纤维增强聚酰亚胺树脂成型体及其制造方法与流程
本发明涉及纤维增强聚酰亚胺树脂成型体及其制造方法,更具体地,涉及具有优异的滑动性能和在成型期间的形状稳定性优异、并且包含分散在聚酰亚胺树脂中的功能性纤维的成型体,和涉及所述成型体的制造方法。
背景技术:
由通过将如碳纤维等功能性纤维与树脂混合获得的纤维增强树脂制成的成型体,由于它们的如耐候性、机械强度和耐久性等特性优异而广泛地用于汽车和飞行器等的运输设备材料、土木工程和建筑材料和运动用品。
例如,以下专利文献1公开了一种包含特定的沥青系碳短纤维混合物和基质树脂的碳纤维增强树脂成型体,并且教导了其有利地用于各种电子部件。
以下专利文献2提出了一种由包含特定的芳香族聚酰亚胺低聚物作为碳纤维用粘结剂的摩擦材料用树脂组合物制成的摩擦材料。专利文献2教导了与在使用已经有利地用作摩擦材料用粘结剂的酚醛树脂的情况相比,该摩擦材料中粘结剂自身的耐热性和机械性质优异并且成型性高。
此外,以下专利文献3提出了一种由包含10-70wt%的具有特定的导热率的碳纤维的碳纤维增强合成树脂制成的滚动体。
当该纤维增强树脂成型体用作如轴承等滑动性构件时,要求包括高的如强度和刚性等机械强度、小的动摩擦系数、少的磨耗量、进一步大的极限PV值(limit PV value)等特性,并且期望机械强度、耐热性和耐久性以及树脂的浸渍性优异的加成反应型聚酰亚胺树脂应当用作基质树脂。
还提出了使得可以通过转移成型(RTM)和树脂注入(RI)来制造碳纤维增强复合材料作为加成反应型聚酰亚胺树脂的高功能的加成反应型聚酰亚胺树脂(专利文献4)。
现有技术文献
专利文献
专利文献1:日本专利No.4538502
专利文献2:JP-A 2009-242656
专利文献3:JP-A 2011-127636
专利文献4:JP-B 2003-526704
技术实现要素:
发明要解决的问题
然而,当加成反应型聚酰亚胺树脂用作纤维增强树脂成型体的基质树脂时,即使获得优异的耐热性、耐久性和机械强度,也存在获得的成型体翘曲并且不能作为滑动性构件投入实际使用的问题。
本发明的发明人为了寻找该原因进行了深入的研究,并且发现以下事实。即,发现由于可以有利地用作如碳纤维等功能性纤维的基质树脂的加成反应型聚酰亚胺树脂在预聚物状态下具有低的熔体粘度,因此当预聚物与功能性纤维混合时,功能性纤维沉降并且不均匀地分布在预聚物中,从而当树脂在该状态下交联固化时,成型体的收缩量根据功能性纤维的存在量而不同,从而导致获得的纤维增强树脂成型体的翘曲。
因此,本发明的目的是提供一种具有优异的滑动性能并且不发生翘曲和在成型期间的形状稳定性优异的纤维增强聚酰亚胺树脂成型体。
本发明的另一目的是提供能够以高形状稳定性成型、具有优异的滑动性能的纤维增强聚酰亚胺树脂成型体的制造方法。
用于解决问题的方案
根据本发明,提供一种包含分散在加成反应型聚酰亚胺树脂中的功能性纤维并且具有3,000kPa·m/s以上的极限PV值的树脂成型体。
在本发明的树脂成型体中,优选,
1.构成树脂成型体的组合物的基质为加成反应型聚酰亚胺树脂,并且上述功能性纤维浸渍在上述聚酰亚胺树脂中;
2.以基于100重量份的加成反应型聚酰亚胺为5-200重量份的量包含上述功能性纤维;
3.上述功能性纤维为选自碳纤维、玻璃纤维、芳香族聚酰胺纤维和金属纤维的任意一种以上;
4.上述功能性纤维为平均纤维长度为50-6,000μm和平均纤维直径为5-20μm的碳纤维;
5.以基于100重量份的加成反应型聚酰亚胺为5-40重量份的量包含增稠剂,并且该增稠剂分散在树脂成型体中;和
6.上述增稠剂为选自石墨、二硫化钼、PTFE(四氟乙烯树脂)、微细碳系材料和金属粉末的至少一种以上。
根据本发明,提供树脂成型体的制造方法,其包括至少以下步骤:
将加成反应型聚酰亚胺树脂的预聚物和功能性纤维在加成反应型聚酰亚胺树脂的熔点(160至170℃)以上且所述树脂的热固化开始温度(约300℃)以下的温度下捏合的分散捏合步骤;和
将所得混合物在加成反应型聚酰亚胺树脂的热固化开始温度以上的温度下成形的成形步骤。
在本发明的树脂成型体的上述第一制造方法中,优选,
1.在上述分散捏合步骤与上述成形步骤之间设置通过将分散捏合步骤获得的捏合产物在加成反应型聚酰亚胺树脂的热固化开始温度以上的温度下保持一定的时间来提高捏合产物的粘度的增稠步骤;
2.上述功能性纤维的含量基于100重量份的加成反应型聚酰亚胺为5-200重量份;
3.在分散捏合步骤后获得的混合物在300至320℃的温度条件下的熔体粘度为10-5,000Pa·s,并且将混合物在冷却、粉碎、混合后加压成形;
4.在上述增稠步骤中,将混合物在300至320℃的温度条件下的熔体粘度调节为10-5,000Pa·s;
5.上述加成反应型聚酰亚胺树脂为具有苯基乙炔基作为加成反应基团的聚酰亚胺树脂;和
6.当上述加成反应型聚酰亚胺树脂为具有苯基乙炔基作为加成反应基团的聚酰亚胺树脂时,在上述增稠步骤中,在310±10℃的温度下保持30-60分钟。
根据本发明,进一步提供包含分散在100重量份的加成反应型聚酰亚胺中的5-200重量份的功能性纤维和5-40重量份的增稠剂的树脂成型体的制造方法,其包括以下步骤:
将上述加成反应型聚酰亚胺树脂的预聚物、功能性纤维和增稠剂在加成反应型聚酰亚胺树脂的熔点以上且该树脂的热固化开始温度以下的温度下捏合的分散捏合步骤;和
将分散捏合步骤后获得的混合物在加成反应型聚酰亚胺树脂的热固化开始温度以上的温度条件下加压成形。
在本发明的树脂成型体的上述第二制造方法中,分散捏合步骤后获得的混合物在300至320℃的温度下的熔体粘度为10-5,000Pa·s。
在本发明的树脂成型体的制造方法中,上述成形步骤优选通过压缩成型来进行。
发明的效果
在本发明的纤维增强聚酰亚胺树脂成型体中,具有优异的耐热性、耐久性和机械强度的加成反应型聚酰亚胺树脂用作基质树脂,并且将5-200重量份的功能性纤维与100重量份的该加成反应型聚酰亚胺混合,从而使得可以实现3,000kPa·m/s以上的极限PV值和优异的滑动性能。另外,由于在功能性纤维均匀地分散在成型体中的同时纤维增强聚酰亚胺树脂交联固化以成型,因而成型体不会如翘曲等变形并且可以有利地用作滑动性构件。极限PV值为通过当摩擦力急剧上升时的表面压力P和速度V的积求得的值并且一般地计算作为用于判断成型体是否适用于滑动性构件的使用环境的指标。在接近于极限PV值的条件下,观察到通过由滑动面的摩擦热使得树脂熔融或燃烧而导致的动摩擦系数和样品的温度的上升、以及材料的异常磨耗,并且该极限PV值高意味着滑动性能高。本发明的纤维增强聚酰亚胺树脂成型体包含功能性纤维浸渍于其中的加成反应型聚酰亚胺和预定量的功能性纤维,具有优异的滑动性能,当该成型体用作滑动性构件时其长期稳定地保持该性能并且使得可以防止由翘曲导致的变形,从而其具有优异的生产性,使得可以减少由长期使用期间的磨耗导致的PV值的变化并且便于交换时间和装置的管理。
在本发明的纤维增强聚酰亚胺树脂成型体的第一制造方法中,在加成反应型聚酰亚胺树脂和功能性纤维的分散捏合步骤后,设置用于提高熔融状态的聚酰亚胺树脂的预聚物(酰亚胺低聚物)的粘度的增稠步骤,从而使得可以保持功能性纤维均匀地分散在预聚物中并且成型而不导致包含均匀分散而不沉降且没有不均匀分布的功能性纤维的纤维增强聚酰亚胺树脂成型体翘曲变形。
此外,在本发明的纤维增强聚酰亚胺树脂成型体的第二制造方法中,由于5-200重量份的功能性纤维和5-40重量份的增稠剂与100重量份的加成反应型聚酰亚胺混合,因而可以将分散捏合步骤后的混合物在300至320℃的温度条件下的熔体粘度调节为10-5,000Pa·s。从而功能性纤维可以保持均匀地分散在预聚物中,并且可以使包含均匀地分散在其中的功能性纤维的纤维增强聚酰亚胺树脂成型体成型而不翘曲变形。
如后所述,优选用于本发明的增稠剂具有优异的滑动性并且可以进一步改进滑动性能。
附图说明
图1为用于说明实施例中极限PV值的测量方法的图;
图2为用于说明实施例中翘曲量的测量方法的图;和
图3为示出不均匀地分布的纤维的不均匀性的图。
具体实施方式
(树脂成型体)
本发明的纤维增强聚酰亚胺树脂成型体为包含分散在后述加成反应型聚酰亚胺树脂中的功能性纤维的树脂成型体,并且具有重要的特征:所述成型体具有3,000kPa·m/s以上的极限PV值,以及耐热性、耐久性、机械强度,和具有大的极限PV值和优异的滑动性能。
[加成反应型聚酰亚胺树脂]
在本发明中,重要的特征是,加成反应型聚酰亚胺树脂用作变为构成纤维增强聚酰亚胺树脂成型体的组合物的基质的聚酰亚胺树脂。
用于本发明的加成反应型聚酰亚胺树脂由在末端具有加成反应基团的芳香族聚酰亚胺低聚物组成并且通过常规已知的制造方法来制备。例如,其可以通过以下而容易地获得:使用芳香族四羧酸二酐,芳香族二胺和分子内具有加成反应基团和酸酐基团或氨基的化合物来确保各酸基的当量总和变为几乎与各氨基的当量总和相等并且使它们优选在溶剂中反应。反应方法的实例包括其中在100℃以下、优选80℃以下进行聚合0.1-50小时从而生成具有酰胺酸键的低聚物,然后用酰亚胺化试剂(imidizing agent)使低聚物化学酰亚胺化的方法;包括在140至270℃的高温下加热和热酰亚胺化的两个步骤的方法;和仅包括在140至270℃的高温下进行聚合/酰亚胺化反应0.5-50小时的一个步骤的方法。
用于这些反应的溶剂的优选实例包括但不限于,如N-甲基-2-吡咯烷酮、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、N,N-二乙基乙酰胺、γ-丁内酯和N-甲基己内酰胺等有机极性溶剂。
在本发明中,如果芳香族酰亚胺低聚物的末端的加成反应基团为当制造树脂成型体时通过加热进行固化反应(加成聚合反应)的基团,则对该加成反应基团没有特别地限定。当考虑可以优选进行固化反应并且获得的固化产物具有高耐热性时,加成反应基团优选为选自由苯基乙炔基、乙炔基、纳迪克酸基(nadic acid group)和马来酰亚胺基组成的组的反应基团,其中苯基乙炔基是特别优选的,因为通过固化反应不产生气体组分并且获得的树脂成型体具有优异的耐热性和机械强度。
通过其中分子内具有加成反应基团和酸酐基团或氨基的化合物优选与芳香族酰亚胺低聚物的末端的氨基或酸酐基团形成酰亚胺环的反应,将这些加成反应基团引入至芳香族酰亚胺低聚物的末端。
在分子内具有酸酐基团或氨基并且具有加成反应基团的化合物优选为4-(2-苯基乙炔基)邻苯二甲酸酐、4-(2-苯基乙炔基)苯胺、4-乙炔基-邻苯二甲酸酐、4-乙炔基苯胺、纳迪克酸酐或马来酸酐。
形成在末端具有加成反应基团的芳香族酰亚胺低聚物的四羧酸组分为选自由2,3,3’,4’-联苯四羧酸二酐、2,2’,3,3’-联苯四羧酸二酐、3,3’,4,4’-联苯四羧酸二酐和3,3’,4,4’-二苯甲酮四羧酸二酐组成的组的至少一种四羧酸二酐,特别优选2,3,3’,4’-联苯四羧酸二酐。
形成在末端具有加成反应基团的芳香族酰亚胺低聚物的二胺组分的实例包括但不限于,如1,4-二氨基苯、1,3-二氨基苯、1,2-二氨基苯、2,6-二乙基-1,3-二氨基苯、4,6-二乙基-2-甲基-1,3-二氨基苯、3,5-二乙基甲苯-2,4-二胺和3,5-二乙基甲苯-2,6-二胺等具有一个苯环的二胺,如4,4’-二氨基二苯醚、3,4’-二氨基二苯醚、3,3’-二氨基二苯醚、3,3’-二氨基二苯甲酮、4,4’-二氨基二苯甲酮、4,4’-二氨基二苯甲烷、3,3’-二氨基二苯甲烷、双(2,6-二乙基-4-氨基苯氧基)甲烷、双(2-乙基-6-甲基-4-氨基苯基)甲烷、4,4’-亚甲基-双(2,6-二乙基苯胺)、4,4’-亚甲基-双(2-乙基、6-甲基苯胺)、2,2-双(3-氨基苯基)丙烷、2,2-双(4-氨基苯基)丙烷、联苯胺、2,2’-双(三氟甲基)联苯胺、3,3’-二甲基联苯胺、2,2-双(4-氨基苯基)丙烷和2,2-双(3-氨基苯基)丙烷等具有两个苯环的二胺,如1,3-双(4-氨基苯氧基)苯、1,3-双(3-氨基苯氧基)苯、1,4-双(4-氨基苯氧基)苯和1,4-双(3-氨基苯氧基)苯等具有三个苯环的二胺,以及如2,2-双[4-[4-氨基苯氧基]苯基]丙烷和2,2-双[4-[4-氨基苯氧基]苯基]六氟丙烷等具有四个苯环的二胺。它们可以单独使用或者以两种以上的组合使用。
从耐热性和成型性的观点,由选自由这些二胺中的1,3-二氨基苯、1,3-双(4-氨基苯氧基)苯、3,4’-二氨基二苯醚、4,4’-二氨基二苯醚和2,2’-双(三氟甲基)联苯胺组成的组的至少两种芳香族二胺组成的混合二胺是优选的,并且特别优选使用由1,3-二氨基苯和1,3-双(4-氨基苯氧基)苯组成的混合二胺,由3,4’-二氨基二苯醚和4,4’-二氨基二苯醚组成的混合二胺,由3,4’-二氨基二苯醚和1,3-双(4-氨基苯氧基)苯组成的混合二胺,由4,4’-二氨基二苯醚和1,3-双(4-氨基苯氧基)苯组成的混合二胺,以及由2,2’-双(三氟甲基)联苯胺和1,3-双(4-氨基苯氧基)苯组成的混合二胺。
用于本发明的在末端具有加成反应基团的芳香族酰亚胺低聚物的重复单元数为大于0且20以下,特别优选1-5。芳香族酰亚胺低聚物的通过GPC测量的苯乙烯换算的数均分子量为10,000以下,特别优选3,000以下。当重复单元数落在上述范围内时,将熔体粘度调节至适当的范围,从而使得可以均匀地混合功能性纤维。此外,芳香族酰亚胺低聚物使得可以提供具有不需要在高温下成型的优异的成型性以及优异的耐热性和机械强度的树脂成型体。
重复单元数的控制可以通过改变芳香族四羧酸二酐、芳香族二胺和分子内具有加成反应基团和酸酐基团或氨基的化合物的比来进行。当分子内具有加成反应基团和酸酐基团或氨基的化合物的比例提高时,低聚物的分子量随着重复单元数的减小而降低,和当上述化合物的比例降低时,低聚物的分子量随着重复单元数的增大而提高。
加成反应型聚酰亚胺树脂可以根据目的树脂成型体的用途基于已知的配方而配混有如阻燃剂、着色剂、润滑剂、热稳定剂、光稳定剂、紫外线吸收剂和填料等树脂添加剂。
[功能性纤维]
在本发明中,作为分散在上述加成反应型聚酰亚胺树脂中的功能性纤维,可以使用如碳纤维、芳香族聚酰胺纤维、玻璃纤维和金属纤维等常规已知的功能性纤维,其中优选使用碳纤维。
可以特别优选使用平均纤维长度为50-6,000μm和平均纤维直径为5-20μm的碳纤维。当平均纤维长度低于上述范围时,不会充分获得碳纤维作为增强材料的效果;和当平均纤维长度超过上述范围时,纤维在聚酰亚胺树脂中的分散性劣化。当平均纤维直径低于上述范围时,纤维的操作性劣化并且所述纤维昂贵;和当平均纤维直径超过上述范围时,功能性纤维的沉降速度增大,功能性纤维会不均匀分布,并且纤维的强度易于降低,从而不会充分获得纤维作为增强材料的效果。
功能性纤维的总含量对树脂成型体的滑动性能和成形期间翘曲的发生具有大的影响。在本发明中,当以基于100重量份的加成反应型聚酰亚胺为5-200重量份、特别是10-150重量份的量包含功能性纤维时,有利地获得具有优异的滑动性能和优异的形状稳定性而不翘曲的成型体。当功能性纤维的总含量少于上述范围时,极限PV值变得小于上述值,从而滑动性会降低。另外,树脂成型体的翘曲发生的可能性会增大。当功能性纤维的总含量超过上述范围时,极限PV值会变得小于当总含量落在上述范围内时获得的值。此外,发生过度的增稠,从而无法进行成形。
在本发明中,选自如炭黑等微细碳系材料、以及如铝粉末和铜粉末等金属粉末的至少一种无机材料可以与上述功能性纤维组合使用。
优选以基于100重量份的加成反应型聚酰亚胺为5-40重量份的量包含上述无机材料。
[增稠剂]
在本发明中,当增稠剂以基于100重量份的加成反应型聚酰亚胺为5-40重量份的量与功能性纤维组合使用时,加成反应型聚酰亚胺树脂的预聚物的粘度可以在不经历增稠步骤的情况下增大,从而功能性纤维可以保持均匀地分散在预聚物中而不沉降。
作为增稠剂,可以使用石墨、二硫化钼、PTFE(四氟乙烯树脂)、氧化镁、氢氧化镁和氢氧化钙,并且石墨、二硫化钼和PTFE是特别优选的,因为它们进一步改进滑动性能。
优选以如上所述基于100重量份的加成反应型聚酰亚胺为5-40重量份的量包含增稠剂。当增稠剂的量少于上述范围时,预聚物的粘度不能充分提高并且功能性纤维的沉降不能被充分抑制,从而不能使没有翘曲变形且包含均匀分散在其中的功能性纤维的树脂成型体成型。当增稠剂的量超过上述范围时,会损害滑动性能,其结果为摩擦系数的增大和耐摩耗性的降低。
(树脂成型体的第一制造方法)
本发明的树脂成型体的第一制造方法包括至少以下步骤:
(A)将加成反应型聚酰亚胺树脂的预聚物(酰亚胺低聚物)和功能性纤维在加成反应型聚酰亚胺树脂的熔点以上且该树脂的热固化开始温度以下的温度下捏合的分散捏合步骤;和
(C)将分散捏合步骤后获得的混合物在反应型聚酰亚胺树脂的热固化开始温度以上的温度下加压成形的成形步骤,和
(B)根据需要在上述分散捏合步骤(A)与上述成形步骤(C)之间设置根据需要通过将从分散捏合步骤获得的捏合产物在反应型聚酰亚胺树脂的热固化开始温度以上的温度下保持一定的时间来提高捏合产物的粘度从而将捏合产物的粘度调节至适当的范围的增稠步骤。
如上所述,由于用于使本发明的树脂成型体成型的加成反应型聚酰亚胺树脂在作为交联固化前的预聚物的状态下具有低粘度,因而当树脂中包含功能性纤维时,功能性纤维沉降,结果,功能性纤维不均匀地分布并且成型体翘曲。在本发明的第一制造方法中,在上述分散捏合步骤(A)后,通过在上述增稠步骤(B)中增大预聚物的粘度来防止功能性纤维的沉降,并且在维持该状态的同时,在成形步骤(C)中使捏合产物成形,从而功能性纤维均匀地分散并且加热固化时均匀地收缩,从而使得可以成型没有翘曲的成型体。
[分散捏合步骤]
将加成反应型聚酰亚胺树脂的预聚物(酰亚胺低聚物)和功能性纤维在加成反应型聚酰亚胺树脂的熔点以上的温度下加热并且在预聚物熔融以混合到一起的同时一起捏合。此时,如上所述,基于100重量份的加成反应型聚酰亚胺,使用5-200重量份、特别是10-150重量份的功能性纤维。此外,可以以上述量使用上述无机材料。尽管不特别必要,但可以以上述量混合上述增稠剂。
预聚物和功能性纤维可以通过使用如亨舍尔混合机、转鼓混合机或螺带式混合机等常规已知的混合机来捏合到一起。然而,由于重要的是功能性纤维不应当被破断并且应当均匀地分散,因而特别优选使用间歇式加压捏合机(捏合机)。
本发明中期望的是,分散捏合步骤后预聚物和功能性纤维的混合物应当冷却以固化并且分裂成具有预定尺寸的块状材料。因而,包含分散在预聚物中的功能性纤维的混合物可以长期贮存,并且操作性得以改进。
[增稠步骤]
然后,当已经熔融捏合到一起的预聚物和功能性纤维的混合物在300至320℃的温度条件下的熔体粘度为10Pa·s以下时,将该混合物在接近于用于混合物的聚酰亚胺树脂的热固化开始温度的310±10℃的温度下保持30-60分钟,从而将在300-320℃的温度条件下的熔体粘度调节至10至5,000Pa·s。
即,预聚物通过将预聚物和功能性纤维的混合物在电炉中在310±10℃的温度下保持30-60分钟而逐渐交联,从而提高粘度。此外,在上述分散捏合步骤中浸渍在预聚物中的功能性纤维由于此粘度的提高可以保持分散在预聚物中而不沉降。通过将加热温度和保持时间设定为上述范围,在没有使预聚物完全交联固化的情况下,仅粘度可以提高至上述范围。因此,增稠步骤在预聚物的热固化开始温度以上且低于预聚物完全交联固化的温度的温度下来进行。
加成反应型聚酰亚胺树脂的反应开始温度取决于加成反应基团,并且本发明具有苯基乙炔基作为优选加成反应基团的聚酰亚胺树脂期望在接近于热固化开始温度的310±10℃的温度下加热30-60分钟。
[成形步骤]
在增稠步骤中已经将其熔体粘度调节至上述范围的预聚物和功能性纤维的混合物在使用时的聚酰亚胺树脂的热固化开始温度以上的温度下成型为具有期望的形状的树脂成型体。
为了进行成形步骤,将处在具有上述范围内的粘度的熔融状态的聚酰亚胺预聚物和功能性纤维的混合物引入至模具,并且在热固化开始温度以上的温度下加热并加压以固化,从而使树脂成型体成型。
尽管成形优选通过其中使引入至模具中的混合物加压压缩并成型的压缩成型或转移成型来进行,但也可以采用注射成型或挤出成型。
(树脂成型体的第二制造方法)
本发明的树脂成型体的第二制造方法包括:
将100重量份的加成反应型聚酰亚胺树脂的预聚物(酰亚胺低聚物)、5-200重量份的功能性纤维和5-40重量份的增稠剂在加成反应型聚酰亚胺树脂的熔点以上且该树脂的热固化开始温度以下的温度下捏合的分散捏合步骤;和
将分散捏合步骤后的混合物在反应型聚酰亚胺树脂的热固化开始温度以上的温度下加压成形的成形步骤。
由于如上所述用于成型本发明的成型体的加成反应型聚酰亚胺树脂在交联固化前的预聚物状态下具有低粘度,因而当包含功能性纤维时,功能性纤维沉降,结果,功能性纤维不均匀地分布并且成型体翘曲。在本发明的第二制造方法中,通过将功能性纤维和预定量的增稠剂与预聚物混合,预聚物的粘度可以在不经历增稠步骤的情况下增大,结果,功能性纤维分散在预聚物中而不沉降,并且在功能性纤维保持分散的同时在成形步骤中使混合物成形,从而随着在热固化时其均匀地收缩使得可以使成型体没有翘曲地成型。
[分散捏合步骤]
将加成反应型聚酰亚胺树脂的预聚物(酰亚胺低聚物)、功能性纤维和增稠剂在加成反应型聚酰亚胺树脂的熔点以上的温度下加热以使预聚物熔融并且捏合到一起,从而使预聚物与功能性纤维混合。此时,如上所述,基于100重量份的加成反应型聚酰亚胺,使用5-200重量份、特别是10-150重量份的功能性纤维和5-40重量份的增稠剂。
可以以与上述第一制造方法相同的方式将预聚物和功能性纤维捏合到一起。
在该方式中,分散捏合步骤的温度为预聚物的熔点以上且交联固化温度以下,特别优选分散捏合步骤后的混合物在300至320℃的温度下具有10-5,000Pa·s的熔体粘度。将此粘度的上升和预聚物向功能性纤维的渗透结合,使得功能性纤维保持分散在预聚物中而不沉降。
在将分散捏合步骤后的预聚物、功能性纤维和增稠剂的混合物冷却以固化之后,期望分裂为具有预定尺寸的块状材料。因而,包含分散在预聚物中的功能性纤维的混合物可以长期贮存,并且操作性得以改进。
[成形步骤]
以与上述第一制造方法相同的方式使在分散捏合步骤后熔体粘度已经调节至上述范围的预聚物、功能性纤维和增稠剂的混合物成形。
实施例
(极限PV值的测量)
基于JIS K 7218(塑料的滑动磨耗试验方法),通过使用推力型磨耗试验机,在图1所示的盘上环系统(ring-on-disk system)中以一定的速度每隔5分钟或10分钟提高表面压力,摩擦力急剧上升发生显著变形、或者产生磨耗粉末的时间取作极限时间,并且极限时间的前一个的表面压力(P)与速度(V)的积取作极限PV值。
极限PV值测量条件
试验速度:0.5m/s,初期表面压力:0.5MPa
表面压力步骤:0.5MPa/10min(~10MPa)
1MPa/10min(10MPa~)
配合材料:S45C环,表面粗糙度Ra为0.8μm,外径为25.6mm,内径为20mm(接触面积为2cm2)
试验环境:23±2℃,50%RH±5%RH
试验机:A&D Company,Limited制的EMF-III-F摩擦磨耗试验机。
(纤维的分散)
不均匀分布的纤维的有无通过目视观察成型体的截面或者通过扫描电子显微镜(Hitachi High-Technologies Corporation制的S-3400N)来确认。○表示纤维分散和×表示纤维沉降。
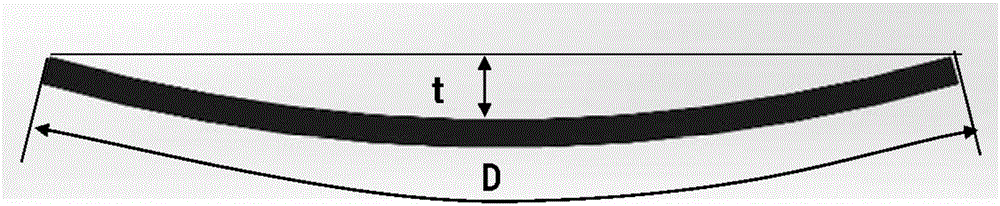
(翘曲量的测量)
通过测量图2所示的试验片的翘曲量“t”(mm)和制品的直径“D”(mm)由下式(1)来计算翘曲/直径比。
翘曲/直径比(%)=t/Dx100
t:试验片的翘曲量(mm),D:制品的直径(mm)
对于判定翘曲/直径比是否可接受,○表示所述比小于1.5%和×表示所述比为1.5%以上。
(熔体粘度的测量)
310℃下的熔体粘度用流变仪(TA Instrument制的ARES)来测量。将测量模式设定为动态频率分散,将角频率设定为0.1-500rad/s,并且测量0.1rad/s下的熔体粘度。
(实施例1)
将11.1重量份的平均单纤维长度为200μm的沥青系碳纤维(Mitsubishi Plastics Inc.制的K223HM)与100重量份的加成聚合型聚酰亚胺(Ube Industries,Ltd.制的PETI-330)混合,并且将所得混合物通过捏合机在大气压下在280℃下熔融捏合30分钟。其后,获得冷却至室温的混合物(块状模制化合物(bulk molding compound),以下缩写为“BMC”)。将获得的BMC分裂为容易操作的尺寸,在电炉内在310℃下保持30分钟,急冷并且再粉碎,从而获得树脂混合物(增稠BMC),然后将其在压缩成型机用模具内在280至320℃下保持一定时间以熔融并均热,然后在对其施加2.4MPa的压力的同时,以3℃/min的升温速度加热至371℃并且在该温度下保持60分钟,并且逐渐冷却,从而获得直径为40mm和厚度为3mm的板。将获得的板材在357℃下固化6小时并且切成期望的尺寸,从而获得试验片。
(实施例2)
将42.9重量份的平均单纤维长度为200μm的沥青系碳纤维(Mitsubishi Plastics Inc.制的K223HM)与100重量份的加成聚合型聚酰亚胺(Ube Industries,Ltd.制的PETI-330)混合,并且将所得混合物通过捏合机在大气压下在280℃下熔融捏合30分钟。其后,获得冷却至室温的BMC。将获得的BMC分裂为适合模具的尺寸,在压缩成型机用模具内在280至320℃下保持一定时间以熔融并均热,然后在对其施加11MPa的压力的同时,以3℃/min的升温速度加热至371℃并且在该温度下保持1小时,并且逐渐冷却,从而获得直径为100mm和厚度为3mm的板。将获得的板材在357℃下固化6小时并且切成期望的尺寸,从而获得试验片。
(实施例3)
除了将碳纤维的配混量改变为100重量份以外,重复实施例2的步骤。
(实施例4)
除了BMC不在电炉内310℃下保持以外,重复实施例1的步骤。由于获得的树脂成型体翘曲,因而刮削前后表面层以获得预定的平行度,从而测量极限PV值。尽管没有测量刮削前的表面的极限PV值,但当观察测量的表面时,与刮削前的表面相比,明显存在大量的碳纤维。从其理解的是,表面上需要预定量的碳纤维。
(比较例1)
除了不使用碳纤维以外,重复实施例2的步骤。
(比较例2)
除了将碳纤维的配混量改变为233重量份以外,重复实施例2的步骤。熔融捏合后获得的BMC的粘度高,在成形步骤中在模具内部分观察到伸展不足,并且无法测量极限PV值。
实施例1-4和比较例1和2获得的试验片的极限PV值的测量结果、增稠步骤的有无、纤维的分散状态和成型品的缺陷的有无示于表1。
[表1]
※压缩成型前在电炉内在310℃下进行增稠30分钟
(实施例5)
除了将BMC在电炉内在310℃下加热45分钟以外,重复实施例1的步骤。
(实施例6)
除了将BMC在电炉内在310℃下加热60分钟以外,重复实施例1的步骤。
(比较例3)
除了将BMC在电炉内在310℃下加热15分钟以外,重复实施例1的步骤。BMC从模具内漏出,并且由于纤维的不均匀分布而发生翘曲变形。
(比较例4)
除了将BMC在电炉内在310℃下加热75分钟以外,重复实施例1的步骤。树脂粘度过高以致树脂不能伸展并且不能成形。
实施例1、5和6以及比较例3和4中获得的各试验片的成形性、纤维的分散状态、翘曲/直径比和熔体粘度的测量结果示于表2。
[表2]
※在增稠步骤中在电炉内在310℃下保持的时间
(实施例7)
将12.5重量份的平均单纤维长度为200μm的沥青系碳纤维(Mitsubishi Plastics Inc.制的K223HM)和12.5重量份的石墨粉末(Wako Pure Chemical Industries,Ltd.制的070-01325)与100重量份的加成聚合型聚酰亚胺(Ube Industries,Ltd.制的PETI-330)混合,并且将所得混合物通过捏合机在大气压下在280℃下熔融捏合30分钟。其后,获得冷却至室温的混合物(块状模制化合物,以下缩写为“BMC”)。将获得的BMC分裂为适合模具的尺寸,在压缩成型机用模具内在280至320℃下保持一定时间以熔融并均热,然后在对其施加2.4MPa的压力的同时,以3℃/min的升温速度加热至371℃并在该温度下保持1小时,并且逐渐冷却,从而获得直径为40mm和厚度为3mm的板。将获得的板材在357℃下固化6小时并且切成期望的尺寸,从而获得试验片。
(实施例8)
除了将碳纤维的配混量改变为28.6重量份和石墨粉末的配混量改变为14.3重量份以外,重复实施例7的步骤。
(实施例9)
将28.6重量份的平均单纤维长度为200μm的沥青系碳纤维(Mitsubishi Plastics Inc.制的K223HM)和14.3重量份的PTFE粉末(Kitamura Limited制的KT-600M)与100重量份的加成聚合型聚酰亚胺(Ube Industries,Ltd.制的PETI-330)混合,并且将所得混合物通过捏合机在大气压下在280℃下熔融捏合30分钟。其后,获得冷却至室温的BMC。将获得的BMC分裂为适合模具的尺寸,在压缩成型机用模具内在280至320℃下保持一定时间以熔融并均热,然后在对其施加11MPa的压力的同时,以3℃/min的升温速度加热至371℃并在该温度下保持1小时,并且逐渐冷却,从而获得直径为200mm和厚度为3mm的板。将获得的板材在357℃下固化6小时并且切成期望的尺寸,从而获得试验片。
(实施例10)
除了将碳纤维的配混量改变为14.3重量份和PTFE粉末的配混量改变为28.6重量份以外,重复实施例9的步骤。
(实施例11)
除了将碳纤维的配混量改变为33.3重量份和PTFE粉末的配混量改变为33.3重量份以外,重复实施例9的步骤。
(比较例5)
在将加成聚合型聚酰亚胺(Ube Industries,Ltd.制的PETI-330)在280至320℃下保持一定时间以熔融并均热后,在对其施加11MPa的压力的同时,以3℃/min的升温速度将其加热至371℃并且在该温度下保持1小时,并且逐渐冷却,从而获得直径为100mm和厚度为3mm的板。将获得的板材在357℃下固化6小时并且切成期望的尺寸,从而获得试验片。
实施例4、7-11和比较例5获得的各试验片的极限PV值的测量结果和纤维的分散状态示于表3。
除了板厚度为1.5mm以外其它与实施例12相同的试验片中纤维的分散状态示于图3。
[表3]
(实施例12)
除了将碳纤维的配混量改变为14.3重量份和石墨粉末的配混量改变为28.6重量份以外,重复实施例6的步骤。
(比较例6)
除了将碳纤维的配混量改变为16.7重量份和石墨粉末的配混量改变为50.0重量份以外,重复实施例6的步骤。
实施例4、7-9和12以及比较例6获得的各试验片的成形性、纤维的分散状态、以及翘曲/直径比和熔体粘度的测量结果示于表4。对于制品的翘曲,在全部实施例和比较例中通过压缩成型获得板材之后,在357℃下6小时的固化之前的状态下测量其是否良好以判定。
[表4]
产业上的可利用性
由于本发明的树脂成型体具有3,000kPa·m/s以上的极限PV值和优异的滑动性,其可以作为汽车、电气和电子领域等的滑动构件而应用于各种用途。
- 还没有人留言评论。精彩留言会获得点赞!
- 一种聚酰亚胺复合材料及其制备方法与制造工艺
- 树脂前体及含有其的树脂组合物、聚酰亚胺树脂膜、树脂薄膜及其制造方法与制造工艺
- 使用了2‑苯基‑4,4’‑二氨基二苯基醚类的清漆、成形性优异的酰亚胺树脂组合物及具有优异断裂伸长率的固化树脂成形体、以及使用了这些的预浸料、酰亚胺预浸料、及耐热性和机械强度优异的纤维强化材料的制造方法与工艺
- 球状氧化铝粉末以及使用该球状氧化铝粉末的树脂组成物的制造方法与工艺
- 用于高TG聚酰亚胺覆铜板的树脂材料配方的制造方法与工艺
- 一种BPADA型13BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种HQDA型14BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种BPADA型14BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种PMDA型双酚A四胺支化聚酰亚胺树脂薄膜及其制备方法与制造工艺
- 一种ODPA型13BDAPB支化聚酰亚胺树脂薄膜及其制备方法与制造工艺