杂烷烃和芳烃的官能化方法与流程
本专利申请要求于2014年8月26日提交的美国临时专利申请号62/042,101和于2014年8月25日提交的美国临时专利申请号62/041,270的权益,其全部内容均通过引用并入全文作为参考。
关于联邦政府资助的研发的声明
本发明是在由美国能源部授予的GQ10044-133945的政府支持下进行的。政府享有本发明的某些权利。
发明背景
在过去几十年中出现的一个重要方法是设计基于C-H活化反应的用于烃的氧化性官能化的分子(均相)催化剂或试剂。这涉及使可再生的M-X催化剂或试剂(M是处于氧化状态的主族元素;X是一个或多个平衡电荷的抗衡离子)与烃(R-H)的C-H键在相对温和的条件下反应,以选择性地产生M-R中间体,其可以在使M-X再生的情况下转化为期望的R-X产物(方程1)。
在均相和非均相催化剂的该研究领域已经做出了巨大努力,并且近年来已取得了实质性的进展。关于均相体系的大部分工作主要基于过渡金属(具有未填充的d壳,d<10),例如Pt、Pd、Rh和Ir。相比之下,对于具有已填充的d壳(d10)的经典主族元素的研究则相对较少。1993年,Periana报道了主族金属阳离子HgII在超强酸溶剂,浓H2SO4和CF3SO3H中用于使甲烷直接转化成甲醇酯的实例(Periana et al.,Science 259,340-343(1993));还参见国际专利申请WO 92/14738)。尽管HgII系统简单,但由于缺乏在更可行的更弱的酸介质如CF3CO2H(TFAH或HTFA)、CH3CO2H(HOAc)或水性酸(其中产物分离是可行的)中的反应,对其没有进行进一步的开发。另一个关键问题是使用HgII体系时乙烷和丙烷的反应没有选择性。
发现TlIII对使甲烷氧化成酯是有活性的。然而,仅在超强酸性介质中且仅在甲烷的情况下研究了该活性。主要由于认识到TlIII(Eo=1.2V)和PbIV(Eo=1.5V)是比HgII(Eo=0.9V)更强的氧化剂和亲电子试剂,所以没有在TFAH中或对高级烷烃研究TlIII或PbIV体系。参见例如A.J.Bard,R.Parsons,J.Jordan,Standard Potentials in Aqueous Solution.(International Union of Pure and Applied Chemistry,New York,NY,1985)。因此,基于当时的一般考虑,这些主族阳离子被理解为比HgII更可能引发与高级烷烃的非选择性自由基反应,并被更弱的更亲核的酸溶剂抑制。这种预期的强的亲电子试剂在弱酸介质中缺乏反应性的模型也似乎与在早期的关于Pt联嘧啶复合物的工作中的观察一致,发现Pt(II)状态在超强酸介质中有活性,而可争论地,更亲电的Pt(IV)状态是非活性的。
类似地,没有将苯转化为苯酚的直接方法。通常通过包括添加丙烯、氯等的间接方法来制备苯酚。直接方法是具有挑战性的,因为诸如苯的芳烃的官能化通常涉及低选择性。
因此,期望提供一种这样的方法,其使诸如杂烷烃和芳烃的化合物被选择性地和直接地官能化,而不产生显著量的副产物和废物,并且不需要昂贵的过渡金属催化剂。
发明概述
本发明提供了制备官能化的化合物的方法,该方法包括:
(a)提供杂烷烃或芳烃化合物,
其中
杂烷烃包含至少一个带有氢原子的sp3-杂化的碳原子和至少一个不同于碳或氢原子的杂原子,且
芳烃包含至少一个带有氢原子的sp2-杂化的碳原子,并且任选包含
(i)一个或多个sp3-杂化的碳原子,
(ii)一个或多个杂原子,或
(iii)(i)和(ii)两者,
(b)使该化合物与以下物质接触以提供初始反应产物,
(i)包含氧化形式的主族元素的氧化性亲电子试剂,或
(ii)氧化剂和氧化性亲电子试剂的还原形式,和
(c)使初始反应产物与官能化反应物接触,其中官能化反应物的官能化部分替代初始反应产物上的氢以提供官能化的化合物。
附图的几个视图的简要说明
图1是所提出的用于烃(RH)通过CH活化反应向相应的酚(ROH)的总转化的反应机理。
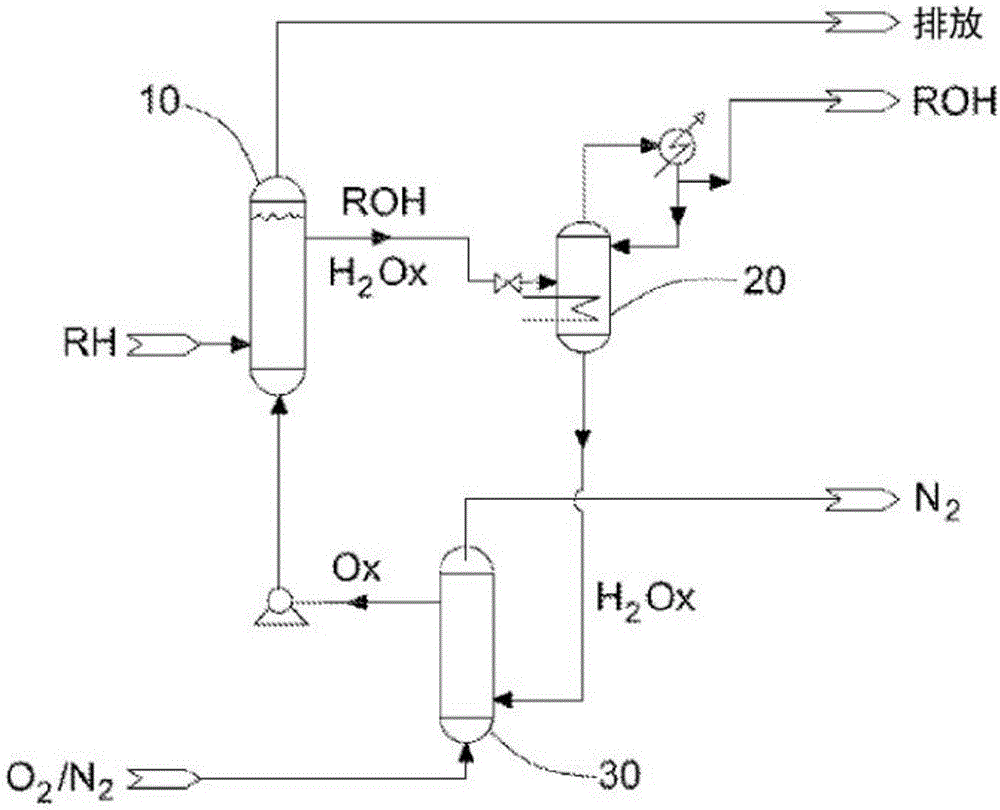
图2是用空气将杂烷烃或芳烃(RH)连续部分氧化成ROH的简化工艺流程图的示例。
图3是在可连续产生产物的切换反应器体系中使用MXn作为可空气再循环的化学计量氧化剂将杂烷烃或芳烃(RH)转化成相应的醇(ROH)的简化工艺图。
图4是显示正丁基TFA与C6F5I(TFA)2的反应结果的核磁共振(NMR)光谱。
发明详述
通过本文公开和要求保护的方法和组合物,提供了可行的工业方法,以将杂烷烃(例如醇、酯、卤代烃、羧酸、羧酰胺等)和芳烃(例如芳基和杂芳基)转化为C-H键替代产物,例如反应底物的羟基化、胺化、卤化或羰基化的衍生物。氧化形式的反应性主族元素(M)(例如MXn)可以带来据信是初始亲电子的C-H键活化反应的反应。该反应可以通过不同的机理(例如通过自由基或其它机理)进行,且产生等同于C-H键反应产物的产物。在形成活化的中间体并转化为官能化的杂烷烃或芳烃后,可以回收还原形式的试剂M,以再循环回反应性的氧化状态(例如MXn)。本文公开和要求保护的试剂和方法的反应性允许该方法在没有昂贵的或难以处理的超强酸的存在下进行,这是迄今为止尚未实现的目标。通过本文所述的发明人的发现,已经设计了具有原位或非原位试剂再循环的低温、低压和可持续的方法,其提供了替代先前已知方法(例如从低级烷烃生产低级醇如甲醇的合成气方法)的经济上和环境上有吸引力的工业规模的方法。
因此,本发明提供了制备官能化的化合物的方法,该方法包括:
(a)提供杂烷烃或芳烃化合物,
其中
杂烷烃包含至少一个带有氢原子的sp3-杂化的碳原子和至少一个不同于碳或氢原子的杂原子,且
芳烃包含至少一个带有氢原子的sp2-杂化的碳原子,并且任选包含
(i)一个或多个sp3-杂化的碳原子,
(ii)一个或多个杂原子,或
(iii)(i)和(ii)两者,
(b)使该化合物与以下物质接触以提供初始反应产物,
(i)包含氧化形式的主族元素的氧化性亲电子试剂,或
(ii)氧化剂和氧化性亲电子试剂的还原形式,和
(c)使初始反应产物与官能化反应物接触,其中官能化反应物的官能化部分替代初始反应产物上的氢以提供官能化的化合物。
氧化性亲电子试剂作为试剂在非超强酸体系中的使用代表了一种惊人的发现,其提供了使用工业上可用的物质的杂烷烃或芳烃氧化体系。
在该方法中,待官能化的化合物是如本文所述的杂烷烃或芳烃。
本反应的“杂烷烃”底物是包括至少一个sp3-杂化的碳原子的分子,其中该碳原子的至少一个取代基是氢原子,使得存在C-H键。杂烷烃的烷烃部分是指含有例如约1至约16个碳原子(例如约1至约12个碳原子,约1至约10个碳原子,约1至约8个碳原子,约1至约6个碳原子或约1至约4个碳原子)的直链或支链烷基取代基。杂烷烃另外包含至少一个“杂原子”,即不是碳或氢的原子。杂原子的实例包括诸如氧、氮、硫、卤素(例如氯)和/或金属(例如锡)的元素的原子。因此,作为本文中使用的术语的杂烷烃底物可以是例如烷基甲醇、烷基胺、卤代烃或有机金属化合物。可用于本发明方法的实践的杂烷烃底物的实例包括醇(例如正丙醇或正丁醇)和包含醚氧、酯或酰胺基团的化合物。例如,本发明的方法可用于提供杂烷烃如丁醇、卤代丁烷和丁酰基化合物如酯和酰胺的反应产物。
“官能化的”杂烷烃是杂烷烃底物的衍生物,其中带有氢原子的sp3-杂化的碳原子在该C-H键处发生反应,以产生官能化的杂烷烃产物,其包含杂烷烃结构,但其中sp3-杂化的碳原子带有官能(即非氢)基团。例如,除了被氧化/水解以产生相应的烷基醇之外,杂烷烃还可以进行其它反应,例如通过与肼或其它含氮试剂反应产生胺。可以从初始反应产物获得其它产物,例如锡烷化、硫醇化、氧膦基化、羰基化、消除和/或卤化反应的产物。
本文所用的术语“芳烃”是指包含至少一个带有氢原子的sp2-杂化的碳原子的有机化合物,其中芳烃化合物可进一步包含(i)一个或多个sp3-杂化的碳原子;(ii)一个或多个杂原子;或(iii)(i)和(ii)两者。术语“芳烃”包括“芳基”和“杂芳基”环体系。
术语“芳基”是指未取代或取代的芳香性碳环部分,如本领域中通常理解的,并且包括单环和多环芳香性化合物,例如苯、联苯、萘、蒽、芘等。芳基部分通常含有例如6至30个碳原子,6至18个碳原子,6至14个碳原子或6至10个碳原子。应当理解,根据Hückel规则,术语芳基包括平面的且包含4n+2个π电子的碳环部分,其中n=1、2或3。芳基可以是取代的或未取代的,如本文所述。
术语“杂芳基”是指芳香性5或6元单环基团,9或10元双环基团和11至14元三环基团,其在至少一个环中具有至少一个杂原子(O、S或N)。含有杂原子的杂芳基的每个环可以含有一个或两个氧或硫原子和/或一至四个氮原子,条件是每个环中杂原子的总数为四个或更少,且每个环具有至少一个碳原子。构成双环和三环基团的稠环可以仅含有碳原子,并且可以是饱和的、部分饱和的或不饱和的。氮和硫原子可以任选地被氧化,并且氮原子可以任选地被季铵化。双环或三环的杂芳基必须包括至少一个完全芳香性的环,但其它稠环可以是芳香性或非芳香性的。杂芳基可以在任何环的任何可用的氮或碳原子处连接。杂芳基的说明性实例是喹啉、吡啶、哒嗪、嘧啶、吡嗪、苯并咪唑、三嗪、咪唑、(1,2,3)-和(1,2,4)-三唑、吡嗪、四唑、呋喃、吡咯、噻吩、异噻唑、噻唑、异噁唑和噁二唑。杂芳基可以是取代的或未取代的,如本文所述。
芳烃可以任选地被一个或多个杂原子,一个或多个烷基等取代。杂原子的实例包括以下诸如氧、氮、硫、卤素(例如氯)和/或金属(例如锡)的元素的原子,例如芳基或杂芳基醇(酚)、烷氧基、酯、胺、硫醇、卤代烃、羧酸和羧酰胺。合适的芳烃底物的实例包括芳基,例如苯、萘、酚、酚醚、苯胺的衍生物、卤代芳基化合物等。合适的芳烃底物的其它实例包括杂芳基,例如吡啶、喹啉、吡咯、吲哚、噻吩等。在一个实施方案中,芳烃是苯、吡啶、喹啉或萘,其各自任选被取代。
“官能化的”芳烃是芳烃底物的衍生物,其中带有氢原子的sp2-杂化的碳原子在该C-H键处进行反应以产生官能化的芳烃产物,即产物包含芳烃底物结构但其中进行反应的sp2-杂化的碳原子现在带有非氢基团。一种可能的机理是,在sp2-杂化的碳原子上的氢原子可以首先被来自氧化性亲电子试剂的主族元素M替代,并且随后被羟基、氨基、卤素、羧酰胺基、C=O加成产物等替代。
在一些实施方案中,该化合物是杂烷烃。优选地,杂烷烃是烷基单酯(例如正丁醇或正丙醇的酯)。在一个具体的实施方案中,化合物可以是正丁醇或正丙醇的酯,并且官能化的杂烷烃产物可以分别是1,4-丁二醇或1,3-丙二醇的二酯。
在其他方面,该化合物是芳烃。优选地,芳烃包括芳环体系和/或芳烃包括杂芳环体系。
在该方法的步骤(b)中,使待官能化的化合物(即杂烷烃或芳烃)与(i)包含氧化形式的主族元素的氧化性亲电子试剂或(ii)氧化剂和氧化性亲电子试剂的还原形式接触。在一些实施方案中,接触步骤(步骤(b))包括使所述化合物与包含氧化形式的主族元素的氧化性亲电子试剂接触。在其它实施方案中,步骤(b)包括使所述化合物与氧化剂和氧化性亲电子试剂的还原形式接触。
本文所述的任何方法的氧化性亲电子试剂包含主族元素。本文中使用的术语“主族元素”是指金属和非金属,包括CAS IIIA、IVA、VA、VIA和VIIA族的元素,它们是后过渡元素,即具有比第一过渡系列的最后一个元素Zn更高的原子序数,即原子序数>30。在一个实施方案中,主族元素是选自CAS IIIA、IVA、VA和VIA族的元素。因此,用于本发明方法的实践的氧化性亲电子试剂包括具有原子序数31-35、49-53和81-83的稳定同位素形式的元素。在优选的实施方案中,氧化性亲电子试剂包括这样的至少一种元素,其是原子序数为31-34、49-52和81-83中任一个的稳定同位素形式。在一些实施方案中,主族元素具有d10的电子构型。然而,在本发明的方法的实践中使用的氧化性亲电子试剂可以具有不同于d10的电子构型。主族元素可以在较高氧化态(在与烷烃C-H键反应的氧化性亲电子试剂中)和较低氧化态(亲电还原产物,可以通过原位或独立的步骤使氧化性亲电子试剂由其再生)之间循环。通过这种方式,可以形成用于杂烷烃或芳烃转化(例如转化成杂烷烃或芳烃的含氧化合物)的仅消耗第二氧化剂(例如过氧化氢、氧或臭氧)的经济上和环境上有利的自给体系。在一个实施方案中,氧化形式的主族元素处于+n的氧化态。在其它实施方案中,对于由氧化性亲电子试剂形成的亲电还原产物,主族元素处于+(n-2)或+(n-1)的氧化态。
如本领域已知的,氧化性亲电子试剂可以被称为软氧化性亲电子试剂。如本文所用的术语“软”亲电子试剂涉及在硬/软酸/碱(HSAB)概念(其被称为Pearson酸碱概念)下的分类,其用术语“硬”或“软”以及术语“酸”或“碱”来划分化学物质。术语“硬”适用于可弱极化的物质,而术语“软”适用于可强极性的物质。参见R.G.Pearson,Chemical Hardness-Applications From Molecules to Solids,Wiley-VCH,Weinheim,1997。
表1是基于Pearson硬和软理论的示例性物质的列表。根据HSAB理论,在本发明方法的实践中使用的氧化性亲电子试剂被分类为软的,并且包括主族元素如Tl、Pb、Bi、Sb、Se、Te和I的形式。作为其盐或其化合物,这些元素的较高氧化态用作用于本发明方法的实践的软的氧化性亲电子试剂。
表1:Pearson硬和软酸的分类
其它软酸是本领域技术人员已知的,并且本领域技术人员可以选择具有合适的氧化态对的元素来实施本发明的方法。
在一些实施方案中,氧化性亲电子试剂包含选自铊、铅、铋、锑、硒、碲、碘及其混合物的主族元素,其各自为氧化形式。在一个具体实施方案中,氧化性亲电子试剂包含选自铊、铅、铋、锑、硒、碲及其混合物的主族元素,它们各自为氧化形式。在Hg、Tl和Pb的情况下,最具活性的氧化形式是具有Xe的电子构型5d10,6s0的那些。然而,这不一定是发生反应的体系的电子构型,因为发现具有Kr的电子构型4d10,5s2,5p2的I(III)对于CH活化是有活性的。在具体实施方案中,氧化性亲电子试剂可以包含铊(III)、铅(IV)、铋(V)、碘(III)、Sb(V)、碘(V)或上述元素的任何混合物。在优选的实施方案中,氧化性亲电子试剂包含铊(III)、铅(IV)、铋(V)、Sb(V)或其任意混合物。在一个实施方案中,氧化性亲电子试剂包含铊(III)。在另一个实施方案中,氧化性亲电子试剂包含铅(IV)。在另一个实施方案中,氧化性亲电子试剂包含铋(V)。在又一个实施方案中,氧化性亲电子试剂包含Sb(V)。
在一些实施方案中,包含氧化形式的主族元素的氧化性亲电子试剂是盐,其中氧化形式的主族元素的抗衡离子是酸的共轭阴离子(例如,一种或多种三氟乙酸盐、乙酸盐、硫酸盐和/或烷基磺酸盐阴离子)。例如,氧化性亲电子试剂可以具有式M+nXn,其中M是氧化态为n的金属或非金属主族元素阳离子,X是阴离子抗衡离子,n是平衡金属离子的n+正电荷所必需的阴离子电荷的数目。阴离子抗衡离子(X)是能够形成亲电还原产物(例如包含三氟乙酸盐、乙酸盐、硫酸盐和/或烷基磺酸盐阴离子中的一种或多种的亲电还原产物)的任何合适的阴离子抗衡离子/配体。不希望受任何特定理论的束缚,本发明人相信M+nXn与杂烷烃或芳烃发生反应以产生式M+(n-2)Xn-2或M+(n-1)Xn-1的亲电还原产物。优选地,这种反应在如本文所述的酸性介质中进行。
在本文所述的任何方法的实施方案中,反应性主族元素与待官能化的化合物(即杂烷烃或芳烃)的反应(例如,步骤(b))在酸性介质,包括水性酸性介质中进行。酸性酸是任何合适的酸,例如无机酸、羧酸、磺酸、其水溶液或其任何组合。酸性介质优选包含含氧酸,例如三氟乙酸、乙酸、甲磺酸或其水溶液。如果期望,可以使酸性介质再循环。不希望受理论束缚,发明人相信形成了活化的X(n-1)MR初始反应产物,其中主族元素可能通过亲电过程替代杂烷烃或芳烃的C-H键的H。令人惊讶地发现,通过使用本文公开的组合物和方法,可以在不存在超强酸的情况下,并且在相对温和的条件下,例如在300℃以下,优选在200℃以下,实现杂烷烃或芳烃的有效反应,以提供活化的中间体(初始反应产物)。
该活化的中间体X(n-1)MR,其中R是杂烷烃或芳烃底物,其中杂烷烃的sp3-杂化的碳原子的氢原子或者芳烃的sp2-杂化的碳原子的氢原子分别被原子M替代,然后其可以与溶剂环境例如与酸例如三氟乙酸、乙酸、甲磺酸等进行反应,以提供可分离的产物R-X,即官能化的杂烷烃或芳烃。例如,可以分别形成杂烷基含氧化合物,例如杂烷基酯,例如三氟乙酸酯、乙酸杂烷基酯、甲磺酸杂烷基酯等。然后可以将官能化的杂烷基酯进一步水解以产生官能化的杂烷烃产物,其中sp3-杂化的碳原子的氢原子已被羟基代替。对于芳烃底物,可以设想类似的机理,其中芳烃的sp2-杂化的碳原子的氢原子优先于sp3-杂化的碳原子的氢原子被官能化。可以从活化的中间体(例如X(n-1)M-R)化合物获得其它产物,例如胺化、锡烷化、硫醇化、氧膦基化、羰基化、消除或卤化反应的产物。
本发明可以提供不需要使用超强酸的用于杂烷烃或芳烃的氧化或官能化的方法,尽管在各种实施方案中,可以有效地使用超强酸以实现所期望的转化。如本文所用的术语超强酸是具有大于或等于浓硫酸的酸度的酸,其具有-12的Hammett酸度函数(H0)。市售的超强酸包括浓硫酸以及三氟甲磺酸(CF3SO3H)和氟硫酸(HSO3F),两者都比浓硫酸强约一千倍(即具有更负的H0值)。
在本发明的方法中避免使用超强酸的优点包括更低的成本、对反应器构建材料的不那么严苛的需求、来自高级烷烃的产物的提高的稳定性以及易于使反应环境的酸性组分再循环。超强酸可以对水具有非常高的亲合力,因此涉及从水中重新浓缩超强酸的任何方法都会导致过高的用于水去除的能量成本。
因此,本发明提供了对先前公开的使用超强酸的方法的改进,因为可以使用比98%的硫酸弱的酸溶剂,例如三氟乙酸、乙酸、甲磺酸、磷酸、水性无机酸或有机酸等,包括其水溶液。在先前公开的汞体系或(bpym)PtX2体系中,需要非常强的酸(超强酸),例如98%的硫酸或CF3SO3H。假定这些体系通过亲电子机理操作。与该假设一致,发现这两种体系在较弱的、更亲核的酸溶剂例如CF3CO2H或乙酸中是无活性的。还提出由于Hg(II)的强氧化性,Hg(II)体系中可能涉及自由基机理。上述信息表明,较强的亲电子试剂和氧化剂,例如Tl(III)、Pb(IV)、Bi(V)等,会比基于Hg(II)或Pt(II)的亲电性较弱的体系具有更差的活性和选择性。实际上,观察到的(bpym)Pt(IV)在较弱酸中无活性的结果似乎也支持这种亲电抑制模型。因此,自从1993年的Hg(II)体系的报告以来,似乎没有关于使用主族阳离子进行烷烃氧化的工作的报道。
有趣的是,研究表明,水与强的亲电子试剂例如Hg(II)和Tl(III)交换的速率可以是亲电子试剂例如Ir(III)、Pt(II)和Pt(IV)的1020倍(F.Basolo,R.G.Pearson,Mechanisms of Inorganic Reactions(Wiley,New York,ed.2,1967))。这种效应在概念上可归因于,具有d10电子构型的阳离子(例如Hg(II)和Tl(III))缺乏配体场稳定化能(LFSE),而具有d<10的电子构型的阳离子的高LFSE。相反地,强的d10亲电子试剂如Hg(II)、Tl(III)、Pb(IV)和Bi(V)的这些高的交换速率向发明人表明,预期中的亲电性的增加和C-H活化速率的降低之间的相关性可能不正确。事实上,发明人的研究表明,增加亲电性实际上增加了d10阳离子中C-H活化的速率,其中Pb(IV)>Tl(III)>>Hg(II)(Hashiguchi et al.,Science,343,1232(2014))。这种关系是有趣的,因为它表明HgII在早期的1993年的研究中在CF3CO2H中不反应,因为亲电性不够高。发明人的这一非常重要的观察结果表明,可以设计其它廉价的、充足的后过渡金属阳离子,用于烷烃在非超强酸性介质中的活化和官能化。鉴于铋、碘、锑等的低毒性和常用性,基于这些阳离子的均相体系的设计是特别有吸引力的。
在本文所述的任何实施方案中,本发明的方法可以进一步包括将官能化的化合物与由氧化性亲电子试剂形成的亲电还原产物分离。
在本文所述的任何实施方案中,来自官能化反应的反应性主族元素副产物相对于在初始步骤中使用的试剂处于还原状态,并且可以被回收并再循环回其反应性的氧化状态。例如,亲电还原产物可以与氧化性再生试剂接触以使氧化性亲电子试剂再生。氧化性再生试剂优选包含过氧化物(例如过氧化氢)、氧、臭氧、硝酸或卤素(例如氯)。如果反应在氧化剂(例如,O2或H2O2)的存在下进行,则还原形式可以被原位再氧化,并且反应性主族元素会作为催化剂起作用。在一个实施方案中,氧化性再生试剂相对于杂烷烃或芳烃以至少化学计量的量存在。在另一方面,包含氧化形式的主族元素的氧化性亲电子试剂相对于杂烷烃或芳烃以小于化学计量的量存在,并且作为催化剂起作用。
在一些实施方案中,亲电还原产物和氧化性再生试剂在氧化性再生催化剂的存在下接触。氧化性再生催化剂可以包含例如铜、银、铁或钒。在一些实施方案中,当使用氧化性再生催化剂时,氧化性再生试剂相对于杂烷烃或芳烃以至少化学计量的量存在。在其它实施方案中,没有氧化性再生试剂与包含氧化形式的主族元素的氧化性亲电子试剂一起存在,并且氧化性亲电子试剂相对于杂烷烃或芳烃以至少化学计量的量存在。在这样的实施方案中,亲电还原产物可以在独立的步骤中被氧化回氧化性亲电子试剂,例如在如本文所述的双反应器体系中,其在化合物的官能化和用第二氧化剂进行的氧化性亲电子试剂的再生之间进行循环。
为了提供官能化的化合物,例如官能化的杂烷烃或官能化的芳烃,通过使杂烷烃或芳烃与氧化性亲电子试剂接触而形成的初始反应产物与官能化反应物接触。官能化反应物的官能化部分替代初始反应产物上的氢,以提供官能化的化合物(例如步骤(c))。官能化反应物是能够形成官能化的化合物的任何合适的试剂。例如,官能化反应物可以是含氧酸、肼、羟胺、氨、伯胺、仲胺、亚锡盐、八面体硫(octasulfur)、烷基硫醇、膦、弱碱、甲醛、一氧化碳或卤化物。在优选的实施方案中,官能化反应物是含氧酸,并且步骤(c)可以被描述为氧化步骤。
如果必要,步骤(c)可以进一步包括使初始反应产物和官能化反应物与水、氧化剂或这两者接触。氧化剂可以是任何合适的试剂,例如氧、臭氧或过氧化物(例如过氧化氢)。
不希望受任何具体理论束缚,发明人认为,将杂烷烃或芳烃RH转化为其相应的羟基化官能化产物ROH的一种可能的反应机理是图1中所示的反应机理。在方案1中给出了所提出的三步法的机理。
方案1:杂烷烃或芳烃(RH)转化为羟基化产物(ROH)
RH+MnXn→RX+HX+M(n-2)Xn-2 (方程2)
M(n-2)Xn-2+2HX+1/2O2→MXn+H2O (方程3)
RX+H2O→ROH+HX (方程4)
净反应:RH+1/2O2→ROH
在本发明的一个实施方案中,氧化性物质MXn与杂烷烃或芳烃(RH)反应以使官能化产物(RX)和氧化剂的还原形式MXn-2选择性地再生,如方程2所示。选择官能化产物RX以使过度氧化为不期望的产物如CO2的过程最小化。因为期望使用氧作为总的氧化剂,所以可以用来自空气的氧使氧化剂的还原形式MXn-2再生,如方程3所示。如方程4所示,RX可以被水解成相应的醇ROH。净反应是杂烷烃或芳烃(RH)向相应的羟基化产物ROH的总转化。如果MXn与杂烷烃或芳烃的反应是容易的话,可以在没有催化剂的情况下进行方程2的反应。然而,也可以使用催化剂。使用MXn盐作为可空气再循环的化学计量的氧化剂而不需要催化剂可以是有利的,以最小化体系的复杂性和对于鉴别与杂烷烃和/或芳烃的反应相容的单独的催化剂和氧化剂的需求。方案1不代表唯一可能的反应方案。例如,在一个反应或反应器中可以组合方程2和4或者方程2和3或者方程1、2和3。然而,为了最大化选择性、安全性和降低成本,方案1中所示的三个步骤可以分别进行。可以使用其它氧化剂例如过氧化氢代替氧。
可以使用四个标准来鉴别用于方案1的方程2的合适的MXn材料:(A)良好氧化剂,其中MXn+2e-+2H+MXn-2+2HX,Eo≈≤-0.5V;(B)与碳形成相对强的共价键;(C)强的亲电性;和(D)与抗衡阴离子X形成相对弱的键。具有这些特性的物质可以被描述为氧化性亲电子试剂(例如“软”的氧化性亲电子试剂)。
使用特定的氧化剂Tl(CF3CO2)3作为实例,在方案2中提出了使杂烷烃或芳烃(RH)羟基化的机理。
方案2
Tl(CF3CO2)3+RH→Tl(CF3CO2)+CF3CO2R+CF3CO2H (方程5)
Tl(CF3CO2)+1/2O2+2CF3CO2H+Tl(CF3CO2)3+H2O (方程6)
CF3CO2R+H2O→ROH+CF3CO2H (方程7)
净反应:RH+1/2O2→ROH
图2是使用便宜的气-液鼓泡柱反应器的连续反应器体系的工艺设计。图2的体系与所提出的方案2的机理的相关性如下:Ox是Tl(CF3CO2)3,H2Ox是Tl(CF3CO2),且方案2的方程5和7在烃氧化反应器(10)中组合。从ROA闪蒸分离器(20)中将最终产物ROH与H2Ox分离。Ox和H2Ox在氧化剂再生器(30)中的烃氧化器(20)之间循环。
使用Ox作为可空气再生的化学计量试剂的另一种可能的方法设计示于图3中。该方案的中心方面是使用两个平行的反应器,其中杂烷烃或芳烃(RH)和氧(1/2O2)反应物在两个反应器之间切换以允许连续处理。如果在两个反应器之间的泵送是不可行的,则可以利用这种工艺设计。该方法可以用引导至图3中所述反应器的反应物以如下引发:A→C,D→B,E→G,和H→F(循环1)。通过与氧化剂MXn反应而使杂烷烃或芳烃转化成期望的产物可以在一个反应器中进行,同时氧化剂的还原形式MXn-2通过空气的再生在另一个反应器中进行。在与杂烷烃或芳烃反应之后,氧化剂MXn被转化为还原形式MXn-2。与该过程同时,还原形式MXn-2用空气氧化为氧化剂MXn。将杂烷烃或芳烃和空气进料切换到反应器以允许用于将杂烷烃或芳烃和空气转化为相应的醇的连续方法。在这些作用的一个实施方案中,溶剂可以是阴离子X的相应的酸HX。然而,反应可以用其它溶剂或潜在地甚至作为高度分散的固体来进行。图3提供了可选的合适循环的实例:
循环1:A→C和D→B
E→G和H→F
循环2:A→B和D→C
E→F和H→G
循环3:A→C和D→B
E→G和H→F
循环4:A→B和D→C
E→F和H→G
等。
杂烷烃和芳烃的官能化的具体实例概述如下。
二官能化的烷烃例如1,4-丁二醇或1,3-丙二醇是高度有价值的物质。如从下面的方案3和4明显看出的,这些物质的合成涉及从简单烷烃开始的多个步骤。这些合成是非常昂贵的过程,其是资金和能量密集型的并产生过多排放物。
方案3.1,4-丁二醇的商业合成
方案4.1,3-丙二醇的商业合成
需要较少能量和资金的将烷烃以较少步骤直接转化为这些二醇的方法是更清洁和更便宜的。例如,这样的通过CH活化反应将烷烃转化成相应的醇的方法如方案5所示。
方案5.通过直接氧化合成1,4-丁二醇和1,3-丙二醇
已报道了基于主族阳离子例如TlX3、PbX4、IX3等的几种潜在的实用体系用于未取代烷烃的转化(Hashiguchi,et al.,Science,343,1232(2014)及其中的参考文献;和Konnick et al.,Angew.Chem.Int.Ed.,53,10490-10494(2014)及其中的参考文献)。这些主族体系的一个关键方面是反应通过亲电的CH活化进行,并且从相应的含氧酸溶剂产生高区域选择性的含氧酯。该结果的主要原因是相应的酸(例如CF3CO2H)的酯基(例如CF3CO2-)是吸电子的。因此,含氧酯(例如CF3CO2-CH3)的α-CH键相对于亲电的CH活化反应的反应性比母体烷烃CH4的CH键低得多。这种效应可以导致高选择性反应。
除了α-CH键之外,β-CH键的反应性较低。然而,这种“保护”效应随着进行反应的CH键与酯基之间的键的数目的增加而减弱。因此,CH键相对于酯基的相对反应性以α<β<γ<δ<ε等的顺序增加。根据这些反应的相对速率,该效应可用于合成1,3-丙二醇或1,4-丁二醇。因此,如方案6所示,正丁基TFA酯(X=TFA=CF3CO2)的反应选择性地生成1,4-二酯。
方案6:根据本发明的方法的杂烷烃的官能化
先前已经通过实验证明就是这种情况。C6F5I(TFA)2可以以高选择性使烷烃(RH)选择性地转化为相应的TFA-酯(Konnick et al.,Angew.Chem.Int.Ed.,53,10490-10494(2014)及其中的参考文献)。图4显示来自正丁基TFA,1与C6F5I(TFA)2在HTFA中的反应的粗反应混合物的NMR光谱。可以看出,反应中生成的唯一物质2是1,4-丁二醇的1,4-二酯。因此,发明人出乎意料地发现,可以提供杂烷烃(例如烷基酯)的区域选择性官能化,以产生烷基二酯,该烷基二酯在远离酯基的烷基链上的碳原子上被选择性地官能化。类似的反应已经应用于芳烃底物。
因此,在某些实施方案中,C-H键活化反应性中间体可与为含氧酸(例如三氟乙酸、乙酸、甲磺酸、其水溶液及其组合)的官能化反应物接触。如果需要,官能化的化合物可以被水解以形成期望的最终产物,例如醇或二醇。
除了醇和二醇之外,可以使用类似的反应顺序以产生氨基醇,例如乙醇胺或丙醇胺,以胺化合物作为前体开始。
通过使初始反应产物与含氮的官能化反应物(例如肼、羟胺、氨、氨、伯或仲胺或等同物)接触,C-H键活化反应性中间体可用于制备起始杂烷烃或芳烃的胺衍生物。肼或羟胺试剂可以是未取代的或可以被例如烷基或芳基等取代。C-H键活化反应性中间体与含氮的官能化反应物的反应可以原位进行,或者可以例如在液-液萃取之后在分离的产物流上进行。可以通过氧化步骤来回收亲电还原产物和使其再循环,并且起始杂烷烃或芳烃的胺衍生物可以被回收和进一步纯化、进一步反应或这两者。
通过使初始反应产物与甲锡烷基化官能化反应物(例如亚锡盐)在氧化剂的存在下反应,可以将C-H键活化反应性中间体转化成有机锡(锡烷)化合物。已知有机锡化合物在多种反应中作为多功能的合成中间体。
在一些实施方案中,可以用含硫官能化反应物如S8或烷基硫醇处理C-H键活化反应性中间体,以提供硫醇化的杂烷烃或芳烃。所获得的有机硫产物可以是硫醇、二硫化物或硫醚。可以通过硫原子的氧化而将这些化合物进一步转化为亚砜和砜。
在进一步的实施方案中,可以进一步处理C-H键活化性反应中间体以提供对应于起始杂烷烃或芳烃的羧酰胺基衍生物。例如,初始反应产物可以任选地在氧化剂存在下与甲酰化试剂例如甲醛和胺反应,以产生具有侧链羧酰胺基的起始杂烷烃或芳烃的同系衍生物,其中额外的碳原子已被添加至底物分子。
C-H键活化反应性中间体可以与膦进行随后的反应,以提供对应于原料的氧膦基化的杂烷烃或芳烃。使用三取代的膦可以提供鏻盐,而使用单-或二取代的膦可以提供类似的膦衍生物。
可以通过用非常弱的碱(例如弱酸的共轭碱,包括乙酸盐和三氟乙酸盐)处理,使杂烷烃的C-H键活化反应性中间体经历消除反应,产生杂烯烃。然后可以将所得的杂烯烃环氧化、转化为二醇等。
C-H键活化反应性中间体例如与一氧化碳的羰基化反应可以提供酰基化合物,其是起始杂烷烃或芳烃的同系物,其具有添加的额外的碳原子。取决于反应条件,所获得的酰基化合物例如醛、羧酸或羧酰胺,可以进行进一步的转化,例如本领域所公知的那些。
C-H键活化反应性中间体可以例如通过使用卤化物和本文所述的氧化剂(例如O2)进行卤化反应以提供起始杂烷烃或芳烃的卤代烃衍生物。
官能化反应(例如步骤(c))可以使用任何合适的条件进行。例如,可以使用与用于使杂烷烃或芳烃与氧化性亲电子试剂接触的反应器分离的反应器,其允许次级反应与初始反应器中存在的试剂分离。在此阶段,可以如本文所述在氧化过程中回收亲电还原产物和使其再循环,以使进行官能化反应(例如步骤(c))所需的高氧化态的氧化性亲电子试剂再生。
通过以下实施方案进一步说明本发明。
(1)一种制备官能化的化合物的方法,所述方法包括:
(a)提供杂烷烃或芳烃化合物,
其中
所述杂烷烃包含至少一个带有氢原子的sp3-杂化的碳原子和至少一个不同于碳或氢原子的杂原子,且
所述芳烃包含至少一个带有氢原子的sp2-杂化的碳原子,并且任选包含
(i)一个或多个sp3-杂化的碳原子,
(ii)一个或多个杂原子,或
(iii)(i)和(ii)两者,
(b)使所述化合物与以下物质接触以提供初始反应产物,
(i)包含氧化形式的主族元素的氧化性亲电子试剂,或
(ii)氧化剂和所述氧化性亲电子试剂的还原形式,和
(c)使所述初始反应产物与官能化反应物接触,其中所述官能化反应物的官能化部分替代所述初始反应产物上的氢以提供所述官能化的化合物。
(2)根据实施方案(1)所述的方法,其中步骤(b)包括使所述化合物与包含氧化形式的主族元素的氧化性亲电子试剂接触。
(3)根据实施方案(1)所述的方法,其中步骤(b)包括使所述化合物与氧化剂和氧化性亲电子试剂的还原形式接触。
(4)根据实施方案(1)-(3)中任一项所述的方法,其中所述氧化性亲电子试剂包含各自为氧化形式的铊、铅、铋、锑、硒、碲或其混合物。
(5)根据实施方案(4)所述的方法,权利要求1-3中任一项所述的方法,其中所述氧化性亲电子试剂包含氧化形式的碘,优选碘(III)或碘(V)。
(6)根据实施方案(1)-(4)中任一项所述的方法,其中所述氧化性亲电子试剂包含氧化形式的碘,优选碘(III)或碘(V)。
(7)根据实施方案(1)-(6)中任一项所述的方法,其中所述氧化性亲电子试剂是盐,所述盐包含氧化形式的主族元素的抗衡离子,并且其中所述抗衡离子是酸的共轭阴离子。
(8)根据实施方案(1)-(7)中任一项所述的方法,其中所述氧化形式的主族元素处于+n的氧化态,并且其中对于由所述氧化性亲电子试剂形成的亲电还原产物,所述元素为+(n-2)或+(n-1)的氧化态。
(9)根据实施方案(1)-(8)中任一项所述的方法,其中所述氧化性亲电子试剂具有式M+nXn,其中M是氧化态为n的金属或非金属主族元素阳离子,X是阴离子抗衡离子,并且n是平衡金属离子的n+正电荷所必需的阴离子电荷的数目。
(10)实施方案(9)所述的方法,其中式M+nXn的氧化性亲电子试剂与杂烷烃或芳烃反应,以产生式M+(n-2)Xn-2或M+(n-1)Xn-1的亲电还原产物。
(11)根据实施方案(1)-(10)中任一项所述的方法,其中所述氧化性亲电子试剂包含三氟乙酸根、乙酸根、硫酸根或烷基磺酸根阴离子中的一种或多种。
(12)实施方案(1)-(11)中任一项所述的方法,其中所述步骤(b)的接触在酸性介质中进行。
(13)实施方案(12)所述的方法,其中所述酸性介质是水性酸性介质。
(14)实施方案(12)或实施方案(13)所述的方法,其中所述酸性介质包含无机酸、羧酸、磺酸或其任意组合。
(15)根据实施方案(1)-(14)中任一项所述的方法,其还包括将官能化的化合物与由氧化性亲电子试剂所形成的亲电还原产物分离。
(16)根据实施方案(15)所述的方法,其还包括使分离的亲电还原产物与氧化性再生试剂接触以使所述氧化性亲电子试剂再生,其中所述氧化性再生试剂优选包含过氧化氢、氧、臭氧、硝酸或卤素。
(17)实施方案(16)所述的方法,其中使所述亲电还原产物和所述氧化性再生试剂在氧化性再生催化剂的存在下接触,其中所述氧化性再生催化剂优选包含铜、银、铁或钒。
(18)根据实施方案(17)所述的方法,其中所述氧化性再生试剂相对于所述化合物以至少化学计量的量存在。
(19)根据实施方案(1)-(18)中任一项所述的方法,其中包含氧化形式的主族元素的所述氧化性亲电子试剂相对于所述杂烷烃或芳烃以小于化学计量的量存在,并且作为催化剂起作用。
(20)根据实施方案(1)-(15)中任一项所述的方法,其中没有氧化性再生试剂与包含氧化形式的主族元素的所述氧化性亲电子试剂一起存在;并且所述氧化性亲电子试剂相对于所述化合物以至少化学计量的量存在。
(21)根据实施方案(1)-(20)中任一项所述的方法,其中步骤(c)还包括使所述初始反应产物和所述官能化反应物与水、氧化剂或这两者接触。
(22)根据实施方案(1)-(21)中任一项所述的方法,其中所述官能化反应物选自由含氧酸、肼、羟胺、氨、伯胺、仲胺、亚锡盐、八面体硫、烷基硫醇、膦、弱碱、甲醛、一氧化碳和卤化物组成的组。
(23)根据实施方案(1)-(22)中任一项所述的方法,其中所述化合物是杂烷烃。
(24)根据实施方案(23)所述的方法,其中所述杂烷烃是烷基单酯。
(25)根据实施方案(24)所述的方法,其中所述杂烷烃是正丁醇或正丙醇的酯,并且官能化的杂烷烃产物分别是1,4-丁二醇或1,3-丙二醇的二酯。
(26)根据实施方案(1)-(22)中任一项所述的方法,其中所述化合物是芳烃。
(27)根据实施方案(26)所述的方法,其中所述芳烃包含芳环体系。
(28)根据实施方案(26)所述的方法,其中所述芳烃包含杂芳环体系。
以下实施例进一步说明本发明,但是当然,其不应被解释为以任何方式限制本发明的范围。
实施例1
本实施例说明了包含氧化形式的主族元素的氧化性亲电子试剂用于使芳烃苯的C-H键活化和官能化的用途。
发现C6F5I(+3)(TFA)2(1)有效地用于在较低温度(125℃)下将苯(PhH)选择性单官能化为PhTFA。将该反应的温度降低至100℃导致1的完全消耗和在溶液中产生新的物质,其通过1H-和19F-NMR初步鉴定为二芳基-λ3-碘[C6F5-IIII-C6H5][TFA](3),并仅观察到痕量的PhTFA和C6F5-II(2)。在125℃下进一步加热该溶液导致3完全转化为PhTFA和2。该观察结果与3在PhH向PhTFA的转化中作为中间体一致(方案7)。
方案7
实施例2
本实施例说明了包含氧化形式的主族元素的氧化性亲电子试剂用于使芳烃甲苯的C-H键活化和官能化的用途。
研究了含有均裂的弱苄基CH键的甲苯与C6F5I(+3)(TFA)2(1)的反应性。使用标准反应条件(例如3h,150℃),观察到p-MeC6H4TFA和o-MeC6H4TFA以约3:1的比率定量产生(基于开始的[1]);且没有形成由自由基路径所预期的任何苄基氧官能化的产物(方案8)。
方案8
显然,相对于甲基的sp3-杂化的碳原子的官能化,芳基环的sp2-杂化的碳原子的官能化是有利的。
实施例3
该实施例说明了包含氧化形式的主族元素的氧化性亲电子试剂用于使芳烃苯的C-H键活化和官能化的用途。
在25℃下使Pb(TFA)4与苯(PhH)在三氟乙酸(TFAH)中反应,从而以80%的产率提供选择性单官能化的产物PhTFA(方案9)。反应非常容易,因为反应几乎在室温下即刻发生。
方案9
实施例4
本实施例说明了包含氧化形式的主族元素的氧化性亲电子试剂用于使芳烃甲苯的C-H键活化和官能化的用途。
研究了含有均裂的弱苄基CH键的甲苯与Pb(TFA)4的反应性。温和的反应条件(例如,室温和TFAH)仅产生p-MeC6H4-TFA产物(>95%产率),没有形成由自由基路径所预期的任何苄基氧官能化的产物(方案10)。
方案10
实施例5
本实施例说明了包含氧化形式的主族元素的氧化性亲电子试剂用于使杂烷烃三氟乙酸正丁酯的C-H键活化和官能化的用途。
向具有玻璃内衬和磁力搅拌棒的两个小(3mL)的高压反应器中各自装入1.0mL的250mM的C6F5-I(TFA)2在100mM的三氟乙酸酐(TFAA)/三氟乙酸(TFAH)中的溶液。向每个反应器中还加入17μL(17.3mg,0.1mmol)的三氟乙酸正丁酯。将反应器用500psig的氩气进行压力脱气8次;然后充入500psig的氩气并密封。然后将反应器在1000rpm搅拌下在150℃下加热3小时。完成后,将反应器冷却至室温,并释放压力。向每种溶液中装入100μL的31.32mM的CH2Cl2在TFAH中的溶液作为内标;并通过1H-NMR分析产物。分析表明生成1,4-二(三氟乙酰氧基)丁烷作为主要产物(5mM,5%产率),剩余的物料为原料三氟乙酸正丁酯。
实施例6
该实施例说明了包含氧化形式的主族元素的氧化性亲电子试剂用于使杂烷烃三氟乙酸正丁酯的C-H键活化和官能化的用途。
向具有玻璃内衬和磁力搅拌棒的两个大(15mL)的高压反应器中各自装入2.0mL的300mM的Tl(TFA)3在TFAH中的溶液。向每个反应器中还加入0.1mL(102mg,0.6mmol,相对于氧化剂为1当量)的三氟乙酸正丁酯。将反应器用500psig的氩气进行压力脱气5次;然后充入500psig的氩气并密封。然后将反应器在1000rpm搅拌下在180℃下加热3小时。完成后,将反应器冷却至室温,并释放压力。向每种溶液中装入0.2mL的300mM的CH2Cl2在TFAH中的溶液作为内标;并通过1H-NMR分析产物。分析表明生成1,4-二(三氟乙酰氧基)丁烷作为主要产物(8mM,3%产率),剩余的物料为原料三氟乙酸正丁酯。
本文引用的所有参考文献,包括出版物、专利申请和专利,在此通过引用并入本文,其程度如同每个参考文献被单独地和具体地视为通过引用并入本文作为参考并且在本文中给出其全部内容。
在描述本发明的上下文(特别是在所附权利要求书的上下文中)中,术语“一个(a)”、“一个(an)”和“该(the)”和“至少一个(at least one)”和类似术语的使用应被解释为覆盖单数和复数这两者,除非本文另有说明或与上下文明显矛盾。使用术语“至少一个”,随后列出一个或多个项目(例如,“A和B中的至少一个”)应被解释为表示所列出的项目中的一个(A或B)或所列项目中的两个或更多个的任何组合(A和B),除非本文另有说明或与上下文明显矛盾。除非另有说明,否则术语“包含(comprising)”,“具有(having)”,“包括(including)”和“含有(containing)”应被解释为开放式术语(即意味着“包括但不限于”)。除非本文中另有说明,否则本文中数值范围的列举仅意在用作单独提及落在该范围内的每个单独值的简写方法,并且每个单独值被并入本说明书中,如同其在本文中单独列举一样。本文所述的所有方法可以以任何合适的顺序进行,除非本文另有说明或与上下文明显矛盾。本文提供的任何和所有实施例或示例性语言(例如,“诸如”)的使用仅旨在更好地说明本发明,而不对本发明的范围构成限制,除非另有声明。说明书中的语言不应被解释为指示任何未要求保护的要素对于实施本发明是必要的。
本文描述了本发明的优选实施方案,包括发明人已知的用于实施本发明的最佳模式。在阅读前面的描述之后,那些优选实施方案的变型对于本领域的普通技术人员来说是显而易见的。发明人期望技术人员适当地采用这样的变化,并且发明人意图以本文具体描述以外的方式实施本发明。因此,本发明包括适用的法律所允许的所附权利要求书中所述主题的所有修改和等同物。此外,除非在本文中另有说明或与上下文明显矛盾,否则本发明涵盖上述要素在其所有可能变型中的任何组合。
- 还没有人留言评论。精彩留言会获得点赞!