(HIC条件与实施例1步骤e中所列相同),经脱盐换液和超滤浓缩,获得抗体-药物偶联物I-3。通过HIC对所得抗体-药物偶联物I-3进行DAR分析,HIC谱图如图9所示。从图9可以看出,抗体-药物偶联物I-3的DAR为2,表明该抗体-药物偶联物中n=2,是一分子抗体上偶联两分子药物的抗体-药物偶联物,并且产物纯净。进一步对抗体-药物偶联物I-3进行轻重链还原液相色谱分析,得到重叠色谱图,如图10所示。在图10中,深色曲线是曲妥珠单抗裸抗的谱图,浅色曲线是抗体-药物偶联物I-3的谱图。在两谱图中,较高的峰指示抗体重链,较低的峰指示抗体轻链。从图10可以看出,与未偶联药物的曲妥珠单抗裸抗相比,抗体-药物偶联物I-3中的抗体轻链因偶联药物分子而疏水性增强,因此保留时间后移。这说明,抗体-药物偶联物I-3中药物分子全部偶联在抗体轻链上。由于实现了定点偶联,可获得优异的产品质量均一性、质量可控性和重现性。实施例4.体内活性测试本实施例评价实施例1~3的抗体-药物偶联物对皮下移植了人肿瘤细胞的小鼠的肿瘤增殖的抑制。具体而言,本实施例考察将实施例1~3的抗体-药物偶联物单次尾静脉注射给皮下移植人胃癌细胞株NCI-N87、乳腺癌细胞株HCC-1954、乳腺癌细胞株BT474、卵巢癌细胞株SK-OV-3的小鼠后,测定肿瘤体积和动物体重变化,计算抗体-药物偶联物对荷瘤小鼠的药效(抑瘤疗效)。1、实验原料1.1.肿瘤细胞胃癌细胞株NCI-N87(ATCC)、乳腺癌细胞株HCC-1954(Concortis)、乳腺癌细胞株BT-474(Concortis)、卵巢癌细胞株SK-OV-3(Concortis)均采用细胞体外单层培养。1.2.肿瘤模型将1.1中得到的肿瘤细胞分别接种至BALB/c裸鼠或NOD-SCID小鼠(SPF级,雌性,16-18g/只,约6~8周龄,北京维通利华实验动物技术有限公司)。在每只荷瘤鼠右侧腋下皮下接种4×106个胃癌细胞株NCI-N87(悬浮于0.1mlPBS中),接种后待肿瘤生长至100~200mm3时,剔除瘤体积过小(小于100mm3)或过大(大于200mm3)的荷瘤鼠,将余下荷瘤鼠随机分组,作为胃癌NCI-N87模型。得到乳腺癌HCC-1954模型、乳腺癌BT-474模型、卵巢癌SK-OV-3模型的方法与上述方法类似。1.3处理品溶液配制称取适量曲妥珠单抗裸抗、T-DM1(阳性对照,(ado-trastuzumabemtansine),罗氏制药)或本发明的抗体-药物偶联物(I-3、I-1或I-2,实施例1~3制备),分别用无菌超纯水配制成一定浓度的母液,轻柔摇匀后,分装于-80℃保存。使用时用生理盐水根据剂量稀释,得到处理品溶液,并采用相同浓度的生理盐水作为溶剂对照。2、实验方法:选择随机分组(根据样品数量决定分组数)的肿瘤体积在100~200mm3的荷瘤鼠(1.2中得到的模型),7只/组。给药体积为10ml/kg。给药途径为单次尾静脉注射。给药后观察8周,每周2次用游标卡尺测量肿瘤直径,并按如下计算公式计算肿瘤体积:V=0.5a2×b,其中a和b分别表示肿瘤的长径和短径。每天观察记录动物死亡情况。3、实验结果肿瘤生长抑制率TGI(%)用于评价抗体-药物偶联物的抑瘤疗效。采用以下公式计算TGI(%):TGI(%)=[1-(VT末-VT始)/(VC末-VC始)]*100%其中VT末:处理组实验结束时肿瘤体积均值VT始:处理组给药开始时肿瘤体积均值VC末:溶剂对照组实验结束时肿瘤体积均值VC始:溶剂对照组给药开始时肿瘤体积均值计算结果见表1~5。表1.乳腺癌BT-474模型组别样品剂量(mg/kg)TGI(%)1溶剂//2T-DM1354.80%3I-13104.75%表2.乳腺癌HCC-1954模型组别样品剂量(mg/kg)TGI(%)1溶剂//2T-DM1379.37%3曲妥珠单抗裸抗1578.94%4I-13104.47%5I-33104.04%表3:胃癌NCI-N87模型表4:胃癌NCI-N87模型组别样品剂量(mg/kg)TGI(%)1溶剂//2T-DM1230.136%3I-12114.99%4I-3287.04%5T-DM1365.28%6I-33102.72%表5:卵巢癌SK-OV-3模型组别样品剂量(mg/kg)TGI(%)1溶剂//2T-DM16-11.04%3I-2654.72%4I-3672.27%从表1至表5可以看出,本发明的抗体-药物偶联物I-1、I-3和I-2在60天的评价周期内,药效均明显优于T-DM1。实施例5.制备冻干粉针剂使用如下表所示原料制备抗体-药物偶联物冻干粉针剂:抗体-药物偶联物I-128g抗坏血酸20g乳酸10g聚乙二醇400063g注射用水2000ml其中抗坏血酸和乳酸在起到助溶剂的同时也起到了抗氧化剂的作用。制备方法:1、取20g抗坏血酸和10g乳酸,加1000ml注射用水,加热至50~55℃,搅拌使其溶解,取28g抗体-药物偶联物I-1加入溶液中,搅拌溶解后继续搅拌15分钟。2、取63g聚乙二醇4000,加800ml注射用水,搅拌15分钟。3、将1、2中所得溶液合并,加注射用水至全量,加0.15%的针用活性炭,搅拌25分钟,过滤脱炭,中间体检查,合格后用0.22μm滤膜过滤除菌。4、将滤液灌入西林瓶中,进行冷冻干燥,得冻干粉针,检验合格后,包装。尽管本发明通过之前的具体实施例得到说明,但应当理解,不应将其解释为受此限制。本发明涵盖之前公开的一般方面,并且本领域技术人员可在不背离本发明的精神和范围的情况下进行多种修饰或改变本发明的各种细节。因此,本说明书仅为说明的目的,而非为限制的目的。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1