官能化硅烷和包含其的电解液组合物及电化学装置的制作方法
在此要求2014年10月3日提交的美国临时申请62/059,663号的优先权,所述申请结合到本文中。
背景
在锂离子电池中的液体电解液(质)通常包含在碳酸亚乙酯(EC)和诸如碳酸二甲酯(DMC)、碳酸二乙酯(DEC)或碳酸甲乙酯(EMC)的一种或多种共溶剂的有机溶剂共混物中的锂盐,通常为LiPF6。遗憾的是,LiPF6在这些碳酸酯溶剂中在高于60℃下以及在超过4.3伏的充电电压下不稳定。在高于这些温度或压力下操作锂离子电池导致电极材料和电池性能快速变劣。另外,当前的锂离子电解液溶剂表现出约35℃的闪点,且是在极端锂离子电池故障期间释放的能量的主要来源。鉴于这些显著的限制,目前的电解液阻碍了用于包括便携式产品、电力驱动车辆(EDV)和公用事业规模使用的所有用途的高级锂离子电池的发展。大型锂离子电池还需要大大降低电池故障率以有效地服务在EDV和电网存储(grid storage)中的应用。
因此,对于能量存储装置诸如锂离子电池中的改善的电解液溶液存在长期且未满足的需求。
发明概述
本文公开了在电化学装置中用作电解液溶剂以及其他用途的有机硅(OS)化合物。
一般来讲,OS化合物是环境友好、不可燃、耐高温的材料。这些特性使得OS材料非常适合用作储能装置中的电解液溶剂、粘合剂和涂料。基于OS的电解液与所有基于锂(Li)的电化学系统相容,所述电化学系统包括一次和可再充电电池(即,锂-离子电池、Li-空气电池)和电容器(即,超级电容器(super/ultra-capacitor))。将基于OS的电解液设计到锂电池的方法涉及在电池设计中的有限变化,且这些电解液可采用现有制造工艺和设备结合到生产操作中。
本文所述的OS化合物可用作替代在传统锂离子电池中的基于碳酸酯的溶剂系统的液体电解液溶剂。所述基于OS的溶剂在Li离子电池中的性能和滥用宽容(abuse tolerance)方面提供显著的改善,包括增加热稳定性以延长在高温下的寿命,增加电解液闪点以改善安全性,增加电压稳定性以允许使用高电压阴极材料并实现更高的能量密度,降低电池故障率以与用于EDV和电网存储应用中的大型锂电池的要求一致,以及与当前在Li离子电池中使用的材料相容以便于在当前设计中采用。也已经证明在采用基于OS的电解液的情况下双电层电容器(EDLC)装置的功能。本文所述的OS化合物可用于基于OS的电解液共混物中以满足在工业、军事和消费产品装置中的具体应用的要求。
所述化合物和电解液制剂的目的和优势将从以下详述和附图中更充分地显现。
因此,本文公开了:
1. 一种化合物,其选自:
其中:
“a”为1至4的整数;“b”为0至(3 x a)的整数;
当“b”= 0时,“Z”不存在,否则
“Z”选自“R”和
其中“R”各自独立地选自卤素;C1-6直链或支链的烷基、烯基或炔基;和C1-6直链或支链的卤烷基、卤烯基或卤炔基;
在式I和II中的“Sp”各自独立地选自C1-15直链或支链的亚烷基和C1-15直链或支链的卤亚烷基;且
在式I中的“Y”各自独立地选自有机极性基团。
2. 权利要求1的化合物,其中“Y”各自独立地为选自以下的有机极性基团:
其中弯曲键表示C2-6亚烷基(akylene)桥连部分。
3. 权利要求2的化合物,其中“Y”各自独立地为选自以下的有机极性基团:
。
4. 权利要求3的化合物,其中“R”各自独立地选自氟和C1-6直链或支链的氟烷基、氟烯基或氟炔基;且
在式I和II中的“Sp”各自独立地选自C1-15直链或支链的氟亚烷基。
5. 权利要求1至4中任一项的化合物,其中“a”为1且“b”为0-3。
6. 权利要求5的化合物,其中“b”为1且“Z”为“-R”。
7. 权利要求5的化合物,其中至少一个“R”为氟且“Sp”各自独立地选自C1-6直链亚烷基和C1-6直链氟亚烷基。
8. 权利要求1至4中任一项的化合物,其中“a”为2且“b”为0-6。
9. 权利要求7的化合物,其中“b”为1且“Z”为“R”。
10. 权利要求7的化合物,其中至少一个“R”为氟且“Sp”各自独立地选自C1-6直链亚烷基。
11. 权利要求1至4中任一项的化合物,其中“a”为3且“b”为0-9。
12. 权利要求11的化合物,其中“b”为1且“Z”为“R”。
13. 权利要求11的化合物,其中至少一个“R”为氟且“Sp”各自独立地选自C1-6直链亚烷基。
14. 权利要求1至4中任一项的化合物,其中“a”为4且“b”为0-12。
15. 权利要求14的化合物,其中“b”为1且“Z”为“R”。
16. 权利要求14的化合物,其中至少一个“R”为氟且“Sp”各自独立地选自C1-6直链亚烷基。
17. 权利要求1至4中任一项的化合物,其中至少一个“R”为氟。
18. 一种电解液组合物,其包含权利要求1至4中任一项所述的化合物以及盐。
19. 权利要求18的电解液组合物,其中所述盐为含锂盐。
20. 一种电化学装置,其包含权利要求18中所述的电解液组合物。
在所述化合物的进一步限定的变型中,“Y”各自独立地为选自以下的有机极性基团:
。
另外,“Y”各自可任选地且独立地为选自以下的有机极性基团:
。
在所述化合物的一个供选变型中,“a”为1,且“b”为0-2;任选地“b”为1且“Z”为R。在该变型中,一个任选的实施方案为,至少一个“R”为氟,且“Sp”各自独立地选自可被氟代的直链或支链的亚烷基。
在所述化合物的另一供选变型中,“a”为2,且“b”为0-6;任选地,“b”为1,且“Z”为R。在该变型中,一个任选的实施方案为,至少一个“R”为氟,且“Sp”各自独立地选自可被氟化的C1-6直链或支链的亚烷基。
在所述化合物的又一供选变型中,“a”为3,且“b”为0-9;任选地,“b”为1,且“Z”为R。在该变型中,一个任选的实施方案是,至少一个“R”为氟,“Sp”各自独立地选自可被氟化的C1-6直链或支链的亚烷基。
在所述化合物的还一供选变型中,“a”为4,“b”为0-12;任选地,“b”为1,且“Z”为R。在该变型中,一个任选的实施方案是,至少一个“R”为氟,且“Sp”各自独立地选自可被氟化的C1-6直链或支链的亚烷基。
本文还公开了包含本文的化合物以及盐的电解液组合物。含锂盐是优选的。本文还公开了包含所述电解液组合物的电化学装置。
由于下标“a”和“b”的性质,以下上位结构明确地在本文公开并要求保护的化合物的范围内:
。
各种R基团如上文对“R”所定义;Sp如上所定义,Y如上所定义。优选地,在所述化合物的所有变型中,Y为可配位Li+的有机极性基团。
当极性基团Y具有二(2)或更大的化合价时,它将具有多于一个取代位点。因此,可存在多于一个通过间隔基团键合到Y的硅原子,如上文对于Z的定义中所述。因此,以下结构也明确地在本文公开并要求保护的化合物的范围内:
等。
在所述化合物的所有变型中,“卤素”包括氟、氯、溴和碘。氟和氯为优选的卤素取代基。术语“盐”在本文中以其常规化学意义使用,是指由酸和碱的中和反应产生的离子化合物。如本文所用,盐被有目的地广泛定义为包括所有的盐,包括但不限于钠盐、锂盐、镁盐、硼酸盐、磷酸盐等。非限制性实例包括六氟磷酸钠、高氯酸钠、双-三氟甲磺酰亚胺钠、六氟磷酸镁、高氯酸镁、双-三氟甲磺酰亚胺镁、四氟硼酸镁、三氟甲基磺酸镁、四氟硼酸四乙基铵(TEA-TFB)、四氟硼酸四丁基鏻、六氟磷酸四丁基鏻、四氟硼酸四丁基铵、六氟磷酸四乙基铵、高氯酸四乙基铵和双三氟甲磺酰亚胺酸四乙基铵。术语“含锂盐”明确包括但不限于LiClO4、LiBF4、LiAsF6、LiPF6、LiCF3SO3、Li(CF3SO2)2N、Li(CF3SO2)3C、LiN(SO2C2F5)2、烷基氟磷酸锂(lithium alkyl fluorophosphate)和双(螯合)硼酸锂。
本文还公开了包含如前述段落中所述的一种或多种有机硅化合物的电解液组合物。本文还公开了包含这种电解液组合物的电化学装置。本文公开的化合物非常适用于配制用于所有种类的电荷存储装置(例如,电池、电池组、电容器等)中的电解液。
与常规碳酸酯溶剂相比较,本文公开的有机硅溶剂分子具有显著且出乎意料的热稳定性。即使当配制成包含诸如LiPF6的锂盐的电解液组合物时,有机硅溶剂在高温暴露和操作期间也表现出显著的耐破坏性。如在实施例和附图中所示,本文公开的有机硅溶剂分子的几种变体已经证明在LiPF6存在下、在高达和超过175℃的温度下的热稳定性。这些数据证明了硅作为有机硅分子设计内的基本稳定元素的固有益处。本文公开的分子的变体包括非氟化、氟化、多氟化、多官能和交替的烷基结构,以及许多不同的Li+配位官能度。所述官能团包括腈、碳酸酯、砜和酯等。气相分解和液相分解两者分别通过EI-MS和NMR测量,以确定在暴露于175℃下60分钟后具有有机硅溶剂和1M LiPF6盐的电解液的破坏。没有观察到OS溶剂的显著气相或液相分解。
无论是否具体公开,本文使用的数值范围都应包括该范围内包含的每个数值和数值子集。此外,这些数值范围应被视为对涉及该范围内的任何数值或数值子集的权利要求提供支持。例如,1-10的公开应被视为支持2-8、3-7、1-9、3.6-4.6、3.5-9.9等的范围。
除非另有说明或在提到的上下文中明确暗示为相反,否则对本发明的单数特征或限制的所有提及都应包括相应的复数特征或限制,反之亦然。不定冠词“一”应意指“至少一个”,除非明确限于单数。
除非另有说明或在提到组合的上下文中明确暗示为相反,否则本文使用的方法或工艺步骤的所有组合可以任何顺序执行。
附图简述
图1A以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有LiPF6、LiBF4或LiTFSI的F1S3MN的氧化稳定性。图1B描述了在图1A中示出的相同数据的特写。
图2A和图2B以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了测量具有LiPF6、LiBF4或LiTFSI的F1S3MN的还原稳定性的双重试验。
图3A以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6的F1S3MN或F1S3M2的氧化稳定性。图3B描述了在图3A中示出的相同数据的特写。
图4以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6的F1S3MN或F1S3M2的还原稳定性。
图5A和图5B描述了具有LiPF6的F1S3MN的热稳定性。图5A描述了在图5B中示出的相同数据的特写。
图6描述了具有LiPF6的F1S3M2的热稳定性。
图7描述了具有LiTFSI的F1S3MN的热稳定性。
图8描述了具有LiBF4的F1S3MN的热稳定性。
图9描述了纯F1S3MN的热稳定性。
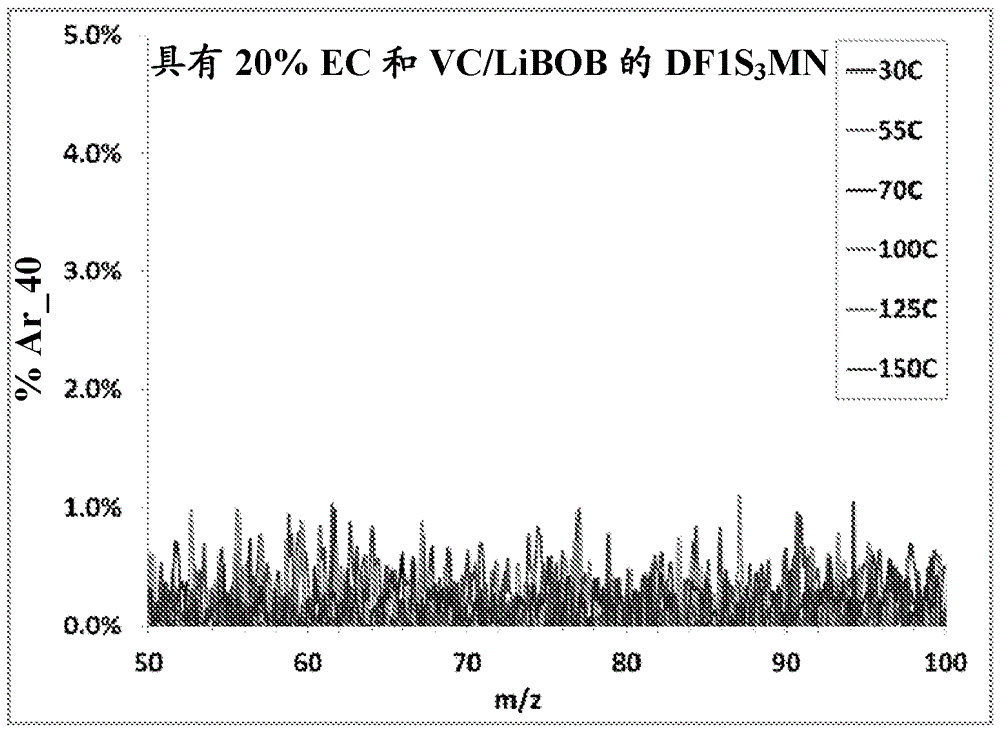
图10描述了具有20% EC和VC/LiBOB的DF1S3MN的热稳定性。
图11描述了当与脱锂NCA阴极材料(de-lithiated NCA cathode)一起加热时,与碳酸酯对照电解液相比,F1S3MN电解液的增强的稳定性。
图12描述了含有各种电解液溶剂的电池在30℃下在各种充电率下的放电容量。
图13描述了测试电池的构造。
图14描述了含有与在图12中所示相同的电解液溶剂的电池在55℃下在各种充电率下的放电容量。
图15描述了EDLC装置的构造。
图16描述了含有具有TEA-BF4的DF1S2MN电解液的EDLC装置的性能。
图17描述了含有具有TBP-PF6的各种电解液溶剂的EDLC装置的性能。
图18以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6或1M LiTFSI的1ND1N的氧化稳定性。
图19以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6或1M LiTFSI的1ND1N的还原稳定性。
图20A和图20B描述了对于使用1ND1N和1M LiPF6或1M LiTFSI从0V至6V和从6V至0V的循环扫描的电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线。图20A描述了第一次循环。图20B描述了第二次循环。
图21A以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6的F1S3MN或1ND1N的氧化稳定性;图21B描述了在图21A中示出的相同数据的特写。
图22A以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiTFSI的F1S3MN或1ND1N的氧化稳定性。图22B描述了在图22A中示出的相同数据的特写。
图23为说明纯1ND1N的热稳定性的质谱。
图24为说明具有LiPF6的1ND1N的热稳定性的质谱。
图25A描述了如关于图24描述的质谱特征的24-30m/z的特写。图25B描述了如关于图24描述的质谱特征的49-55m/z的特写。
图26描述了具有LiTFSI、碳酸亚乙烯酯(VC)和双(草酸根合)硼酸锂(LiBOB)的1ND1N的热稳定性。
图27描述了具有LiBF4的1ND1N的热稳定性。
图28描述了含有各种电解液的电池在各种充电率下的放电容量。
图29描述了含有各种其他电解液溶剂的电池的放电容量,将第一次循环与第五十次循环相比较。
图30A描述了含有基于1ND1N-LiPF6的电解液的电池在各种充电率下的放电容量;图30B描述了含有基于1ND1N-LiTFSI的电解液的电池在各种充电率下的放电容量。
图31为具有峰归属的1ND1N的1H-ΝΜR光谱(在CDCl3中)。
图32为具有峰归属的F1S3MN的1H-ΝΜR光谱(在CDCl3)中。
图33为具有峰归属的DF1S2MN的1H-NΜR光谱(在CDCl3)中。
图34为具有峰归属的DF1S3MN的1H-NΜR光谱(在CDCl3)中。
图35为具有峰归属的F1S3cMN的1H-NΜR光谱(在CDCl3)中。
图36为具有峰归属的1S3MN的1H-NΜR光谱(在CDCl3)中。
图37描述了在175℃下60分钟之后具有1M LiPF6的F1S3MC、F1S3cMN和1S3DN的气相分解。该图示出在高达175℃时没有显著的气相分解。
图38描述了在175℃下60分钟之后具有1M LiPF6的DF1S2MN、TF1S2MN和TF1S3MN的气相分解。该图示出在高达175℃时没有显著的气相分解。
发明详述
在整个说明书中,将使用许多简要缩写以更容易地命名各种有机硅化合物。使用以下约定:
FnSnMN化合物具有通式:
其中R1、R2和R3相同或不同且独立地选自C1-C6直链或支链的烷基、烯基、炔基或卤素(优选F),“间隔基”为C1-C6直链或支链的亚烷基(优选C1-C6直链二价亚烷基),且Y为如先前描述的极性有机部分。
本文公开的化合物可通过许多不同的路径制得。可用于制造所述化合物的通用方法如下:
。
R1、R2和R3基团如本文对于R所定义;R4具有与Y相同的定义;“n”为正整数。
本文公开的化合物还可经由以下方法制造:
。
同样,R1、R2和R3基团如本文对于R定义;R4具有与Y相同的定义。
本文公开的化合物还通过包括以下反应流程的许多具体路径制得:
。
LiTFSI,即双(三氟甲烷)磺酰亚胺锂盐(Sigma-Aldrich目录号449504),为由若干国际供应商供应的商业产品:
。
无论是否明确地描述,本文所述的要素和方法步骤都可以任何组合使用。
除非另有说明或由所提到组合的上下文明确暗示为相反,否则本文使用的方法步骤的所有组合都可以任何顺序执行。
除非内容另有明确规定,否则本文使用的单数形式“一”和“该”包括复数个指示物。
本文引用的所有专利、专利出版物和同行看过的出版物(即,“参考文献”)都通过引用明确地结合,其结合程度如同每个单个参考文献都被具体地且单独地表示为通过引用结合。在本公开与所结合的参考文献之间有冲突的情况下,以本公开内容为准。
应当理解,本文公开的化合物和组合物不限于本文说明并描述的零件的特定构造和布置,而是包括落入权利要求范围内的其改进形式。
本发明公开的化合物为具有以一个或多个末端或内部极性有机取代基或部分形式的共有结构特征的有机硅化合物。本文使用的术语“极性有机取代基”和“极性有机部分”可互换使用。所述术语明确包括但不限于以下官能团:
。
优选的化合物包括以下结构等:
。
上述结构都描述为具有末端氰基。这仅是为了简洁的目的。具有代替氰基部分的如上所述的内部和/或末端极性部分的类似化合物明确在本公开的范围内。同样地,卤代化合物在上文描述为氟代化合物。具有代替氟原子的其他卤素取代基(氯、溴和/或碘)的类似化合物明确在本公开的范围内。对于所列出的每种化合物,提供两种供选的系统名称(每对名称中的第一个指定基本核心为腈;第二个指定基本核心为硅烷。)此外,给出了每种化合物的速记命名,其中DF = 二氟,TF =三氟,“Sn”表示在硅原子和末端氰酸酯、异氰酸酯或硫氰酸酯部分之间的亚烷基间隔基,且“n”表示在间隔基中的碳原子数。所选择的有机硅(OS)化合物的物理性质提供在表1中。
可使用命名为1NMS、F1S3MS和DF1S3MS的化合物的以下说明性合成来完成类似砜的合成。注意,落入本文所包含的更宽泛公开内容内的其他砜可通过相同的路径通过简单地相应改变起始试剂而制得。
1NMS的合成如下进行:将2-(甲基磺酰基)乙醇与0.5摩尔当量的六甲基二硅氮烷和作为催化剂的约1%摩尔当量的Al(H2PO4)3在没有溶剂的情况下混合。将该混合物保持在约80℃下过夜,并蒸馏两次,以得到纯的1NMS。
F1S3MS的合成如下进行:将烯丙基甲基硫醚溶解在乙醇中且将其与4摩尔当量的H2O2混合。加入约3%摩尔当量的七钼酸铵作为氧化催化剂。第二天,将该溶液用NaHCO3溶液中和,且用CH2Cl2萃取,弃去水部分,且蒸发有机层并蒸馏,以产生烯丙基甲基砜。烯丙基甲基砜使用卡斯特(Karstedt)催化剂用二甲基氯硅烷来氢化硅烷化。产物在约150℃下使用NaFHF氟化,过滤,蒸馏两次,且经分子筛干燥,以产生纯的F1S3MS。
。
DF1S3MS的合成如下进行:将3-巯基丙基甲基二甲氧基硅烷溶解于乙醇中,且将其与一(1)摩尔当量的NaOH在水中混合。随后向该混合物中加入一(1)摩尔当量的Me2SO4并回流过夜。滤出固体。粗产物用4摩尔当量的H2O2和3%摩尔当量的七钼酸铵氧化。蒸发溶剂,且加入2摩尔当量的HF的水溶液。产物用CH2Cl2萃取并蒸馏。
这些砜化合物的另外的表征数据包含在附于本文并结合在本文中的附录(appendix)中。
降低的粘度、更高的电导率和在添加氟的情况下降低的闪点以及降低的间隔基长度如在表1中所示。DF1S2MN具有最低的粘度和最高的电导率。
代码:
。
纯1ND2、1ND1、1ND1N、F1S3MN、DPF1S3MN、F1S3MC、F1S3ME、F1S3MA、F1S3MS、1NMS、F1S3M2和F1S3cMN以及含有这些溶剂的电解液溶液的物理性质示于表2中:
表2:溶剂和电解液的物理性质
代码:
。
除了本文公开的有机硅化合物之外,本发明的电解液组合物还可包含常规的非硅共溶剂。例如,本发明的电解液组合物可包含腈和碳酸酯,诸如乙腈、碳酸亚乙酯(EC)、碳酸二甲酯(DMC)、碳酸二乙酯(DEC)或碳酸甲乙酯(EMC)。本发明的电解液组合物可包含宽范围浓度(包括但不限于约1重量%-约40重量%)的非硅共溶剂。合适的共溶剂浓度的实例包括约1重量%、约5重量%、约10重量%、约15重量%、约20重量%、约25重量%、约30重量%、约40重量%或在之间并包括任何前述量的范围。
实施例
F1S3MN的合成:
流程1描述了F1S3MN的合成流程。[F]表示氟化剂,诸如HF、NH4FHF或其他氟化剂。NH4FHF优选用作实验室规模合成的氟化剂。如果使用HF,唯一的副产物是HCl。合成的F1S3MN化合物用己烷从固体盐洗涤,蒸馏,用CaO干燥,并再次蒸馏。
流程2描述了使用NH4FHF作为氟化剂的F1S3MN的合成流程。使用卡斯特催化剂(铂(0)-1,3-二乙烯基-1,1,3,3-四甲基二硅氧烷络合物溶液,目录号479519,Sigma-Aldrich, St. Louis, MO),在仲碳上发生约3%取代,产生isoF1S3MN。isoF1S3MN具有比F1S3MN低的沸点,且其大部分可通过分级蒸馏分离。
流程3描述了使用Cl1S3MN中间体的F1S3MN的供选的更短的合成流程。Cl1S3MN中间体可由Gelest,Inc.获得(产品代码SIC2452.0, 11 East Steel Road, Morrisville, PA)。使用Cl1S3MN中间体减少了合成期间花费的时间。
流程4描述了F1S3MN的又一合成流程。与流程1一样,[F]表示氟化剂,诸如HF、NH4FHF或其他氟化剂。在该合成流程中使用HF作为氟化剂不会产生固体副产物,因此不需要己烷萃取和过滤固体。唯一的副产物是HCl。
流程5描述了F1S3MN的又一合成流程。与流程1一样,[F]表示氟化剂,诸如HF、NH4FHF或其他氟化剂。
F1S3MN的合成:
在一个优选的路径中,使用少量卡斯特催化剂,将烯丙基氰加热到约100℃。逐滴添加二甲基氯硅烷并回流4小时。在冷却至室温之后,在室温下,混合物使用1摩尔当量的氟化氢铵进行氟化。向混合物中加入冷己烷,滤出固体,且蒸发溶剂。将氧化钙加到粗产物中,并将其在45-55℃和0.4托下真空蒸馏,以产生所需产物F1S3MN。
F1S3ME的合成
在一个优选的路径中,将乙酸烯丙酯与二甲基氯硅烷混合,并加入少量的卡斯特催化剂。使混合物保持反应过夜。第二天,在强烈搅拌下,使用0.33摩尔当量的三氟化锑在室温下将混合物氟化过夜。第二天,滤出固体,且在25-30℃下在0.3托下真空蒸馏产物,以产生所需产物F1S3ME。
该酯适合作为电解液组合物中的电化学溶剂。F1S3ME对1M LiPF6具有良好的溶解性,并在室温下容易地溶解。其66℃的闪点是可接受的,同样可接受的还有其为6.6的介电常数(Diel.)。其CV氧化/还原行为同样适合于锂离子电荷存储单元。使用80% F1S3ME电解液的全电池循环稳定,具有良好的容量。其在30℃下在处于或低于5V的电势下保持非常低的电流密度。F1S3ME通过基本的Pt氧化和石墨还原在电化学上是可行的。其在第一次循环中表现出正常的还原行为,且在第二次循环中观察到钝化。F1S3ME在经历26次循环的NMC/石墨电极系统中在80%电解液制剂中循环良好(数据未示出)。
DF1S3DN的合成:
将双(氰基丙基)二氯硅烷与2摩尔当量的HF水溶液在室温下混合,并保持反应过夜。粗产物用二氯甲烷萃取,蒸发溶剂,且产物在165℃下在0.1托下真空蒸馏,以产生所需产物DF1S3DN。
1S3DN的合成:
将DF1S3DN溶解于THF中并用冰浴冷却,随后加入2摩尔当量的甲基溴化镁,并使混合物保持反应直至缓慢达到室温。将混合物用乙醇淬灭,粗产物用二氯甲烷萃取,蒸发溶剂,且产物在125℃下在0.2托下真空蒸馏,以产生所需产物1S3DN。
TF1S3MN的合成:
将氰基丙基三氯硅烷在冰浴中冷却且逐滴添加4摩尔当量的HF水溶液。使混合物保持反应不超过2小时。将粗产物用二氯甲烷萃取,蒸发溶剂,且将产物在25℃下在0.2托下真空蒸馏以产生所需产物TF1S3MN。
DPF1S3MN的合成:
将双(异丙基)氰基丙基氯硅烷溶解于THF中且在冰浴中冷却,随后逐滴添加1摩尔当量的HF水溶液,并保持在室温下反应过夜。第二天,蒸发溶剂,且将产物在真空中在60℃下在0.2托下蒸馏,以产生所需产物DPF1S3MN。
制备DPF1S3MN以探究在硅原子上的较大烷基如何影响分子的物理特性和电子特性。该化合物对LiPF6具有良好的溶解性,在室温下容易地溶解。它在高达约5V的循环伏安法(CV)中表现出稳定氧化行为。其CV还原行为也良好,在第二次循环时钝化。该化合物适用于在高温(闪点=118℃)下和在高达约5V的电势下的电化学应用。DPF1S3MN通过基本的Pt氧化和石墨还原在电化学上是可行的。
DF1S3MSO的合成:
根据所提到的程序,在DF1S3MS的合成期间获得作为不完全氧化的副产物的DF1S3MSO。DF1S3MSO通过在40℃下在0.1托下真空蒸馏纯化。
F1S3MA的合成:
在该合成中,将2摩尔当量的二甲基烯丙基胺与1,1,3,3-四甲基二硅氧烷和少量的卡斯特催化剂混合。将混合物加热到100℃并保持反应过夜。第二天,在强烈搅拌下,使用0.67摩尔当量的三氟化锑在150℃下将混合物氟化过夜。第二天,滤出固体,且将产物在20-25℃下在0.2托下真空蒸馏以产生所需产物F1S3MA。
F1S3MC的合成:
使用催化量的磷酸二氢铝在80℃下将烯丙基醇用六甲基二硅氮烷甲硅烷基化过夜。在氢化硅烷化之前,将中间体烯丙氧基三甲基硅烷蒸馏以将其纯化。氢化硅烷化用1摩尔当量的二甲基氯硅烷和少量的卡斯特催化剂在80℃下进行过夜。第二天,在室温下在强烈搅拌下使用2摩尔当量的氢氟酸水溶液将混合物氟化并水解,得到(3-羟丙基)二甲基氟硅烷,将其在下一步骤之前蒸馏。将最后的中间体溶解于己烷中,加入1摩尔当量的三甲胺,将混合物在冰浴中冷却,且向混合物中逐滴添加氯甲酸甲酯,随后使其在室温下保持反应过夜。第二天,滤出固体,蒸发溶剂,且产物在28℃下在0.2托下真空蒸馏以产生所需产物F1S3MC。
合成F1S3MC以比较通常使用的碳酸酯与甲硅烷基类似物的电化学性能。显示F1S3MC的CV氧化和还原适合电化学应用。该合成非常好,产生纯度> 99.8%的F1S3MC。F1S3MC在室温下容易地溶解在1M LiPF6中。其在30℃下表现出可接受的循环且容量良好。F1S3MC在高达约5V的电势下也表现出良好的氧化稳定性。在CV中,还原在第一次循环中显示两个峰(在1.7V和0.6V处),且在第二次循环中显示钝化。
有机硅材料的电化学稳定性的确定:
使用计算机化学方法来计算各种有机硅分子的电化学性质。我们使用由爱荷华州立大学Gordon研究小组开发的GAMESS程序进行密度函数理论(DFT)分子轨道计算。在B3LYP/DZV水平下计算与化合物的还原和氧化电位相关的HOMO(最高占据分子轨道)能级和LUMO(最低未占分子轨道)能级。使用线性扫描伏安法(LSV)或循环伏安法(CV)在3-电极电池中测定含有机硅溶剂的电解液的氧化稳定性。使用铂微电极作为工作电极,使用锂金属作为反电极和参比电极。在10mV/s的扫描速率下,系统的电势从开路电压(OCV)增加到6或8V(相对于Li/Li+)。记录在每个电势下的所得电流密度(mA/cm2),较高的电流表示氧化反应(即,较低的氧化稳定性)。对于线性扫描伏安法,使用8V作为最终电势,以评估材料在较宽电压范围内的基本氧化稳定性。对于循环伏安法,使用6V以评估在与传统电池应用更相关的电势下在多次扫描中的材料。在循环伏安实验中进行多次扫描以确定所观察到的任何反应的可逆性/不可逆性。
在3-电极电池中使用线性扫描伏安法(LSV)测定含有机硅溶剂的电解液的还原稳定性。使用玻璃碳电极作为工作电极,锂金属作为反电极和参比电极。在10mV/s的扫描速率下,系统的电势从开路电压(OCV,通常为3V)减小到0.1V(相对于Li/Li+)。记录在每个电势下的所得电流密度(mA/cm2),较大的电流表示还原反应(即,更低的还原稳定性)。进行两次扫描以评估还原过程是可逆的还是不可逆的(即,钝化)。
F1S3MN的电化学稳定性:
F1S3MN和F1S3M2的分子轨道图(未示出)揭示,就最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)之间的能量差而言,F1S3MN(9.07eV)比F1S3M2 (8.20eV)更大。F1S3MN还具有比F1S3M2(-6.84eV)更高的氧化电位(-8.75eV)。
图1A和图1B以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有LiPF6、LiBF4或LiTFSI的F1S3MN的氧化稳定性。氧化稳定性在室温下在Pt作为工作电极,Li作为反电极,Li/Li+作为参比电极且扫描速率为10mV/s的情况下测试。图1B描述了在图1A中所示的相同数据的特写。F1S3MN-LiPF6电解液表现出最佳的氧化稳定性,在7.3V下具有1mA/cm2的电流密度,相比之下,F1S3MN-LiBF4和F1S3MN-LiTFSI分别在6.8V和6.2V下具有1mA/cm2的电流密度。
图2A和图2B以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有LiPF6、LiBF4或LiTFSI的F1S3MN的还原稳定性。还原稳定性在室温下在Pt作为工作电极,Li作为反电极,Li/Li+作为参比电极且扫描速率为10mV/s的情况下测试。图2A和图2B为两次单独的扫描。F1S3MN-LiPF6电解液表现出最佳的还原稳定性。
图3A和图3B以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6的F1S3MN或F1S3M2的氧化稳定性。氧化稳定性在室温下在Pt作为工作电极,Li作为反电极,Li/Li+作为参比电极且扫描速率为10mV/s的情况下测试。图3B描述了在图3A中所示的相同数据的特写。F1S3MN证实了相对于F1S3M2改善的氧化稳定性。
图4以在两次单独扫描中电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了与具有LiPF6的碳酸酯对照电解液相比,具有1M LiPF6的F1S3MN或F1S3M2的还原稳定性。还原稳定性在室温下在Pt作为工作电极,Li作为反电极,Li/Li+作为参比电极且扫描速率为10mV/s的情况下测试。与F1S3M2相比较,F1S3MN证实更低的耐还原性。
纯溶剂和配制的电解液的热稳定性的确定:
纯有机硅溶剂和电解液组合物两者的热稳定性如下测定:将约0.75mL的液体样品在氩气吹扫下在密封的小室中加热。将氩气吹扫进行至常压取样质谱仪,在质谱仪中可使用电子轰击质谱法(EI-MS)以非常低的水平检测任何气相杂质和/或分解产物。将样品在与电池应用相关的预定温度水平(30、55、70、100、125、150、175和200℃)下保持1小时。通过将从EI-MS获得的碎裂图案和NIST标准比较来鉴定气相分解产物。在加热实验(和检测/收集所有气相产物)之后,剩余的液体样品经由NMR光谱分析以定量分析分解的程度。检查多个核以全面分析系统的所有组分,包括有机硅溶剂、任何碳酸酯共溶剂、所有添加剂和锂盐(如果存在的话)。
F1S3MN的热稳定性:
图5A和图5B描述了具有LiPF6的F1S3MN的热稳定性。将F1S3MN-LiPF6电解液(批次ZP815-01)暴露于30℃-175℃的温度,并分别通过电子轰击质谱法(EI-MS)和核磁共振光谱法(NMR)分析气体和液体分解产物。标注了突出峰出现的温度。F1S3MN在高达175℃时没有显示出明显的气相和/或液相分解。Me2SiF2在100-125℃的温度下在81m/z处出现,且MeSiF3在150-175℃的温度下在85m/z处出现。然而,81m/z峰和85m/z峰在100-175℃下不一致地出现。此外,1H NMR分析显示加热至175℃后没有分解。因此,F1S3MN在高达175℃时没有显示出一致的分解。图5A描述了在图5B中所示的相同数据的特写。
图6描述了具有LiPF6的F1S3M2的热稳定性。将F1S3M2-LiPF6电解液暴露于30℃-150℃的温度并通过质谱分析分解产物。标注了突出峰出现的温度。F1S3M2在≥ 125℃的温度下显示分解。分解产物包括Me2SiF2和1,4-二噁烷。1H NMR分析显示在150℃下约6%分解。这些结果与关于图5A、图5B论述的那些结果相结合,表明F1S3MN比F1S3M2更加热稳定。
图7描述了具有LiTFSI的F1S3MN的热稳定性。将F1S3MN-LiTFSI电解液暴露于30℃-185℃的温度,并通过质谱分析分解产物。标注了突出峰出现的温度。在≥ 150℃的温度下观察到气相峰。对于F1S3MN-LiBF4电解液和纯溶剂观察到在117和102匹配图案处的峰(见图8和图9)。
图8描述了具有LiBF4的F1S3MN的热稳定性。将F1S3MN-LiBF4电解液暴露于30℃-200℃的温度,并通过质谱分析分解产物。标注了突出峰出现的温度。在≥ 175℃的温度下观察到气相峰。对于纯溶剂和F1S3MN-LiTFSI电解液观察到在117和102匹配图案处的峰(见图7和图9)。1H NMR分析显示没有氟化分解产物和< 0.5%的非氟化水解产物。
图9描述了纯F1S3MN的热稳定性。将F1S3MN电解液暴露于30℃-195℃的温度,并通过质谱分析分解产物。标注了突出峰出现的温度。在≥ 150℃的温度下观察到气相峰。在150℃,观察到Me2SiF2 (96/81m/z),但其他峰与该产物无关。1H NMR分析显示没有氟化分解产物且< 0.5%水解。
上述数据显示,在具有LiPF6的情况下,F1S3MN为最热稳定的OS溶剂。
DF1S3MN的合成:
将商购的3-氰基丙基二氯甲基硅烷(CAS号1190-16-5;Sigma Aldrich, St.Louis, MO, US)在室温下用二氟化铵氟化。随后将冷己烷加入混合物中。滤出固体并蒸发溶剂。向粗产物中加入氧化钙。在35-45℃下且在0.4托下真空蒸馏溶剂,以产生纯度非常高(约99.8%)的所需产物且产率为约90%。
DF1S2MN的合成:
将丙烯腈与N,N,N',N'-四甲基乙二胺和氧化铜(I)在烧瓶中混合并加热至60℃。随后逐滴添加二氯甲基硅烷并回流过夜。在冷却至室温之后,将混合物真空蒸馏(43℃,0.2托)以产生二氯中间体(DCl1S2MN)。使用1.2摩尔当量的氟化氢铵在室温下或1.2摩尔当量的氟化氢钠在130℃下将中间体氟化。随后加入二氯甲烷并滤出固体。蒸发溶剂,且真空蒸馏粗产物。将三乙胺和分子筛加到产物中并在25℃-33℃之间在0.1托下真空蒸馏,以大约75%的产率产生极高纯度(> 99%)的所需产物。
DF1S3MN的热稳定性:
图10描述了具有LiPF6的DF1S3MN的热稳定性。将DF1S3MN-LiPF6电解液(ZP990-01)暴露于30℃-150℃的温度,并分别通过电子轰击质谱(EI-MS)和核磁共振光谱(NMR)分析气体和液体分解产物。DF1S3MN显示高达150℃时没有显著气相和/或液相分解。
热滥用宽容的差示扫描量热(DSC)评估:
DSC测量在脱锂阴极材料存在下用基于F1S3MN和碳酸酯的电解液进行,以评估潜在的热滥用宽容效应,热滥用宽容效应可转换为在全电池型式(full cell format)中的安全优势。较高的开始温度、较低的总热输出和较低的峰值热输出为暗示在全型式电池中改善的热滥用行为的所有效应。
图11描述了具有LiPF6和各种碳酸酯共溶剂的F1S3MN的热稳定性,且与具有LiPF6的碳酸酯对照电解液相比较。将含有各种电解液的电池充电至4.25V,且随后拆开。用碳酸亚乙酯冲洗锂镍钴铝氧化物(NCA)阴极并使其干燥。将含有5mg活性物质和2mg新鲜电解液的各个样品气密地密封在不锈钢DSC盘中。以2℃/min的速率DSC扫描显示,碳酸酯对照电解液在比任何所述有机硅电解液共混物低得多的开始温度下反应。此外,其中有机硅替代EMC的电解液具有比对照电解液低得多的峰值热输出。
电解液的制备:
电解液的共混在无水分(< 5ppm)和无氧(< 20ppm)的氩气手套箱中完成。包括溶剂、盐和添加剂的所有电解液组分在共混之前已经适当地干燥并储存在手套箱中。溶剂水分通过Karl Fischer测定定期监测,以确保水分水平维持在< 20ppm下。通常,首先将溶剂称重到单独的小瓶中并混合直到均匀。将70%的溶剂加入容量瓶中。缓慢加入锂(或其他)盐,并通过磁力搅拌棒搅拌直至完全溶解。随后缓慢加入任何其他添加剂(即,VC、LiBOB)并搅拌直至溶液均匀。除去搅拌棒,且加入一部分剩余溶剂以完成体积需求。将搅拌棒放回容量瓶中,且搅拌电解液直至均匀。在共混完成之后,将电解液分配到干燥的小瓶或备用容器中以便储存。
F1S3MN在锂离子电池中的性能:
图12描述了含有各种电解液溶剂的电池在30℃下的放电容量。在包含石墨阳极、锂镍钴铝氧化物(NCA)阴极和来自Celgard, LLC (Charlotte,NC)的“2500”型隔板的2032型纽扣电池组件(如在图13中堆叠的组件)中在锂离子电池中在不同充电率(C-rate)下经一系列循环测试三种不同的电解液溶剂。这三种电解液溶剂是:(1) 包含1:1(体积计)的碳酸亚乙酯(EC)和碳酸二乙酯(DEC)的对照EPA6碳酸酯电解液(三角形);(2) 包含79% F1S3MN、20% EC、1M LiPF6和固体电解质界面膜(solid electrolyte interphase,SEI)-形成添加剂的基于F1S3MN的电解液(正方形);和(3) 包含79% F1S3M2、20% EC、1M LiPF6和SEI形成添加剂的基于F1S3M2的电解液(圆)。如在图12中所示,基于F1S3MN的电解液在4C充电率下与EPA6等效。
图14描述了含有与图12中显示且对于图12所述相同的电解液的电池在55℃下的放电容量。电池以相同的方式组装并以C/2充电率循环。如在图14中所示,与碳酸酯对照物和基于F1S3M2的电解液两者相比较,基于F1S3MN的溶剂在55℃下具有改善的循环稳定性。
F1S3MN和DF1S2MN在双电层电容器电池中的性能:
将对称的双电层电容器(EDLC)组装成如在图15中所描述的CR2032纽扣电池。玻璃纤维隔板(AP40,Merck Millipore)夹在两片AC(活性炭)布电极之间,其中将100微升电解液加到隔板上。四氟硼酸四乙基铵(TEA-BF4,Alfa Aesar,99%)和六氟磷酸四丁基鏻(TBP-PF6,Sigma Aldrich,≥ 99.0%)作为盐使用。有机硅溶剂F1S3MN (99.4%)和DF1S2MN (99.8%)由Silatronix制造。使用乙腈(AN,Sigma Aldrich,无水,99.8%)作为共溶剂。
来自Calgon碳的Zorflex FM10 100%活性炭(AC)布用作两个电极。FM10具有1000-2000m2/g的表面积、0.5毫米的厚度和120g/m2的面积密度。将AC布冲压成15mm直径的盘,并直接用作电极,而没有任何粘合剂或导电添加剂。
使用Biologic BMP300恒电位仪通过循环伏安法(CV)测试EDLC电池的性能。在烘箱中的受控温度的变化为±0.1℃。在0V和3V之间以10mV/s的扫描速率进行EDLC电池的循环伏安(CV)响应。根据以下等式[1,2]导出归一化比电容C:
其中i为电流,v为扫描速率,m为一个电极的质量。
图16显示具有含有TEA-BF4盐的OS电解液的EDLC电池的循环伏安图。电解液ZX1193包含溶解在70体积% DF1S2MN和30体积%乙腈中的1.0M TEA-BF4。电解液ZX1190包含溶解在体积比为60:40的共混DF1S2MN和乙腈溶剂中的0.8M TEA-BF4。具有两种电解液制剂的EDLC电池显示相对于0水平轴规则且对称的特点,表明电池的非氧化还原或感应电流性质。
图17示出具有含TBP-PF6盐的ZX1170电解液和ZX1184电解液的EDLC电池的循环伏安图。电解液ZX1170具有溶解到F1S3MN中的1.2M TBP-PF6且电解液ZX1184具有溶解到DF1S2MN中的1.2M TBP-PF6。还可以从具有电解液ZX1170和ZX1184制剂两者的EDLC电池中观察到非氧化还原或感应电流性质。
1ND1N的合成:
流程16描述了1ND1N的合成流程。1ND1N不能用钠(Na)、氧化钙(CaO)或氢化钙(CaH2)化学干燥。
1ND1N的电化学稳定性:
1ND1N和1ND1的分子轨道图(未示出)揭示1ND1N的最高占据分子轨道(HOMO)和最低未占分子轨道(LUMO)之间的能量差为7.88eV (LUMO = 0.21eV;HOMO = -7.88 eV)且1ND1的最高占据分子轨道(HOMO)和最低未占分子轨道(LUMO)之间的能量差为8.36eV (LUMO = 1.63eV;HOMO = -6.73eV)。1ND1N具有很好的氧化稳定性,但具有比1ND1低的耐还原性。
图18以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6或1M LiTFSI的1ND1N的氧化稳定性。氧化稳定性在室温下测试,工作电极为铂,反电极为锂,参考电极为Li/Li+,且扫描速度为10mV/s。
图19以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6或1M LiTFSI的1ND1N的还原稳定性。还原稳定性在室温下测试,工作电极为铂,反电极为锂,参考电极为Li/Li+,且扫描速度为10mV/s。对于每种电解液示出了两次单独扫描。
图20A和图20B针对在用1ND1N和1M LiPF6或1M LiTFSI的情况下从0V到6V和从6V到0V的循环扫描描述了电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线。图20A描述了第一次循环。图20B描述了第二次循环。
图21A和图21B以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiPF6的F1S3MN或1ND1N的氧化稳定性。氧化稳定性在室温下测试,工作电极为铂,反电极为锂,参考电极为Li/Li+,且扫描速度为10mV/s。图21B描述了在图21A中所示的相同数据的特写。F1S3MN-LiPF6电解液在7.3V下具有1mA/cm2的电流密度,且1ND1N-LiPF6电解液在7.2V下具有1mA/cm2的电流密度。
图22A和图22B以电流密度(mA/cm2)与电压(V,相对于Li/Li+)的关系曲线描述了具有1M LiTFSI的F1S3MN或1ND1N的氧化稳定性。氧化稳定性在室温下测试,工作电极为铂,反电极为锂,参考电极为Li/Li+,且扫描速度为10mV/s。图22B描述了在图22A中所示的相同数据的特写。F1S3MN-LiTFSI电解液在6.2V下具有1mA/cm2的电流密度,且1ND1N-LiTFSI电解液在6.5V下具有1mA/cm2的电流密度。
1ND1N的热稳定性:
图23描述了纯1ND1N的热稳定性。将1ND1N暴露于30℃-189℃的温度,并通过质谱分析分解产物。1ND1N在高达189℃下未示出液相或气相分解产物。1H NMR显示约5%分解。
图24描述了具有LiPF6的1ND1N的热稳定性。将1ND1N-LiPF6电解液暴露于30℃-150℃的温度并通过质谱分析分解产物。标注了突出峰出现的温度。1ND1N显示气相分解≥ 70℃,但是高达150℃也没有观察到剧烈反应。在125-150℃的温度下出现Me2SiF2 (81m/z)(96g/mol)和在52/53 m/z处的被怀疑为丙烯腈(53g/mol)的峰。在150℃下没有观察到1,4-二噁烷气体。1H NMR分析显示,在125℃下剩余50.6% 1ND1N且在150℃下剩余58% 1ND1N。在125℃下,观察到存在39.7%的氟化产物F1NM1N(相比之下,在未加热样品中为2.3%)、1.6% Me2SiF2(相比之下,在未加热样品中为0%)和2.95%水解物(相比之下,在未加热样品中为5.5%)。在150℃下,观察到存在41%的氟化产物F1NM1N(相比之下,在未加热样品中为2.3%)、1.7% Me2SiF2(相比之下,在未加热样品中为0%)和5.0%水解物(相比之下,在未加热试样中为5.5%)。
为了鉴定在125-150℃下加热1ND1N-LiPF6时在52/53 m/z处观察到的峰,将加热的1ND1N-LiPF6的质谱特征与美国标准与技术研究所(NIST)关于2-丙烯腈和氰化氢的标准物的质谱特征进行比较。图25A描述了如关于图24所描述的质谱特征从24至30m/z的特写。图25B描述了如关于图24所描述的质谱特征从49至55m/z的特写。标注了在图25A和图25B中出现突出峰的温度。在图25B中在51、52和53m/z处的峰表示可能存在丙烯腈。由于在NIST光谱中在26和27m/z处存在峰,HCN的存在不能被无疑地证实或者未证实。在图25A中的光谱显示与27m/z相比在26m/z处的峰强度更大,其支持丙烯腈的存在。然而,在27m/z处的峰的大小大于对于单独的丙烯腈的预期。
图26描述了具有LiTFSI、碳酸亚乙烯酯(VC)和双(草酸根合)硼酸锂(LiBOB)的1ND1N的热稳定性。将1ND1N-LiTFSI-VC-LiBOB暴露于30℃-185℃的温度,并通过质谱分析分解产物。1ND1N-LiTFSI-VC-LiBOB在高达185℃下没有显示出气相分解产物。1H NMR显示,水解从3%(在未加热的样品中)增加到18.7%(加热后),这可能是由于在执行NMR分析之前的延迟。
图27描述了具有LiBF4的1ND1N的热稳定性。将1ND1N-LiBF4暴露于30℃-125℃的温度,并通过质谱分析分解产物。标注了突出峰出现的温度。气相产物在≥ 30℃下释出。如所预期,观察到Me2SiF2 (81m/z)(96g/mol)。未观察到丙烯腈。1H NMR显示,3.7%的水解物和34.2%的氟化产物(3组峰)。19F NMR显示在系统中的所有F都键合到硅。没有留下BF4。没有足够的F来完全分解1ND1N (约5M 1ND1N vs. 4M F)。
虽然通过质谱在加热的1ND1N-LiBF4样品中没有观察到丙烯腈,但在未加热的对照物中观察到(70ppm)。这表明1ND1N在具有LiBF4的情况下在室温下不稳定。NMR分析揭示加热几乎不增加分解,如在下表中所示:
1ND1N在电池中的性能:
图28描述了含有各种电解液的电池在各种充电率下的放电容量。电解液溶剂为:(1) 1ND1N;(2) 具有20%碳酸亚乙酯(EC)共溶剂的1ND1N (1ND1N_EC);和(3) 具有20% EC共溶剂的1ND2 (1ND2_EC)。所有制剂还含有SEI形成添加剂和1M LiPF6盐。如在图28中所示,20% EC共溶剂改善1ND1N的性能。与1ND2相比,在用20%EC共溶剂的情况下,1ND1N在所有充电率下都显示降低的性能。
图29描述了含有各种其他电解液溶剂的电池的放电容量。所述电解液溶剂为:(1) 具有20% EC共溶剂、1M LiPF6和SEI形成添加剂的1ND1N (1ND1N-EC-LiPF6,在图29中示为1ND1N_EC);(2) 具有20% EC共溶剂、1M LiTFSI和SEI形成添加剂的1ND1N (1ND1N-EC-LiTFSI,在图29中示为1ND1N_T);及(3) 具有20% EC共溶剂、1M LiPF6和SEI形成添加剂的1ND2 (1ND2-EC-LiPF6,在图29中示为CP597-07)。1ND1N-EC-LiPF6组合和1ND1N-EC-LiTFSI组合显示与1ND2-EC-LiPF6组合相媲美的性能。
图30A和图30B分别描述了在各种充电率下含有基于1ND1N-LiPF6的电解液或基于1ND1N-LiTFSI的电解液的电池的放电容量。对于每个实验,使用具有Saft America (Cockeysville, MD) NCA阴极、石墨阳极和来自Celgard, LLC (Charlotte, NC)的2500隔板的CR2032纽扣电池。电池用恒定电流/恒定电压(CCCV)程序以C/5、C/2、1C或2C充电率充电到4.1V。电池以与充电相同的倍率在恒定电流下每个循环放电到3.0V。在图30A中,基于1ND1N-LiPF6的电解液溶液包含1M LiPF6和1ND1N(批次ZP780-01),且在30℃或55℃下执行充电/放电。在图30B中,基于1ND1N-LiTFSI的电解液溶液包含1M LiTFSI和1ND1N(批次ZT781-01),并且在30℃、55℃或70℃下执行充电/放电。如在图30A和38B中所示,基于1ND1N-LiTFSI的电解液表现出比基于1ND1N-LiPF6电解液好的倍率性能(rate capability)。
OS溶剂和电解液溶液的物理性质:
上表1示出了作为纯溶剂或配制的电解液溶液形式的所选有机硅(OS)化合物(1S3MN、F1S3MN、F1S3cMN、DF1S3MN、DF1S2MN和F1S3M2)的物理性质。上表2示出纯1ND2、1ND1、1ND1N F1S3MN、DPF1S3MN、F1S3MC、F1S3ME、F1S3MA、F1S3MS、1NMS、F1S3M2和F1S3cMN和含有它们的各种电解液组合物的物理性质。在两个表中,电导率的单位为mS/cm,粘度的单位为cP,且闪点是以摄氏度计。
在CDCl3中得到的1ND1N、1ND1N、DF1S2MN、DF1S3MN、F1S3cMN和1S3MN的质子(1H) NMR谱分别提供于图31-36。对于所选含有氟原子的化合物,19F-NMR数据在CDCl3和DMSO-d6中收集。结果如下:
在CDCl3中的19F-NMR
在DMSO-d6中的19F-NMR
OS电解液溶液的热稳定性:
图37和图38描述了具有1M LiPF6的F1S3MC、F1S3cMN、1S3DN DF1S2MN、TF1S2MN和TF1S3MN的气相分解。将每种电解液暴露于30℃-175℃的温度(在每个步骤60分钟),并分别通过电子轰击质谱(EI-MS)和核磁共振光谱(NMR)分析气体和液体分解产物。高达175℃时没有观察到显著的气相和/或液相分解。
结论:
F1S3MN和1ND1N两者都适合用作锂离子电池中的电解液溶剂。已经证明F1S3MN和DF1S2MN充当在EDLC装置中的电解液溶剂。F1S3MN对于所测试的所有盐显示很高的热稳定性(通过1 H-NMR测量)。F1S3MN在具有LiPF6的任何OS中显示最高热稳定性(175℃),没有观察到分解。F1S3MN作为纯溶剂、与LiBF4一起和与LiTFSI一起产生气相产物。这些气相产物可归因于低水平的F1S3MN蒸发。F1S3MN显示与F1S3M2相比较增加的电压稳定性(在宽窗中的较高氧化电位)。在高达4C的充电率下,F1S3MN仍提供与EPA6相当的性能。LiBOB在F1S3MN中在没有共溶剂的情况下具有有限的溶解度(< 0.03M),但在使用共溶剂(即,20%EC)的情况下,LiBOB溶解度改善(> 0.1M)。F1S3MN的分解产物为Me2SiF2和MeSiF3,这两者都是气体。
1ND1N作为纯溶剂或与LiTFSI电解液组合时高达185-190℃也没有显示出气相分解。1ND1N与LiTFSI电解液的组合在高达70℃和更高下表现出是有希望的。具有LiPF6的1ND1N比具有LiPF6的1ND1或1ND2更加热稳定。它在高于125℃时形成丙烯腈。与其他无间隔基化合物一样,1ND1N在室温下与LiBF4反应。然而,没有足够的F来完全分解1ND1N,并且它不形成丙烯腈。1ND1N的倍率性能略低于1ND2。
F1S3MN、DPF1S3MN和F1S3MC都适合用作锂离子电池中的电解液溶剂。证明F1S3MC、F1S3cMN、1S3DN、DF1S2MN、TF1S2MN和TF1S3MN与LiPF6热稳定(175℃),没有观察到分解。
- 还没有人留言评论。精彩留言会获得点赞!