使用有机光氧化还原催化剂实现无金属光调节的受控自由基聚合的制作方法
相关申请案的交叉参考
本申请案要求于2014年10月6日提交的美国临时申请案第62/060,414号的优先权,所述美国临时申请案的公开内容以全文引用的方式并入本文中。
本公开内容提供使用有机光氧化还原催化剂的丙烯酸和/或苯乙烯系单体的受控自由基聚合的方法,其中聚合通过光介导以及调节。
背景技术:
受控自由基聚合(crp),如氮氧化物介导聚合(nmp)、可逆-加成断裂链转移聚合(raft)和原子转移自由基聚合(atrp)已彻底改变聚合物化学领域,允许具有优异官能团容许性的定义明确的大分子结构的合成。更重要的可能为方法的便捷性和温和反应条件,这允许非专家得到功能材料。最近,对活性自由基聚合的附加控制已通过由外部刺激调节链生长方法实现。该辅助控制为本领域的主要进展,并且进一步创新的可能是显著的。举例来说,电化学atrp已经用于在表面上的图案聚合物刷,以及对水性聚合的增益控制。
可理解地,已经存在多种努力以增加聚合方法的工业适用性,例如通过使用外部刺激调节激活和去激活步骤的策略。在考虑可能外部刺激的广范围中,光提供许多有吸引力的特性,如容易可用的光源,便利使用以及空间和时间控制。在此基础上,大量工作已致力于发展光引发和光调节的自由基聚合(即,光控制的raft、atrp、有机催化、钴-介导以及碲-介导方法)。还已经公开通过基于ir的光氧化还原催化剂催化的甲基丙烯酸酯和丙烯酸酯的光控制的自由基聚合。然而,使用昂贵基于ir的催化剂限制了这种体系的实用性以及其在多种应用(如电子材料)中的使用。在此基础上,在没有甚至百万分之一(ppm)金属的情况下执行这些光控制的自由基聚合的能力应延伸这种方法的范围至多种应用,以及使该方法更加经济可行。
技术实现要素:
本公开内容的方法在聚合物合成中提供精确控制分子量和分子量分布以及增益顺序和架构控制的能力。在广义方面,本公开内容的方法提供使用有机光氧化还原催化剂的多种单体的受控光介导自由基聚合。本公开内容的聚合方法提供通过光的有效激活和去激活,其中在黑暗中未观测到聚合,并且在再暴露于光时发生快速再激活。本公开内容的有机光氧化还原催化剂有利地提供较大单体容许性,然后是基于过渡金属的体系。此外,本公开内容的方法允许嵌段共聚物的合成,以及与其它聚合方法组合以制备定义明确的材料的能力。
在一方面,本公开内容提供用于制备聚合物组合物的方法,其包含:
使一种或多种(甲基)丙烯酸酯、(甲基)丙烯酰胺、(甲基)丙烯腈、苯乙烯、丙烯腈、乙酸乙烯酯、乙烯基咔唑、乙烯基吡啶和氯乙烯单体与引发剂和有机光氧化还原催化剂组合以获得反应混合物;以及
通过用光源照射反应混合物使单体聚合,
其中有机光氧化还原催化剂在激发态下还原。
在另一方面,本公开内容提供用于制备式i的聚合物组合物的方法:
其中
m和n独立地为约3到约1500的整数;
p为1到6的整数;
x为卤素、黄原酸酯、双硫酯、三硫代碳酸酯、二硫代氨基甲酸酯或氮氧化物;
各个r1和r1′独立地为-oh、c1-c20烷氧基、氨基、单(c1-c20烷基)氨基或二(c1-c20烷基)氨基、芳氧基或烷氧基芳基,其中各个烷基或芳基任选被一个或多个基团取代,所述一个或多个基团独立地为c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基或卤代(c1-c6)烷氧基;
r2为-cn、-ch=ch2、-c(o)c1-c6烷氧基、-cooh、-(ch2)1-2c(o)c1-c6烷氧基、-(ch2)1-2cooh、-(ch2)1-3c1-c6烷氧基、-(ch2)1-3oh、c1-c6烷基、芳基或杂芳基;
r3和r4独立地选自氢、-cn、c1-c6烷基和苯基;以及
各个r5和r5'独立地为h或甲基;
该方法包含:
使一种或多种单体与引发剂和有机光氧化还原催化剂组合以获得反应混合物;以及
通过用光源照射反应混合物使单体聚合,
其中有机光氧化还原催化剂在激发态下还原。
在某些方面,本公开内容提供用于制备预定分子量和多分散性聚合物的方法。在其它某些方面,本公开内容提供用于制备具有选择的长度和/或选择的分子量和/或选择的分子量分布和/或选择的架构的聚合物的方法。在其它某些方面,本公开内容提供用于制备具有约1.0与约2.5之间的多分散指数的聚合物的方法。在其它某些方面,本公开内容提供用于制备具有大于2.0的多分散指数的聚合物的方法。这些方法包含如上所述的步骤。
在某些方面,本公开内容提供用于制备预定分子量和多分散性的丙烯酸和/或苯乙烯类聚合物的方法。在其它某些方面,本公开内容提供用于制备具有选择的长度和/或选择的分子量和/或选择的分子量分布和/或选择的架构的丙烯酸和/或苯乙烯类聚合物方法。在其它某些方面,本公开内容提供用于制备具有约1.0与约2.5之间的多分散指数的丙烯酸和/或苯乙烯类聚合物的方法。在其它某些方面,本公开内容提供用于制备具有大于2.0的多分散指数的丙烯酸和/或苯乙烯类聚合物的方法。这些方法包含如上所述的步骤。
附图说明
图1示出当使反应循环暴露于光并且监测(a)转化速率;(b)分子量对比转化率,以及(c)自由基浓度时,使用10-苯基吩噻嗪(pth)的甲基丙烯酸苯甲酯(bnma)的聚合。实验程序在实例3中描述。
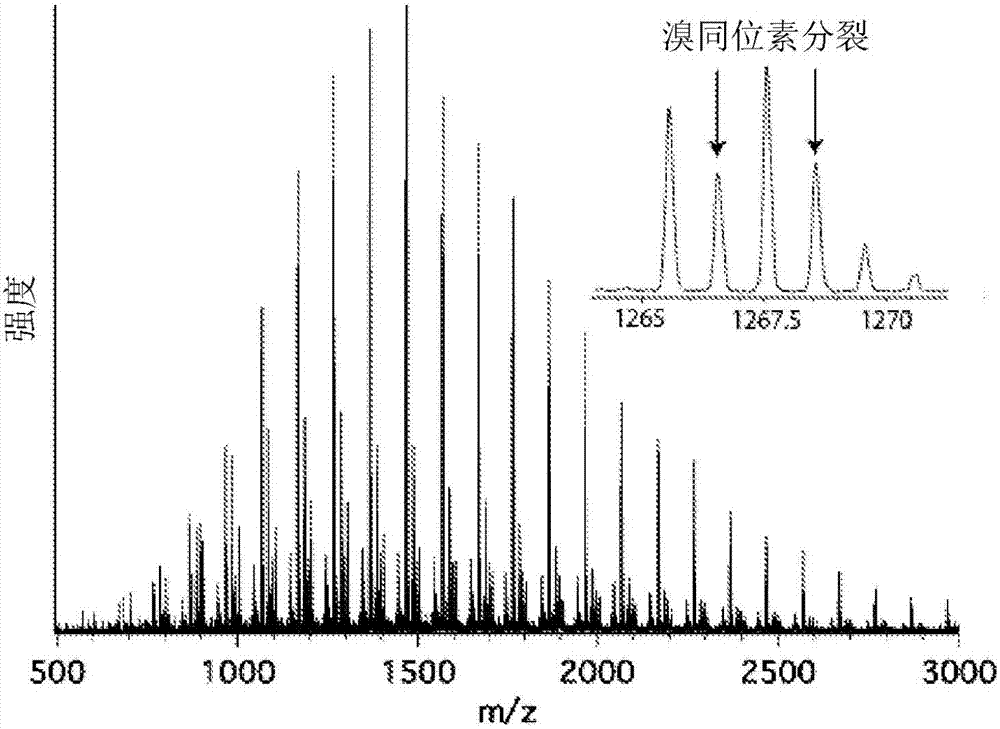
图2示出在本公开内容反应条件下产生的聚(甲基丙烯酸甲酯)(pmma)的esi-ms,其中主要峰放大(插图)以指示溴同位素分裂。
图3示出具有sec迹线(a)pmma-b-pbnma;(b)pmma-b-pma;以及(c)ptbuma-b-pdmaema的嵌段共聚物合成(在各个图的右侧处的迹线表示均聚物的产生,并且在各个图左侧的迹线表示嵌段共聚物的产生)。
图4示出用光掩模图案化20μm×200μmpmma。
图5提供具有对于有机催化的光氧化还原聚合的提出的机制和必需属性的流程。
具体实施方式
在描述所公开的方法和材料之前,应理解,本文所描述的方面不限于具体实施例、方法、设备或配置,并且因此,当然可变化。也应理解,本文所用的术语仅用于描述特定方面的目的,并且除非在本文中具体定义,否则不打算进行限制。
鉴于本公开内容,本文所描述的方法可由本领域的普通技术人员配置以符合期望的需要。一般来说,所公开的方法提供具有聚合的有效激活和去激活的聚合物组合物的聚合的改进,其中通过仅使用有机(即,非过渡金属)催化剂控制分子量和分子量分布。该方法的基本优点还在于,在不存在照射的情况下,链端部以休眠烷基溴化物形式搁置,保护免受有害自由基反应,但是当再暴露于光时可用于再激活。因此,本公开内容的方法提供聚合的改进的有效链封端和再引发。本公开内容的有机光氧化还原催化剂有利地提供较大单体容许性,然后是基于过渡金属的体系。此外,本公开内容的方法允许嵌段共聚物的合成,以及与其它聚合方法组合以制备定义明确的材料的能力。
虽然光氧化还原催化的出现的领域已经主要采用过渡金属(即铱和钌),但是使用有机催化剂来进行适用的变换也是已知的。但是,这些的催化剂中大部分已用作通过激发态的还原猝灭的电子接受体。在本公开内容的方法中,需要可进行氧化猝灭(电子供体)的催化剂以便增益对链-生长方法的控制,如图5所示。因此,本公开内容提供通过使一种或多种单体与引发剂和有机光氧化还原催化剂组合来制备聚合物组成物的方法,所述有机光氧化还原催化剂在激发态下还原。术语“在激发态下还原”意指催化剂在激发态下具有单个电子转移事件,其中电子从催化剂供给到底物。激发态还原电势可由以下方程估算:
e1/2*=eox-e0,0
其中,e0,0在以下方程中由催化剂的发射开始估算:
e0,0=h×c/λ开始
其中,h为普朗克常量,c为光速。在一个实施例中,在激发态下还原的催化剂使自由基阳离子物质在激发态下稳定。
举例来说,在某些实施例中,有机光氧化还原催化剂可选自吩噻嗪衍生物、啡噁嗪衍生物、9,10-二氢吖啶衍生物、咔唑衍生物、芳基胺衍生物、二芳基胺衍生物、三芳基胺衍生物和大的π-扩展的所有碳衍生物,如(但不限于)红萤烯、芘石墨烯和碳纳米管。
在某些其它实施例(例如,实施例1)中,有机光氧化还原催化剂具有式a-z,其中
各个a独立地为:
其中
各个y独立地为键,o、s、nr14或c(r14)2;
o和q独立地为零或1到4的整数;
r11和r12独立地选自由以下组成的群组:卤素、氰基、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、单芳基氨基或二芳基氨基、c1-c6烷基、卤代(c1-c6)烷基、c1-c6烷氧基、卤代(c1-c6)烷氧基、c3-c7环烷基、芳基、芳氧基、烷氧基芳基和杂芳基,其中各个烷基、环烷基、芳基和杂芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基;
各个r14独立地为氢、c1-c6烷基、芳基或芳基(c1-c6烷基),其中各个烷基和芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基;以及
各个z为r13、-b-(a)2、-b-(r13)2、-b(a)(r13)、-nh-(a)、-n-(a)2、-nh-(r13)、-n-(r13)2、-n(a)(r13)、-si-(r13)3、-sih(r13)2、-sih2(r13)、-sih(a)2、-sih2(a)、-si(a)3、-si(a)2(r13)、-si(a)(r13)2、-sih(a)(r13)、-ph2、-p-(a)2、-p-(r13)2、-p(r13)(a)或-p(o)-(or13)2,其中
其中r13为h、c1-c20烷基、-c(o)c1-c20烷基、c3-c7环烷基、芳基、芳基(c1-c6烷基)、杂芳基或杂芳基(c1-c6烷基),其中各个烷基、环烷基、芳基和杂芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基、卤代(c1-c6)烷氧基、c3-c7环烷基、芳基、芳氧基、烷氧基芳基、杂芳基、杂芳氧基或基团a。
实施例2涵盖实施例1的光氧化还原催化剂,其中各个z为r13、-b-(a)2、-b-(r13)2、-b(a)(r13)、-nh-(a)、-n-(a)2、-nh-(r13)、-n-(r13)2、-n(a)(r13)、-si-(r13)3、-sih(r13)2、-sih2(r13)、-sih(a)2、-sih2(a)、-si(a)3、-si(a)2(r13)、-si(a)(r13)2或-sih(a)(r13)。具体来说,在实施例2中,各个z为r13、-b-(r13)2、-nh-(r13)、-n-(r13)2、-si-(r13)3、-sih(r13)2或-sih2(r13)。具体来说,在实施例2中,各个z为r13、-b-(a)2、-b(a)(r13)、-nh-(a)、-n-(a)2、-n(a)(r13)、-sih(a)2、-sih2(a)、-si(a)3、-si(a)2(r13)、-si(a)(r13)2或-sih(a)(r13)。具体来说,在实施例2中,各个z为r13、-b-(a)2、-b-(r13)2、-nh-(a)、-n-(a)2、-nh-(r13)、-n-(r13)2、-si-(r13)3、-sih(r13)2、-si(a)(r13)2或-sih(a)(r13)。实施例3涵盖实施例1的光氧化还原催化剂,其中各个z为r13、-b-(a)2、-b-(r13)2、-b(a)(r13)、-nh-(a)、-n-(a)2、-nh-(r13)、-n-(r13)2或-n(a)(r13)。具体来说,在实施例3中,各个z为r13、-b-(a)2、-b-(r13)2、-nh-(a)、-n-(a)2、-nh-(r13)或-n-(r13)2。实施例4涵盖实施例1的光氧化还原催化剂,其中各个z为-si-(r13)3、-sih(r13)2、-sih2(r13)、-sih(a)2、-sih2(a)、-si(a)3、-si(a)2(r13)、-si(a)(r13)2、-sih(a)(r13)、-ph2、-p-(a)2、-p-(r13)2、-p(r13)(a)或-p(o)-(or13)2。具体来说,在实施例4中,各个z为-si-(r13)3、-sih(r13)2、-sih2(r13)、-ph2、-p-(r13)2、或-p(o)-(or13)2。具体来说,在实施例4中,各个z为-sih(a)2、-sih2(a)、-si(a)3、-si(a)2(r13)、-si(a)(r13)2、-sih(a)(r13)、-p-(a)2、-p(r13)(a)或p(o)-(or13)2。在实施例5中,本公开内容提供实施例1的光氧化还原催化剂,其中各个z为-si-(r13)3、-sih(r13)2、-sih2(r13)、-sih(a)2、-sih2(a)、-si(a)3、-si(a)2(r13)、-si(a)(r13)2或-sih(a)(r13)。在实施例6中,本公开内容提供实施例1的光氧化还原催化剂,其中各个z为-ph2、-p-(a)2、-p-(r13)2、-p(r13)(a)或-p(o)-(or13)2。实施例7涵盖实施例1的光氧化还原催化剂,其中各个z为r13、-b-(a)2或-b-(r13)2。具体来说,在实施例7中,各个z为r13或-b-(a)2。
在本公开内容的实施例8中,可用于本公开内容的方法的光氧化还原催化剂为其中z为r13,并且此类化合物由下式表示:
实施例8的特定化合物包括实施例9的那些化合物,即,其中y为o、s、nr14或c(r14)2的光氧化还原催化剂。在实施例10中,本公开内容提供实施例8或9的光氧化还原催化剂,其中y为o、s或nr14。在实施例11中,本公开内容提供实施例8或9的光氧化还原催化剂,其中y为o或s。在实施例12中,本公开内容提供实施例8或9的光氧化还原催化剂,其中y为s。在实施例13中,本公开内容提供实施例8或9的光氧化还原催化剂,其中y为c(r14)2。实施例14涵盖实施例8的光氧化还原催化剂,其中y不存在。
实施例15涵盖实施例8到14中的任一者的光氧化还原催化剂,其中o和q独立地为0。
在实施例16中,本公开内容提供实施例8到14中的任一者的光氧化还原催化剂,其中o和q中的至少一个为1。实施例17涵盖实施例16的光氧化还原催化剂,其中r11和r12独立地选自由以下组成的群组:氢、卤素、氰基、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、c1-c6烷基和c1-c6烷氧基。具体来说,在实施例17中,r11和r12独立地选自由以下组成的群组:氢、卤素、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、c1-c6烷基和c1-c6烷氧基。
基于实施例8到17的特定实施例包括实施例18的那些,即,其中r13为c1-c20烷基、-c(o)c1-c20烷基、c3-c7环烷基、芳基、芳基(c1-c6烷基)、杂芳基或杂芳基(c1-c6烷基)的光氧化还原催化剂,其中各个烷基、环烷基、芳基和杂芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。具体来说,在实施例18中,r13为c1-c20烷基、c3-c7环烷基、芳基、芳基(c1-c6烷基)、杂芳基或杂芳基(c1-c6烷基),其中各个烷基、环烷基、芳基和杂芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。
基于实施例8到17的特定实施例包括实施例19的那些,即,其中r13为c1-c20烷基、芳基、芳基(c1-c6烷基)、杂芳基或杂芳基(c1-c6烷基)的光氧化还原催化剂,其中各个烷基、芳基和杂芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。
基于实施例8到17的特定实施例包括实施例20的那些,即,其中r13为c1-c20烷基、芳基或芳基(c1-c6烷基)的光氧化还原催化剂,其中各个烷基、芳基和杂芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。
基于实施例8到17的其它特定实施例包括实施例21的那些,即,其中r13为c1-c20烷基或芳基的光氧化还原催化剂,各个c1-c20烷基或芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。
基于实施例8到17的一些其它特定实施例包括实施例22的那些,即,其中r13为c1-c6烷基或芳基的光氧化还原催化剂,各个c1-c6烷基或芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。
在实施例23中,本公开内容提供实施例8到17中的任一者的光氧化还原催化剂,其中r13为c1-c4烷基或苯基,各个c1-c4烷基或苯基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。实施例24提供实施例23的光氧化还原催化剂,其中r13为甲基、苯基或2-(三氟甲基)苯基、3-(三氟甲基)苯基或4-(三氟甲基)苯基。
本公开内容的另一个实施例,即实施例25,涵盖实施例8的光氧化还原催化剂,其中
y为o或s;
o和q独立地为0、1或2的整数;
r11和r12独立地选自由以下组成的群组:氢、卤素、氰基、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、c1-c6烷基、c1-c6烷氧基、c3-c7环烷基、芳基或杂芳基,其中各个烷基、环烷基、芳基和杂芳基任选被1到4个基团取代,该1到4个基团独立地为c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基或卤代(c1-c6)烷氧基;以及
r13为c1-c6烷基或芳基,各个c1-c6烷基或芳基任选被1到4个基团取代,该1到4个基团独立地为c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基或卤代(c1-c6)烷氧基。
在实施例26中,实施例25的光氧化还原催化剂,其中y为s。
本公开内容的另一个实施例,即实施例27,提供为如下各者的光氧化还原催化剂:
在本公开内容的实施例28中,可用于本公开内容的方法的光氧化还原催化剂为其中z为-b-(a)2,并且此类化合物由下式表示:
实施例28的特定化合物包括实施例29的那些,即,其中y为o、s、nr14或c(r14)2的光氧化还原催化剂。在实施例30中,本公开内容提供实施例28或29的光氧化还原催化剂,其中y为o、s或nr14。在实施例31中,本公开内容提供实施例28或29的光氧化还原催化剂,其中y为o或s。在实施例32中,本公开内容提供实施例28或29的光氧化还原催化剂,其中y为s。在实施例33中,本公开内容提供实施例28或29的光氧化还原催化剂,其中y为c(r14)2。实施例34涵盖实施例28的光氧化还原催化剂,其中y不存在。
实施例35涵盖实施例28到34中的任一者的光氧化还原催化剂,其中o和q独立地为0。
在实施例36中,本公开内容提供实施例28到34中的任一者的光氧化还原催化剂,其中o和q中的至少一个为1。实施例37涵盖实施例26的光氧化还原催化剂,其中r11和r12独立地选自由以下组成的群组:氢、卤素、氰基、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、c1-c6烷基和c1-c6烷氧基。具体来说,在实施例37中,r11和r12独立地选自由以下组成的群组:氢、卤素、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、c1-c6烷基和c1-c6烷氧基。
本公开内容的另一个实施例,即实施例38,涵盖实施例28的光氧化还原催化剂,其中y为o或s;o和q独立地为0、1或2的整数;并且r11和r12独立地选自由以下组成的群组:氢、卤素、氰基、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、c1-c6烷基、c1-c6烷氧基、c3-c7环烷基、芳基或杂芳基,其中各个烷基、环烷基、芳基和杂芳基任选被1到4个基团取代,该1到4个基团独立地为c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基或卤代(c1-c6)烷氧基。在实施例39中,实施例38的光氧化还原催化剂,其中y为s。
本公开内容的另一个实施例,即,实施例40,提供为如下的光氧化还原催化剂:
实施例41的特定化合物包括实施例1到7中的任一者的那些,即,其中y为o、s、nr14或c(r14)2的光氧化还原催化剂。在实施例42中,本公开内容提供实施例1到7中的任一者的光氧化还原催化剂,其中y为o、s或nr14。在实施例43中,本公开内容提供实施例1到7中的任一者的光氧化还原催化剂,其中y为o或s。在实施例44中,本公开内容提供实施例1到7中的任一者的光氧化还原催化剂,其中y为s。在实施例45中,本公开内容提供实施例1到7中的任一者的光氧化还原催化剂,其中y为c(r14)2。实施例46涵盖实施例1到7中的任一者的光氧化还原催化剂,其中y不存在。
实施例46涵盖实施例1到7中的任一者的光氧化还原催化剂,其中o和q独立地为0。
在实施例47中,本公开内容提供实施例1到7中的任一者的光氧化还原催化剂,其中o和q中的至少一个为1。实施例48涵盖实施例47的光氧化还原催化剂,其中r11和r12独立地选自由以下组成的群组:氢、卤素、氰基、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、c1-c6烷基和c1-c6烷氧基。具体来说,在实施例48中,r11和r12独立地选自由以下组成的群组:氢、卤素、羟基、氨基、单(c1-c20)烷氨基或二(c1-c20)烷氨基、c1-c6烷基和c1-c6烷氧基。
基于实施例1到7和实施例41到48的特定实施例包括实施例49的那些,即,其中r13(如果存在)为c1-c20烷基、芳基、芳基(c1-c6烷基)、杂芳基或杂芳基(c1-c6烷基)的光氧化还原催化剂,其中各个烷基、芳基和杂芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。基于实施例1到7和实施例41到48的其它特定实施例包括实施例50的那些,即,其中r13为c1-c20烷基或芳基的光氧化还原催化剂,各个c1-c20烷基或芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。基于实施例1到7和实施例41到48的一些其它特定实施例包括实施例51的那些,即,其中r13为c1-c6烷基或芳基的光氧化还原催化剂,各个c1-c6烷基或芳基任选被独立地选自以下的一个或多个基团取代:c1-c6烷基、c1-c6烷氧基、卤素、羟基、氨基、单-(c1-c6)烷氨基或二-(c1-c6)烷氨基、卤代(c1-c6)烷基和卤代(c1-c6)烷氧基。实施例52提供实施例51的光氧化还原催化剂,其中r13为甲基、苯基或2-(三氟甲基)苯基、3-(三氟甲基)苯基或4-(三氟甲基)苯基。
如本领域技术人员所理解的,上述任何光氧化还原催化剂(即,式ii或式iii的光氧化还原催化剂或根据实施例1到52中的任一者光氧化还原催化剂)可用于本公开内容的方法中。
本文所描述的方法可在相对低光氧化还原催化剂水平下操作。因此,本文所描述的方法可以显著较低成本执行同时维持良好转化速率和对聚合的分子量控制。举例来说,在某些实施例中,光氧化还原催化剂以相对于使用的单体的量约0.00001mol%到约20mol%存在。在一个实施例中,本公开内容的方法在相对于使用的单体的量约0.005mol%到约20mol%的光氧化还原催化剂下操作。在某些此类实施例中,光氧化还原催化剂以相对于使用的单体的量约0.005mol%到约15mol%,或0.005mol%到约10mol%,或0.005mol%到约7.5mol%,或0.005mol%到约5mol%,或0.01mol%到约20mol%,或0.01mol%到约15mol%,或0.01mol%到约10mol%,或0.01mol%到约7.5mol%,或0.01mol%到约5mol%,或0.01mol%到约4mol%,或0.01mol%到约3mol%,或0.01mol%到约2mol%,或0.01mol%到约1mol%,或0.01mol%到约0.9mol%,或0.01mol%到约0.8mol%,或0.01mol%到约0.7mol%,或0.01mol%到约0.6mol%,或0.01mol%到约0.5mol%,或0.01mol%到约0.4mol%,或0.01mol%到约0.3mol%,或0.01mol%到约0.2mol%,或0.01mol%到约0.1mol%,或0.005mol%到约0.1mol%,或0.01mol%到约0.07mol%,或0.03mol%到约0.07mol%,或0.04mol%到约0.06mol%的光氧化还原催化剂存在。在一些实施例中,光氧化还原催化剂以约0.01mol%到约0.1mol%存在。在一些实施例中,光氧化还原催化剂以约0.05mol%到约0.5mol%存在。在其它实施例中,光氧化还原催化剂以约0.05mol%存在。在一些其它实施例中,光氧化还原催化剂约0.1mol%存在。在另外其它实施例中,光氧化还原催化剂以约0.01mol%存在。当然,本领域的普通技术人员将理解的是,在某些实施例和应用中,所使用的光氧化还原催化剂的量可不同于本文所具体描述的那些。
本公开内容的方法可在溶剂体系中进行以获得反应混合物;并且通过用光源照射反应混合物使单体聚合。如本领域的普通技术人员将理解,本公开内容的方法可根据期望的需要在任何合适的溶剂中进行。本发明的实施例可适用于使用本领域中已知的多种溶剂,例如二甲基甲酰胺(dmf)、甲苯、1,4-二噁烷、二甲苯、苯甲醚、二甲亚砜(dmso)、n,n-二甲基乙酰胺(dma)、四氢呋喃(thf)、水、甲醇、乙腈、氯仿等。
另外,在本发明的一些实施例中,在聚合方法中的一种或多种反应物(例如单体)起溶剂作用。
在一些实施例中,该方法可在包含n,n-二甲基乙酰胺(dma)的溶剂体系中进行,从而获得优异的聚合控制。本领域的技术人员将认识到dma可纯净使用或可与一种或多种附加溶剂混合。这些附加溶剂包括(但不限于)二甲基甲酰胺(dmf)、甲苯、1,4-二噁烷、二甲苯、苯甲醚、二甲亚砜(dmso)、四氢呋喃(thf)、水、甲醇、乙腈、氯仿等。当dma与一种或多种附加溶剂混合时,包含dma的溶剂体系包含以溶剂的总体积计大于约10vol%的dma,或大于约25vol%的dma,或大于约50vol%的dma,或大于约60vol%dma,或大于约70vol%的dma,或大于约80vol%的dma,或大于约90vol%的dma,或大于约95vol%的dma,或大于约98vol%的dma,或大于约99vol%的dma。在本公开内容的方法的一个实施例中,溶剂体系基本上由dma组成。在一个实施例中,用于本公开内容的方法的dma不与任何其它溶剂混合。因此,在本公开内容的方法的一些实施例中,溶剂体系由dma组成。
如本领域的普通技术人员将理解,本公开内容的方法可在反应混合物中的单体的任何合适浓度中进行。举例来说,反应混合物可以在反应混合物中至少约1.0m的单体的浓度。本领域的技术人员将理解,上限将取决于所使用的具体单体,但将不大于在反应混合物中约10m,或9.0m,或8.0m,或7.0m,或6.0m,或5.0m浓度的单体。在一个实施例中,反应混合物以在反应混合物中至少约1.5m单体的浓度。在另一个实施例中,反应混合物以在反应混合物中至少约2.0m单体的浓度。在另一个实施例中,反应混合物以在反应混合物中至少约2.1m,或至少约2.2m,或至少约2.3m,或至少约2.4m,或至少约2.5m,或至少约2.6m,或至少约2.7m,或至少约2.8m或至少约2.9m单体的浓度。在另一个实施例中,反应混合物以在反应混合物中至少约3.0m单体的浓度。特别适用于丙烯酸酯单体的浓度为至少约2.7m,或至少约3.0m,或至少约3.2m或甚至是至少约3.5m的那些浓度。特别适用于甲基丙烯酸酯单体的浓度为至少约1.0m,或至少约1.3m,或至少约1.5m,或至少约1.7m,或至少约2.0m或甚至至少约2.4m的那些浓度。特别适用于(甲基)丙烯酰胺或(甲基)丙烯腈单体的浓度为至少约1.0m,或至少约1.5m,或至少约2.0m,或至少约2.5m或甚至至少约3.0m的那些浓度。
本公开内容的引发剂化合物在自由基聚合过程中与单体反应以形成能够与附加单体连续键联以形成(一个或多个)聚合物链的中间化合物。不受特定理论限制,据相信,生长聚合物链的数目可通过引发剂确定。举例来说,引发越快,终止和转移越少,产生窄分子量分布的增长链的数目越一致。一般来说,在本公开内容的方法中,有机卤化物可用作引发剂。举例来说,可使用烷基卤化物和拟卤化物。合适的烷基卤化物包括烷基溴化物和烷基氯化物。除烷基卤化物或拟卤化物外可用于本公开内容的实施例的引发剂包括(但不限于)烯丙基卤化物、黄原酸酯、硫酯、硫羰酯(thionoesters)、二硫基酯、三硫代碳酸酯和氮氧化物。在本公开内容的一些实施例中,可选择引发剂的形状或结构以影响聚合物的架构。举例来说,在单个核上具有多个烷基卤化物基团的引发剂可产生星状聚合物形状。另外,可基于期望的聚合物的具体端官能团选择合适的引发剂。举例来说,适用于本公开内容的方法的引发剂可含有反应性官能团,如(但不限于)羧基、邻苯二酰亚胺基、氰基、n-羟基丁二酰亚胺酯、五氟苯基酯等。适用于本公开内容的方法的引发剂可任选地结合到基板,如硅基板。适合用于本公开内容的方法的引发剂的实例包括(但不限于):
在一个实施例中,适合用于本公开内容的方法的引发剂为2-溴丙酸乙酯、2-溴丙酸苯甲酯、α-溴异丁酸乙酯或α-溴异丁酸苯甲酯。在另一个实施例中,引发剂为2-溴丙酸乙酯或2-溴丙酸苯甲酯。在又另一个实施例中,引发剂为2-溴丙酸乙酯。在又另一个实施例中,引发剂为2-溴丙酸苯甲酯。
如本领域中已知,单体涵盖一类化合物,主要是可与相同或其它化合物的其它分子反应以形成非常大的分子或聚合物的有机化合物。通常用于聚合反应中的单体包括具有可使增长自由基稳定的取代基的分子。本文所描述的方法可使用例如本领域技术人员可获得的(甲基)丙烯酸酯、(甲基)丙烯酰胺、(甲基)丙烯腈、苯乙烯、乙酸乙烯酯、乙烯基吡啶和氯乙烯单体进行,并且可根据所期望的产物变化。在所公开的方法中进行自由基聚合的单体包括(但不限于)进行传统的自由基聚合的典型烯烃单体。实例包括(但不限于)甲基丙烯酸烷基酯(例如,甲基丙烯酸甲酯)、丙烯酸酯(包括各种丙烯酸烷酯)、苯乙烯、甲基丙烯酸、丙烯酸、甲基丙烯酸苯甲酯、丙烯酸苯甲酯、丙烯酰胺、烷基丙烯酰胺、二烷基丙烯酰胺(例如,n,n-二甲基丙烯酰胺)、甲基丙烯酰胺、烷基甲基丙烯酰胺、二烷基甲基丙烯酰胺、丙烯腈、甲基丙烯腈、乙酸乙烯酯、乙烯基吡啶、氯乙烯、丙烯酰胺等。单体可进一步用一种或多种反应性官能团官能化,反应性官能团包括(但不限于)羧酸、胺、酰胺、醇、酮、醛、炔烃、氟化物、氯化物、溴化物、碘化物、醚、酯、羟胺、亚胺、叠氮化物、腈、异氰酸酯、异氰化物、亚硝酸盐、亚硝基化合物、硫醇、硫醚、亚砜、磺酸、硫氰酸酯、异硫氰酸酯、硫酮、硫醛、膦、磷酸、磷酸酯、硼酸、硼酸酯、烃基硼酸、杂芳族化合物和杂环。
在本公开内容的一个实施例中,单体为甲基丙烯酸酯单体。在另一个实施例中,甲基丙烯酸酯可选自甲基丙烯酸、甲基丙烯酸甲酯、甲基丙烯酸乙酯和甲基丙烯酸丁酯。在另一个实施例中,甲基丙烯酸酯包含混合物,例如单体中的一者为甲基丙烯酸酯且单体中的一者为丙烯酸酯。在本公开内容的一个实施例中,单体为丙烯酸酯单体。在另一个实施例中,丙烯酸酯可选自丙烯酸、丙烯酸甲酯、丙烯酸乙酯和丙烯酸丁酯。
本公开内容的方法允许聚合物生长的精确控制以便形成具有选择的长度和/或选择的分子量(例如,在选择的mw范围内)和/或选择的分子量分布和/或选择的架构的聚合物。在本公开内容的某些实施例中,聚合物链具有选自由以下组成的群组的结构:嵌段共聚物;无规共聚物;梯度共聚物;周期聚合物;交替聚合物;统计聚合物;线性聚合物;支化聚合物;星形聚合物;刷状聚合物;梳状聚合物;接枝聚合物和环状聚合物。在一个实施例中,聚合物由至少两种或更多种单体形成。在另一个实施例中,通过本公开内容的方法制备的丙烯酸聚合物为由至少两种不同单体组成的嵌段、无规、梯度、交替、刷状或星状共聚物。在另一个实施例中,通过本公开内容的方法制备的丙烯酸聚合物为由至少两种单体组成的嵌段、无规或梯度共聚物。在一些其它实施例中,通过本公开内容的方法制备的丙烯酸聚合物由相同单体组成。
因为在所公开的方法的实施例中,聚合物形成的速率与光照的量成正比,任选地,例如在反应混合物中聚合物链长度通过控制反应混合物暴露于光的时间的量;和/或通过控制达到反应混合物的光的强度来调节。本发明的实施例可包含多次将反应混合物暴露于光以便精确调整聚合物组合物的一种或多种特征。举例来说,在本公开内容的一些方法中,反应混合物首先暴露于光一段时间使得自由基聚合过程被激活;接着是一段时间,在此期间该反应混合物免受光照,使得自由基聚合过程去激活;以及然后将反应混合物再暴露于光一段时间,使得自由基聚合过程再激活等。本领域的技术人员将理解,光源可为本领域中已知的广泛多种光源中的一种。
本文公开的方法可用于形成具有包括例如相对低多分散性的多种期望质量的聚合物材料。任选地,在本发明的实施例中,聚合物链呈现多分散指数,使得mw/mn在约1.0与约2.5之间。在一些实施例中,多分散指数在约1.0与约2.0之间,或在约1.0与约1.9之间,或在约1.1与约1.9之间,或在约1.0与约1.8之间,或在约1.1与约1.8之间,或在约1.0与约1.7之间,或在约1.1与约1.7之间,或在约1.2与约1.7之间,或在约1.2与约1.6之间,或约1.0,或约1.1,或约1.2,或约1.3,或约1.4,或约1.5,或约1.6,或约1.7,或约1.8,或约1.9,或甚至约2.0。在某些实施例中,聚合物呈现在约1.0和约1.5之间的mw/mn的多分散性。在一些其它实施例中,聚合物呈现在约1.2与约1.4之间的mw/mn的多分散性。在本公开内容的某些实施例中,聚合物链的多分散指数可大于2.0,并且可例如大于约5,或约10,或约25,或约50,或约100,或约150,或约200。
除了移去光源之外,本领域的技术人员将认识到聚合可通过本领域中已知的任何方法中断。举例来说,聚合可通过添加本领域中已知的淬灭剂中断。在此实施例中,光源可或可不再存在。最终,本领域的技术人员将认识到在耗尽可用于反应的单体时,聚合可中断。
如上所述,本公开内容的方法可用于制备式i的聚合物。在特定实施例中,式i的化合物为其中x为-br的那些化合物。在其它实施例中,各个r1和r1′独立地为oh或c1-c6烷氧基。在一个实施例中,各个r1和r1′为c1-c6烷氧基。在示例性实施例中,各个r1和r1′选自甲氧基、乙氧基和丁氧基。在另一个实施例中,各个r1和r1′中的一个为c1-c6烷氧基,并且另一个为-oh。在某些实施例中,各个r5和r5′独立地为h。在一些其它实施例中,各个r5和r5′独立地为甲基。在一些其它实施例中,r5和r5′中的一个为h,并且另一个为甲基。本领域中的技术人员将认识到r2、r3和r4将取决于使用的特定引发剂。
本文所描述的方法可使用如本领域技术人员已知的标准脱气程序在惰性氛围下进行。这些方法还可在含氧氛围下进行,例如在在没有用于去除氧的特别预防措施的情况下包含常压水平的氧的密封反应器中。对于包含常压水平的氧的系统,使用0.5mol%光氧化还原催化剂是有利的。
定义
在整个本说明书中,除非上下文另外要求,否则词“包含(comprise)”和“包括(include)”以及变化形式(例如“包含了(comprises)”、“包含着(comprising)”、“包括了(includes)”、“包括着(including)”)将理解为暗示包括陈述的组件、特征、要素或步骤或者组件、特征、要素或步骤的群组,但不排除任何其它整数或步骤或者整数或步骤的群组。
除非上下文另外明确规定,否则如说明书和所附权利要求书中所用的单数形式“一个(种)(a/an)”和“所述(the)”包括多个指示物。
范围在本文中可表述为从“约”一个特定值和/或到“约”另一个特定值。当表述此类范围时,另一方面包括从所述一个特定值和/或到所述另一个特定值。类似地,当值通过使用前置词“约”被表述为近似值时,应理解,特定值形成另一方面。进一步应理解,无论与另一个端点相关还是另一个端点不相关,每一个范围的端点都是有意义的。
如本文所用,术语“组合”包括将一种或多种物料添加到反应混合物。
如本文所用,术语“分散性”、“多分散性”、“多分散指数”、“pdi”和“mw/mn”可互换使用,并且是指关于分子质量的分布的聚合物均一性的量度。分散性可通过将重均分子量(mw)除以数均分子量(mn)计算(即,mw/mn)。在某些实施例中,分散性可根据聚合度计算,其中分散性等于xw/xn,其中xw为重均聚合度,并且xn为数均聚合度。
除非另外说明,否则本文中的所有百分比、比率和比例都是按重量计。除非特意相反的说明,否则组分的重量百分比(重量%,还为wt%)以其中包括组分的组合物的总重量计(例如,以反应混合物的总量计)。
如本文所用,术语“烷氧基”意指通过氧原子连接到母体分子部分的如本文所定义的烷基。烷氧基的代表性实例包括(但不限于)甲氧基、乙氧基、丙氧基、2-丙氧基、丁氧基、叔丁氧基、戊氧基和己氧基。
除非另外规定,否则如本文所用,术语“烷基”意指含有1到20个碳原子的直链或支链烃。烷基的代表性实例包括(但不限于)甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、正己基、3-甲基己基、2,2-二甲基戊基、2,3-二甲基戊基、正庚基、正辛基、正壬基和正癸基。术语“亚烷基”是指二价烷基,其中烷基如本文所定义。
如本文所用,术语“芳基”意指苯基(即,单环芳基),或含有至少一个苯环的双环系统或在芳香族双环系统中仅含有碳原子的芳香族双环,或含有至少一个苯环的多环系统。双环芳基可是奥基、萘基或稠合到环烷基、环烯基或杂环基的苯基。双环或多环芳基通过双环或多环系统的苯基部分内含有的任何碳原子或萘基、奥基、蒽或芘环情况下的任何碳原子连接到母体分子部分。
如本文所用,术语“芳氧基”意指通过氧原子连接到母体分子部分的如本文所定义的芳基。芳氧基的代表性实例包括(但不限于)苯氧基和萘氧基。
术语“环烷基”是指单环或双环环烷基环系统。单环系统是含有3到8个碳原子的环烃基,其中此类基团可是饱和或不饱和的,但不是芳香族的。环烷基的实例包括环丙基、环丁基、环戊基、环己基、环庚基和环辛基。更优选的为c3-c6环烷基。在某些实施例中,环烷基是完全饱和的。单环环烷基的实例包括环丙基、环丁基、环戊基、环戊烯基、环己基、环己烯基、环庚基和环辛基。双环环烷基环系统桥接单环或稠合双环。桥接的单环含有单环环烷基环,其中单环的两个非相邻碳原子通过一个与三个附加碳原子之间的亚烷基桥(即,形式-(ch2)-的桥连基,其中w为1、2或3)连接。双环系统的代表性实例包括(但不限于)双环[3.1.1]庚烷、双环[2.2.1]庚烷、双环[2.2.2]辛烷、双环[3.2.2]壬烷、双环[3.3.1]壬烷和双环[4.2.1]壬烷。稠合双环环烷基环系统含有稠合到苯基、单环环烷基、单环环烯基、单环杂环基或单环杂芳基的单环环烷基环。桥接或稠合双环环烷基通过单环环烷基环含有的任何碳原子连接到母体分子部分。
如本文所用,术语“卤素”意指-cl、-br、-i或-f。
术语“卤代烷氧基”是指被一个或多个卤素原子取代的烷氧基,其中各个卤素独立地为f、cl、br或i。“卤代烷氧基”包括全卤烷氧基,如ocf3或ocf2cf3。
术语“卤代烷基”是指被一个或多个卤素原子取代的烷基,其中各个卤素独立地为f、cl、br或i。
如本文所用,术语“杂芳基”意指单环杂芳基或含有至少一个杂芳环的双环或多环系统。单环杂芳基可是5或6元环。5元环由两个双键和一个、两个、三个或四个氮原子和任选地一个氧或硫原子组成。6元环由三个双键和一个、两个、三个或四个氮原子组成。5或6元杂芳基通过杂芳基内含有的任何碳原子或任何氮原子连接到母体分子部分。双环或多环杂芳基由稠合到苯基、环烷基、环烯基、杂环基或杂芳基的杂芳基组成。杂芳基的代表性实例包括(但不限于)呋喃基、咪唑基、异噁唑基、异噻唑基、噁二唑基、噁唑基、吡啶基、哒嗪基、嘧啶基、吡嗪基、吡唑基、吡咯基、四唑基、噻二唑基、噻唑基、噻吩基、三唑基、三嗪基、苯并咪唑基、苯并呋喃基、苯并噻吩基、苯并噁二唑基、苯并噁噻二唑基、苯并噻唑基、噌啉基、5,6-二氢喹啉-2-基、5,6-二氢异喹啉-1-基、呋喃并吡啶基、吲唑基、吲哚基、异喹啉基、萘啶基、喹啉基或嘌呤基。
实例
本公开内容的方法通过以下实例进一步示出,所述实例不应理解为在范围或精神方面将本公开内容限于其中描述的具体程序和化合物。在所有情况下,除非另外规定,否则柱色谱法使用硅胶固相进行。
材料和设备
所有聚合在氩气氛围下进行。无水n,n-二甲基乙酰胺购自西格玛-奥德里奇公司(sigma-aldrich)并且按原样使用。甲基丙烯酸甲酯购自西格玛-奥德里奇公司并且在使用之前穿过碱性氧化铝的栓塞。亚甲基蓝、伊红y、α-溴苯基乙酸乙酯、10-甲基吩噻嗪、吩噻嗪、ruphos和氯苯购自西格玛-奥德里奇公司并且按原样使用。ruphos预催化剂购自施特雷姆化学品公司(stremchemicalsinc)。
在瓦里安(varian)400mhz、瓦里安500mhz或瓦里安600mhz仪器上记录核磁共振光谱。除非另外陈述,否则所有1hnmr实验都以δ单位,百万分率(ppm)报告,并且相对于氘化溶剂中的残余氯仿的信号(7.26ppm)测量。除非另外陈述,否则所有13cnmr光谱都相对于氘代氯仿(77.23ppm)以ppm为单位报告,并且都在1h解耦下获得。在具有沃特斯(waters)2414折射率检测器的沃特斯2695分离模块上在具有0.25%三乙胺的氯仿中进行凝胶渗透色谱法(gpc)。数均分子量(mn)和重均分子量(mw)相对于用于计算mw/mn的线性聚苯乙烯标准物计算。除非另外指出,否则分子量(mn)使用1hnmr通过比较在引发剂中的乙基峰的积分与在聚合物侧链中的甲基峰来计算。
led条带(380nm)购自elementalled(参见www.elementalled.com)并且如以下展示(图s1)使用。反应物在剧烈搅拌下紧靠380nm灯放置,同时用压缩空气冷却。测量光强度为0.65μw/cm2。
实例1:10-苯基吩噻嗪(pth)的合成:
以下程序采纳自maiti等人(化学科学(chem.sci.)2010,2,57)。向装备有磁性搅拌棒的瓶中添加naotbu(134mg,1.4mmol)、吩噻嗪(199mg,1mmol)、ruphos预催化剂(14mg,02mmol,2mol%)和ruphos(8mg,0.02mmol,2mol%)。在添加干燥二噁烷(1ml)之前用氩气抽空并且回填瓶3次。最后,添加无水氯苯(143μl,1.4mmol)。然后将瓶放入110℃油浴中,并且使反应进行5h。然后,将瓶冷却到室温,用ch2cl2稀释,用水、盐水洗涤,用mg2so4干燥,并且穿过二氧化硅栓塞(5%etoac/己烷)。然后在减压下干燥产物以产生267mg白色固体(97%产率)。1hnmr(600mhz,cdcl3)δ:7.60(t,j=8hz,2h),7.49(t,j=8hz,1h),7.40(d,j=7hz,2h),7.02(d,j=8hz,2h),6.86-6.79(m,4h),6.20(d,j=8hz,2h)ppm.13cnmr(151mhz,cdcl3)δ:144.5,141.2,131.1,130.9,128.4,127.0,126.9,122.7,120.4,116.3ppm。
测试的催化剂的化学结构:
实例2
可商购的10-甲基吩噻嗪(me-pth)用于在与公开于2014年4月7日提交的美国临时申请案第61/976,178号中的基于ir的体系类似的条件下聚合甲基丙烯酸甲酯,所述申请案以其全文并入本文中。简单地说,向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸甲酯(401μl,3.75mmol)、光催化剂(0.1mol%)和二甲基乙酰胺(1ml)。将反应混合物用三个冷冻-泵送-解冻循环脱气。然后将瓶用氩气回填,并且将α-溴苯基乙酸苯甲酯(6.6μl,0.0375mmol)经由注射器注入。反应物在380nmled的前方剧烈搅拌,同时用压缩空气冷却以维持环境温度。允许反应进行到大约mma的50%转化率,如由1hnmr监测。取出等分试样并且使用gpc分析以得到聚合物的分子量(mn)和分子量分布(mw/mn)。
表1.使用有机光氧化还原催化剂的光介导的甲基丙烯酸甲酯聚合[a]
[a]mn=数均分子量;mw=重均分子量)。使用尺寸排阻色谱(sec)或1hnmr确定mn和mw/mn;[b]使用甲基丙烯酸苯甲酯;[c]反应在黑暗中进行;并且[d]用可见光(50w萤光灯泡)照射反应。
聚合观测具有理论分子量与实验分子量之间的良好的一致性,尽管具有对分子量分布(表1,第1项)的差的控制。使用吩噻嗪衍生物进一步使自由基阳离子物质稳定,产生烷基溴化物物质的更高效修复和得到对聚合过程的更大控制。为此目的,在一个步骤中由可商购的材料制备10-苯基吩噻嗪(pth,ered(pth+./pth*)=-2.4v对sce),并且在此前报道的反应条件下测试。获得低多分散性同时维持实验mn与理论mn之间的良好一致性。另外,通过调节引发剂浓度,以各种分子量为目标,同时维持优异控制(表1,第2项-第6项)。这些实验第一次确定,原子转移自由基聚合(atrp)类过程可在无金属催化剂体系下发生。当反应在优化条件但没有光的情况下进行时,未观测到聚合(表1,第7项),表明该方法实际上是光介导的方法。此外,当采用在激发态下氧化的其它有机类光氧化还原催化剂(伊红y和亚甲基蓝)时,未发生反应(表1,第8项和第9项)。
实例3
将配备有磁性搅拌棒和装配有橡胶隔板的瓶中装入甲基丙烯酸甲酯(2.4ml,22.5mmol)、10-苯基吩噻嗪(6.2mg,0.1mol%)和二甲基乙酰胺(6ml)。将反应混合物用三个冷冻-泵送-解冻循环脱气。然后将瓶用氩气回填,并且将α-溴苯基乙酸苯甲酯(39μl,0.225mmol)经由注射器注入。反应物在380nmled的前方搅拌,同时用压缩空气冷却以维持环境温度。反应物在光的前方搅拌2.5h(14%转化率),并且然后通过在铝箔中将其缠绕放入黑暗中。缠绕在铝箔中的注射器用于将在黑暗中的反应混合物转移到己烷的搅拌溶液(50ml,也缠绕在铝箔中)中。将倾析白色沉淀,并且在再次沉淀到己烷中之前,再溶解于二氯甲烷中以产生280mg的白色粉末。mn=2,900g/mol,mw/mn=1.33。
实例4
在没有光的情况下没有反应表明这种新型体系可以类似于基于ir的体系的可逆的和反应性方式通过光激活/去激活。在优化反应条件下使10-苯基吩噻嗪(pth)与甲基丙烯酸苯甲酯(bnma)组合,并且首先放入黑暗中1小时。在这个时间段(图1a)期间未观测到转化后,将反应物暴露于光,在1小时内达到18%转化率。随后,然后将反应物放入黑暗中,未观测到转化,指示有效去激活。重复此循环若干次,在黑暗中无聚合并且在照射时链端的有效再激活达到高转化率(88%)。对于相同实验,监测分子量对比转化率(图1b),并且观测到线性增加。此外,贯穿反应过程观测一级动力学,表明在光存在下维持恒定自由基浓度(图1c)。此数据指示当将光从系统除去时,链端被高效氧化成稳定和休眠的烷基溴化物,并且在再暴露于光时,链端被高效再激活。这确定使用10-苯基吩噻嗪作为催化剂允许用光优异调节聚合过程。
实例5
2-(二甲氨基)乙基甲基丙烯酸酯(dmaema)由于其刺激-反应特性而为在工业和学术界中广泛采用的单体。当在此前报告的条件下使ir(ppy)3与dmaema组合时,观测到非常广的多分散性(表2,第1项)。不受特定理论限制,据相信,ir(ppy)3的氧化电势(e1/2ox=0.77v对sce)导致胺氧化,产生不受控的聚合。通过移动到较低氧化10-苯基吩噻嗪(pth)(e1/2ox=0.68v对sce),单体氧化可被抑制,并且可获得受控聚合。因此,当pth与dmaema组合并且在标准条件下照射时,观测到极低的多分散性(表2,第2-3项)。这举例说明通过转换成这种有机催化剂,可实现新反应性,这产生比ir(ppy)3更宽的单体范围。
表2.使用有机光氧化还原催化剂的二甲氨基乙基甲基丙烯酸酯的光介导的聚合[a]
[a]反应条件:dmaema(1当量)、光催化剂(0.1mol%)、2-溴-2-苯基乙酸乙酯(1)(0.01当量)、dma(2m的dmaema),在室温下用380nmled照射3-7h(mn=数均分子量;mw=重均分子量)。使用尺寸排阻色谱(sec)或1hnmr确定mn和mw/mn;并且[b]使用0.005mol%ir(ppy)3。
实例6
良好的链端保真性是受控的自由基的重要特征,并且能够合成定义明确的大分子。因此,为了提供溴基链端的证据,在实例1中所描述的条件下合成低分子量聚(甲基丙烯酸甲酯)(pmma)(mn=2,600g/mol,mw/mn=1.33),并且使用电喷射电离质谱(esi-ms)分析(图2)。在波谱中主峰的分裂图案明确示出溴同位素的存在(图2,插图),和匹配理论的质量。
实例7
进行嵌段共聚以显示具有活性特征的体系是适当的。具体地说,使0.1mol%10-苯基吩噻嗪(pth)、2-溴-2-苯基乙酸乙酯(1)和甲基丙烯酸甲酯(mma)在dma中组合,并且照射以产生低分子量聚(甲基丙烯酸甲酯)(pmma)均聚物(mn=3,600g/mol,mw/mn=1.19)。接下来,在优化条件下使均聚物与甲基丙烯酸苯甲酯组合,并且照射4h以合成pmma-b-pbnma嵌段共聚物(图3a)。尺寸排阻色谱(sec)明确示出了到较高分子量物质的偏移,其中在均聚物能谱中有极少拖尾。
为了进一步探索有机催化体系的范围和链端保真性,生成pmma均聚物并且使用此前开发的条件采用ir(ppy)3用丙烯酸甲酯扩展链。发现这两种方法完全正交,产生单分散嵌段共聚物(图3b),其中在sec迹线的均聚物能谱中几乎没有拖尾,表明所公开的体系与其它聚合方法组合来制备定义明确的材料的能力。
为了呈现用这种方法合成功能材料的能力,制备ptbuma均聚物并且用dmaema扩展链以产生定义明确的、刺激-反应性嵌段共聚物(mn=22,500g/mol,mw/mn=1.30)(图3c)。
实例8:
向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸苯甲酯(551μl,3.77mmol)、pth(0.9mg,0.1mol%)和二甲基乙酰胺(400μl)。在另一烧瓶中,将400μl的二甲基乙酰胺添加到聚(甲基丙烯酸甲酯)大分子引发剂(86mg,0.014mmol)。将两个反应混合物用三个冷冻-泵送-解冻循环脱气。然后使用注射器将单体和催化剂转移到含大分子引发剂的烧瓶。反应物在380nmled的前方搅拌,同时用压缩空气冷却以维持环境温度。在4h之后(59%转化率),通过向空气开放停止反应并且沉淀到甲醇(20ml)中。将沉淀物过滤并且在再沉淀到甲醇中之前再溶解于ch2cl2中。通过1hnmr和gpc分析产物。(产生:180mg白色粉末)mn=27,400g/mol,mw/mn=1.49。
实例9
向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸甲酯(401μl,3.75mmol)、光催化剂(0.05mol%)、raft剂2(4.1mg,0.0188mmol)和二甲基乙酰胺(1ml)。在黑暗中将反应混合物用三个冷冻-泵送-解冻循环脱气。然后将瓶用氩气回填,并且在380nmled前方剧烈搅拌,同时用压缩空气冷却以维持环境温度。反应转化率通过1hnmr监测。取出等分试样并且使用gpc分析以得到聚合物的分子量(mn)和分子量分布(mw/mn),其中结果示出在表3中。
表3.使用通常采用的raft剂的mma的受控聚合
实例10
向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸甲酯(401μl,3.75mmol)、p-cf3-pth(1.3mg,0.1mol%)和二甲基乙酰胺(1ml)。将反应混合物用三个冷冻-泵送-解冻循环脱气。然后将瓶用氩气回填,并且将α-溴苯基乙酸苯甲酯(6.6μl,0.0375mmol)经由注射器注入。反应物在380nmled的前方剧烈搅拌4h,同时用压缩空气冷却以维持环境温度。通过1hnmr确定mma的转化率为35%。取出等分试样并且使用gpc分析以得到聚合物的分子量(mn=4,500g/mol)和分子量分布(mw/mn=1.36)。
实例11
向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸甲酯(401μl,3.75mmol)、b(pth)3(1.1mg,0.05mol%)和二甲基乙酰胺(1ml)。将反应混合物用三个冷冻-泵送-解冻循环脱气。然后将瓶用氩气回填,并且将α-溴苯基乙酸苯甲酯(6.6μl,0.0375mmol)经由注射器注入。反应物在380nmled的前方剧烈搅拌5h,同时用压缩空气冷却以维持环境温度。通过1hnmr确定mma的转化率为34%。取出等分试样并且使用gpc分析以得到聚合物的分子量(mn=4,500g/mol)和分子量分布(mw/mn=1.38)。
实例12
将配备有磁性搅拌棒和装配有橡胶隔板的瓶中装入甲基丙烯酸甲酯(2.4ml,22.5mmol)、p-cf3-pth(7.7mg,0.1mol%)和二甲基乙酰胺(6ml)。将反应混合物用三个冷冻-泵送-解冻循环脱气。然后将瓶用氩气回填,并且将α-溴苯基乙酸苯甲酯(39μl,0.225mmol)经由注射器注入。反应物在380nmled的前方搅拌,同时用压缩空气冷却以维持环境温度。反应物在光的前方搅拌2.5h(17%转化率),并且然后通过将其缠绕在铝箔中放入黑暗中。缠绕在铝箔中的注射器用于将在黑暗中的反应混合物转移到己烷的搅拌溶液(50ml,也缠绕在铝箔中)中。倾析白色沉淀,并且在再次沉淀到己烷中之前再溶解于二氯甲烷中以产生280mg的白色粉末。mn=3,300g/mol,mw/mn=1.33。
实例13
向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸苯甲酯(595μl,3.51mmol)、p-cf3-pth(1.2mg,0.1mol%)和二甲基乙酰胺(500μl)。在另一烧瓶中,将500μl的二甲基乙酰胺添加到聚(甲基丙烯酸甲酯)大分子引发剂(49mg,0.031mmol)。将两个反应混合物用三个冷冻-泵送-解冻循环脱气。然后使用注射器将单体和催化剂转移到含大分子引发剂的烧瓶。反应物在380nmled的前方搅拌,同时用压缩空气冷却以维持环境温度。在4h之后,通过向空气开放停止反应并且沉淀到meoh(20ml)中。过滤产物,再溶解于ch2cl2中,并且再次沉淀到meoh(20ml)中。(产生:201mg白色粉末)mn=10,800g/mol,mw/mn=1.44。
实例14
向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸苯甲酯(639μl,3.77mmol)、pth(1mg,0.1mol%)和二甲基乙酰胺(500μl)。在另一烧瓶中,将500μl的二甲基乙酰胺添加到聚(甲基丙烯酸甲酯)大分子引发剂(86mg,0.033mmol)。将两个反应混合物用三个冷冻-泵送-解冻循环脱气。然后使用注射器将单体和催化剂转移到含大分子引发剂的烧瓶。反应物在380nmled的前方搅拌,同时用压缩空气冷却以维持环境温度。在5.5h之后,通过向空气开放停止反应并且沉淀到己烷(20ml)中。然后将己烷倾析掉,并且在再溶解于ch2cl2中之前蒸发残余溶剂。然后,再溶解的聚合物沉淀到己烷(20ml中。将此过程再重复一次。(产生:228mg白色粉末)mn=10,800g/mol,mw/mn=1.44。
实例15
向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸苯甲酯(635μl,3.75mmol)、光催化剂0.5mol%)和二甲基乙酰胺(1ml)。在不采取任何脱气预防措施的情况下封端反应混合物。最后,添加α-溴苯基乙酸乙酯(5.8μl,0.033mmol)。反应物在380nmled的前方剧烈搅拌,同时用压缩空气冷却以维持环境温度。允许反应进行到大约甲基丙烯酸苯甲酯的50%转化率,如通过1hnmr监测。取出等分试样,并且用gpc分析:mn=17,600g/mol,mw/mn=1.26。
实例16
将配备有磁性搅拌棒和装配有橡胶隔板的瓶中装入甲基丙烯酸甲酯(2.4ml,22.5mmol)、10-苯基吩噻嗪(31mg,0.5mol%)和二甲基乙酰胺(6ml)。在不采取任何脱气预防措施的情况下封端反应混合物。最后,添加α-溴苯基乙酸乙酯(39μl,0.225mmol)。反应物在380nmled的前方搅拌,同时用压缩空气冷却以维持环境温度。反应物在光前方搅拌3.5h(29%转化率),并且通过沉淀到己烷(250ml)中停止。倾析白色沉淀,并且在再次沉淀到己烷(250ml)之前,再溶解于二氯甲烷中以产生517mg的白色粉末。mn=4,200g/mol,mw/mn=1.12。
实例17
向配备有磁性搅拌棒和装配有铁氟龙螺帽隔板的瓶中装入甲基丙烯酸苯甲酯(617μl,3.64mmol)、pth(5mg,0.5mol%)、聚(甲基丙烯酸甲酯)大分子引发剂(90mg,0.0214mmol)和二甲基乙酰胺(1ml)。在不采取任何脱气预防措施的情况下封端反应混合物。反应物在380nmled的前方搅拌,同时用压缩空气冷却以维持环境温度。在4h之后(33%转化率),通过向空气开放停止反应并且沉淀到甲醇(20ml)中。将沉淀物过滤并且在再沉淀到甲醇中之前再溶解于ch2cl2中。通过1hnmr和gpc分析产物。(产生:94mg白色粉末)mn=17,200g/mol,mw/mn=1.23。
实例18:用光掩模图案化
向瓶中装入1mgpth(0.00375mmol)、0.1mldma,并且脱气。向单个瓶中装入无抑制剂mma(400μl,3.75mmol)并且脱气。将瓶带入氮气氛围手套箱中,在将单体/催化剂溶液吹吸到硅基板上之前组合,硅基板已经用基溴化物引发剂均匀地官能化,直到它被完全覆盖。然后,将测量20μm×200μm的透明矩形的光掩模放置在溶液顶端上以形成与基板接触的薄层。将晶片放入光源下大约3cm处,然后用380nm灯照射。在照射之后,从手套工作箱取出聚合物刷涂布的基板,并且在使其干燥之前用二氯甲烷洗涤。在光学显微镜下观测聚合物刷的矩形区域,并且在图4中示出。
应理解,本文所描述的实例和实施例仅为了说明性目的,并且根据其的各种修改或变化将由本领域的技术人员想到并且并入在本申请案的精神和范围以及所附权利要求书的范围内。本文所引用的所有出版物、专利以及专利申请案都出于所有目的以引用的方式并入本文中。
- 还没有人留言评论。精彩留言会获得点赞!