卡洛芬的合成方法与流程
本发明涉及生产卡洛芬及其衍生物的方法。
发明背景
卡洛芬属于包括布洛芬、萘普生和酮洛芬在内的丙酸类非甾体抗炎药(NSAID)。卡洛芬通过抑制COX-2和其他炎症生物靶源来减轻炎症。卡洛芬已经被FDA批准用作兽药,其用于缓解骨关节炎相关的疼痛和炎症和用于控制狗软组织和整形手术相关的术后疼痛。卡洛芬以商品名Rimadyl、Novox和Veprofen,每片或囊片25mg、75mg和100mg卡洛芬的剂量提供。
卡洛芬为白色、结晶化合物。卡洛芬25℃时几乎不溶于水,但易溶于乙醇。卡洛芬为取代咔唑,6-氯-α-甲基-9H-咔唑-2-乙酸的非专利名称。经验式为C15H12ClNO2,分子量273.72。卡洛芬的化学结构为:
卡洛芬的合成首先于20世纪70年代在美国专利号4,158,007中由霍夫曼罗氏有限公司(Hoffmann-La Roche Inc.)的一组科学家报道。根据该专利,可以使用环己烯酮和2-甲基丙二酸作为起始材料制备卡洛芬,总结如下:
起始材料环己烯酮昂贵。作为参考点,西格玛-奥德里奇公司(Sigma-Aldrich)以每0.1升120.50美元将其出售。因此,上面的方法不适合工业规模生产卡洛芬。
之后在EP 0,151,423中霍夫曼罗氏有限公司的科学家报道了他们认为更好的通过从咔唑起始制备卡洛芬的方法,总结如下:
然而,该方法也不适合工业生产,因为其需要使用有毒的三甲基硅烷基氰化物作为试剂,并且其在氯化反应步骤及随后步骤期间形成同分异构体。此外,咔唑为昂贵的起始材料,目前西格玛-奥德里奇公司的售价为每0.2克111.50美元。
此后在EP 0,247,222中霍夫曼罗氏有限公司的科学家报道了制备卡洛芬及其衍生物的不同的,改进的化学方法,其也从咔唑起始。
上面的化学方法消除了使用有毒的三甲基硅烷基氰化物的需求。如CN 101492412中报道,该化学方法由一组中国科学家进一步优化。然而,该化学方法仍不理想,因为其中的氯化反应步骤太复杂,其需要在分子两个特异位点进行区域选择性氯化反应。
美国专利号4,150,031和4,264,500中报道了制备卡洛芬的一些其他合成途径。类似的,它们也使用咔唑或其衍生物作为起始材料。例如,美国专利号4,150,031公开了:
上面方法冗长的合成步骤和严格的反应条件使其不适合大规模生产。
WO 87/00519公开了从7-氯-3-溴咔唑开始通过偶联反应和氧化反应制备卡洛芬的方法,如下所示:
该方法对工业生产卡洛芬不实际,因为起始材料,7-氯-3-溴咔唑,自身需要多步骤合成。
因此,工业上有需要提供经济,高效并可行的,适用于工业规模生产的卡洛芬制备方法。
发明简述
一方面,本发明提供了制备卡洛芬及衍生物的新方法,其环境友好,经济,且工业可行。
根据一个实施方式,卡洛芬及其类似物可以通过以下步骤制备:
(1)在有或无酸性催化剂下,在第一有机溶剂中加热环己酮和第一碱以形成烯胺;其中第一碱为有机二级碱,其可以为,但不限于,吡咯烷或哌啶;其中酸性催化剂可以包括,但不限于,甲苯磺酸、分子筛、和大孔磺酸树脂;
(2)在第二有机溶剂中使烯胺与α-取代羧酸酯反应以提供2-取代的环己酮;
(3)在第二碱存在下使2-取代环己酮与甲酸酯反应以形成2,2’-双取代环己酮;其中第二碱可以为,但不限于,甲醇钠。
(4)第三有机溶剂中的2,2’-双取代环己酮与取代苯基重氮盐的水溶液反应以提供取代肼化合物,其中肼化合物的一个氮与取代的苯基基团相连,而肼化合物另一个氮与环己酮环相连,形成“-N=C-C-(O)-”结合物;其中取代苯基重氮盐可以为,但不限于,其盐酸盐;以及
(5)对取代肼化合物施加Fisher吲哚反应条件以形成三环化合物,随后非吲哚环芳构化以形成卡洛芬或其衍生物。酸性催化剂,如盐酸或硫酸,可以用于环闭合和芳构化反应中。
根据另一个实施方式,卡洛芬及其类似物可以通过以下步骤制备:
(1)按照之前实施方式的第一步从环己酮形成烯胺;
(2)可选地用催化剂,在有机溶剂中使烯胺与α取代羧酸酯反应,随后碱处理,以提供2-取代环己酮烯胺;其中催化剂可以为,但不限于,碘化钠;且其中碱可以为,但不限于DIEA(二异丙基乙胺),DBU(1,8-二氮杂双环[5.4.0]十一碳-7-烯),氢化钠,钠醇盐,碳酸钾或碳酸铯。
(3)使有机溶剂中的2-取代环己酮烯胺与取代苯基重氮盐的水溶液反应以提供取代腙盐,其中腙盐的一个氮与取代的苯基基团相连,而腙盐另一个氮与环己酮烯胺环相连,形成“-N=C-C-(O)-”结合物;其中取代苯基重氮盐可以为,但不限于,四氟硼酸盐;以及
(4)对取代的腙盐施加Fisher吲哚反应条件以形成三环化合物,随后非吲哚环芳构化以使得三环均芳构化并结合。酸性催化剂可以用于Fisher吲哚反应中。适合的酸性催化剂包括,但不限于,多磷酸,甲磺酸中7.7重量%的五氧化二磷,以及二噁烷中的浓硫酸。可以通过加入有机盐并在200-200℃对得到的混合物加热来进行芳构化反应。有机盐可以为,但不限于,盐酸吡啶。芳构化反应期间,烯胺功能基团被同时去除,这最终得到了卡洛芬或其衍生物。
本方法的一个优点为起始材料环己酮便宜且容易获得。
本方法的另一个优点为可以避开对过程中形成的许多中间体的纯化。也就是说,中间体可以不纯化直接用于随后的反应。因此,本方法是高效的。
进一步的优点为本方法安全且环境友好。本方法没有危险的副产品产生。
本发明的方法适合制备卡洛芬类似物(即衍生物)。
另一个方面,本发明提供了上述方法制备的新化学品和中间体。
附图简述
图1显示了根据本发明一个实施方式制备卡洛芬的一种合成方案。
图2显示了根据本发明另一个实施方式制备卡洛芬的另一种合成方案。
图3显示了根据本发明一个进一步的实施方式制备卡洛芬的又另一种合成方案。
发明详述
一方面,本发明提供了从环己酮或其衍生物制备卡洛芬的新方法。
根据图1(方案1),在有或没有酸性催化剂,如甲苯磺酸、分子筛、和大孔磺酸树脂存在下,通过加热回流有机溶剂,如甲苯,环己烷和苯中的式1的环己酮或其衍生物或其他环酮与0.5-2当量(“equiv”)式2的吡咯烷或哌啶或其他环或无环有机二级碱的混合物,合成式3的烯胺来起始合成过程。作为例子,图1显示了通过使用哌啶作为二级碱,甲苯作为溶剂,和分子筛作为催化剂来制备式3的烯胺。
随后用式4的α-取代羧酸酯(更具体地,α-取代丙酸酯)在有机溶剂,如甲醇或乙腈中加热下处理式3的烯胺,以提供式5的2-取代环己酮。R1可以选自包含氢,甲基,乙基,和C3-C6烷基的组;而X可以为氯或溴。
随后在低温,式6的甲酸酯存在下,通过与碱,如甲醇钠反应,将式5的2-取代环己酮转化为式7的化合物。以式9的重氮盐水溶液处理式7的化合物在有机溶剂如甲醇中的溶液以提供式10的化合物。通过将式8的苯胺类似物与盐酸和水中的亚硝酸钠反应,从式8的苯胺类似物(如苯胺,4-氯苯胺,4-溴苯胺,或3-氯苯胺)制备式9的重氮盐。如图1中所示,优选的苯胺为4-氯苯胺。优选地,取代苯基重氮盐新鲜制备并立即使用。尽管式9的重氮盐被显示为盐酸盐,本方法并不局限于盐酸盐,可以使用其他取代苯基重氮盐。因此,适合的重氮盐可以表示为式9’,其中Y可以为任何种类的典型阴离子,包括氯和四氟硼酸阴离子:
随后在有或没有酸性催化剂,如盐酸或硫酸存在下,在Fisher吲哚合成条件下,通过加热有机溶剂,如乙腈,乙酸,甲醇,或二噁烷中的式10的化合物混合物来将式10的化合物转化为式11的化合物。加热式11的化合物与有机盐,如盐酸吡啶的混合物,来以高产率提供卡洛芬。
本领域技术人员会理解可以使用其他类似的起始材料和试剂来在相同的条件下进行方案1的方法,这得到具有相同核心骨架的不同中间体(如式10’和11’的化合物)。例如,代替环己酮1,可以使用其他酮(其可能得到式10’和11’的化合物中不同的“n”);替代式4的α-取代丙酸甲酯,可以使用不同羧酸的不同酯(其可能得到式10’和11’的化合物中不同的R1,R3);和替代式8的苯胺,可以使用苯环上有不同取代基的任何苯胺化合物(其可能得到式10’和11’的化合物中不同的R2)。因此,本方法适合制备式10’的化合物和式11’的化合物:
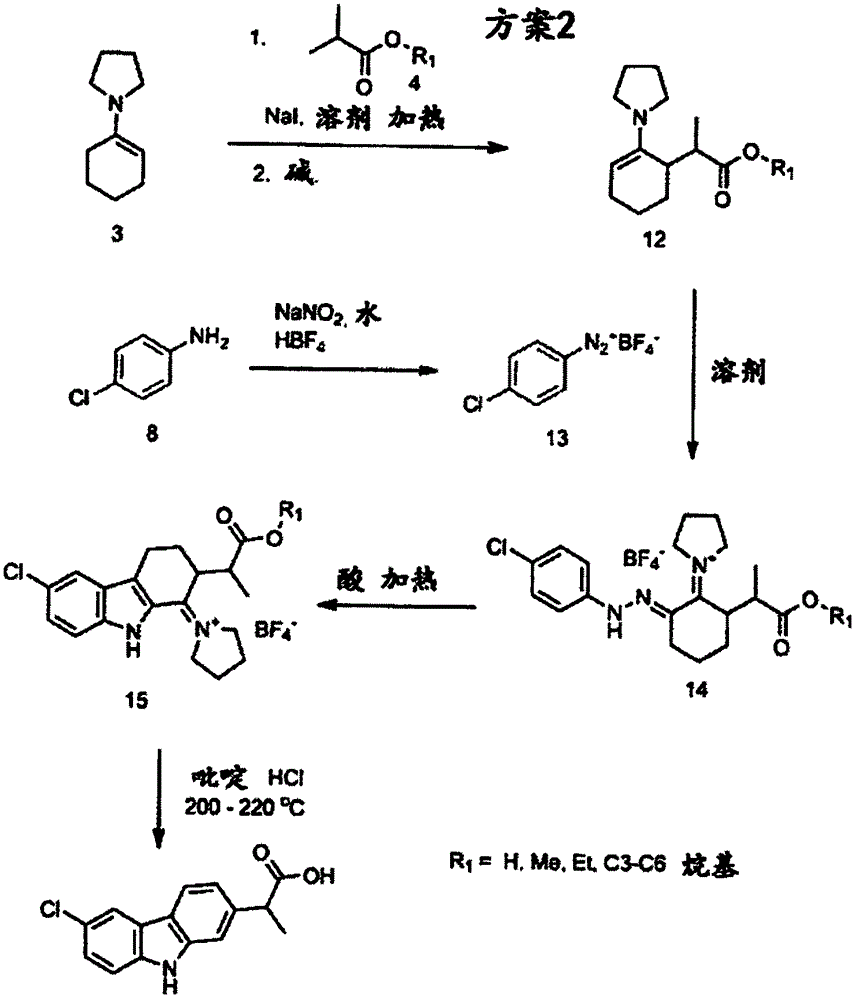
根据图2(方案2),在有或没有催化剂,如碘化钠存在下,加热下在有机溶剂,如甲醇、甲苯、二噁烷、THF、或乙腈中将式4的α-取代羧酸(丙酸)酯与式3的烯胺反应,随后直接用碱,如DIEA,DBU,氢化钠,钠醇盐,碳酸钾或碳酸铯处理,来提供式12的烯胺。可以如方案1所述,从回流式1的环己酮和式2的吡咯烷的混合物来制备式3的烯胺。作为例子,图2显示了通过以乙腈作为溶剂和以碘化钠作为催化剂来制备式12的烯胺。
随后在低温下,以有机溶剂如THF(四氢呋喃)或乙腈中的式13的重氮盐处理式12的烯胺,以提供式14的化合物。在环境温度下,以酸性水介质中的亚硝酸钠,HBF4(四氟硼酸)处理式8的苯胺或其类似物之一,从式8的苯胺或其类似物之一(如苯胺,4-氯苯胺,4-溴苯胺,3-氯苯胺)制备式13的重氮盐。发现重氮化合物的四氟硼盐很稳定。尽管式14的重氮盐被显示为四硼酸盐,本方法并不局限于产生四硼酸盐。可以从使用为式9’的化合物,其中Y可以为任何种类的典型阴离子,包括氯和四氟硼酸阴离子,来生产其他重氮苯基重氮盐,如式14”的化合物:
随后在Fisher吲哚条件下,通过将其在无水和酸性环境,如PPA(多磷酸),Eaton’s试剂(甲磺酸中7.7重量%的五氧化二磷),二噁烷中的浓硫酸中加热,将式14的化合物转化为式15的三环化合物。可选地,式11的化合物在Fisher吲哚条件下可以从式14的化合物形成(图中未显示)。注意无水非质子和酸性溶剂和试剂条件更可能将式14的化合物转化为式15的化合物;酸性水溶剂系统,如盐酸,更可能将式14的化合物转化为式11的化合物。其他酸性质子溶剂系统,如无水乙酸,提供式11和15的化合物的混合物。尽管式15的化合物被显示为四氟硼酸盐,本方法并不局限于产生三环化合物的四氟硼酸盐。取决于所用的式9’和14”的化合物,可以产生三环化合物的其他盐。因此,三环化合物的适合盐可以表示为式15”,其中Y可以为任何种类的典型阴离子,包括氯和四氟硼酸阴离子:
随后通过将其与有机盐,如盐酸吡啶在例如200-220℃加热,将式11和/或15的化合物转化为卡洛芬。
本领域技术人员会理解可以使用其他类似的起始材料和试剂来在相同的条件下进行方案2的方法,这得到具有相同核心骨架的不同中间体(如式14’和15’的化合物)。例如,代替环己酮烯胺3,可以使用其他烯胺(其可能得到式14’和15’的化合物中不同的“n”);替代式4的α-取代丙酸甲酯,可以使用不同羧酸的不同酯(其可能得到式14’和15’的化合物中不同的R1,R3);和替代式8的苯胺,可以使用苯环上有不同取代基的任何苯胺化合物(其可能得到式14’和15’的化合物中不同的R2)。因此,本方法适合制备式14’和式15’的化合物:
图3(方案3)显示了又另一个,更简化的,卡洛芬合成化学方法。方案3与方案2的主要区别在于过程中方案3的数种中间体不被分离和/或纯化;而是将粗制中间体直接用于随后的反应。结果是,其非常类似“一锅””反应。进行方案3的方法更简单且更高效。在方案3中,方括号用于表示其中所含式在其被用于下一反应步骤前没有被分离和/或纯化。
本方法以在有或没有酸性催化剂存在下,环己酮1或其衍生物或其他环酮与0.5-2当量的吡咯烷2或哌啶或其他环或无环有机二级碱在有机溶剂,如甲苯,环己烷或苯中的反应起始。酸性催化剂可以为甲苯磺酸,分子筛,或大孔磺酸树脂。作为例子,图3显示了通过使用哌啶作为二级碱和甲苯作为溶剂来制备式3的烯胺。反应完成后,简单过滤并蒸发反应混合物提供粗制式3的烯胺,其可以不做进一步纯化直接用于下一反应步骤。
不做进一步纯化,在有或没有催化剂,如碘化钠存在下,加热下在有机溶剂,如甲醇或乙腈中以式4的α-取代羧酸酯处理粗制式3的烯胺,随后直接用碱,如DIEA、DBU、氢化钠、钠醇盐、碳酸钾或碳酸铯处理,之后简单蒸发来提供粗制式12的烯胺。
随后不做进一步纯化,通过在低温下,无水溶剂加乙腈或THF中与式13的重氮盐的四氟硼盐反应,将粗制式12的烯胺直接转化为式14的化合物。按方案2所述,从式8的苯胺或其类似物制备式13的重氮盐。通过简单的有机溶剂萃取,水洗和蒸发获得粗制式14的化合物。
不做进一步纯化,在酸性溶剂系统,如浓HCl-AcOH(盐酸-乙酸),浓HCl-二噁烷(盐酸-二噁烷),HCl-MeOH(盐酸-甲醇),6MH2SO4-二噁烷(硫酸-二噁烷),和浓H2SO4-二噁烷中直接加热处理粗制式14的化合物以提供式11或15的粗制化合物。不做进一步纯化,以方案2所述相同的方式将式11或15的粗制化合物转化为卡洛芬。
如方案1-3所描述,本发明一个特征为使用环己酮作为起始材料。环己酮可以以合理的价格容易地获得。西格玛-奥德里奇公司以每2升105.00美元将其出售。更大量的环己酮可以以更便宜的单位价格购得。
本发明的另一个特征为方法被设计成使得,大多数中间体在随后反应中使用其之前的纯化是可选的。结果是,可以进行连续的反应步骤,一步接一步,而不纯化其中形成的中间体,这极大简化了方法并得到更高的产率。这样的反应步骤甚至可以在一个反应容器中进行,使其成为“一锅”反应。
术语“纯化”,意思为使用常规纯化方法,如结晶或经闪蒸塔层析,来获得有适合纯度的化合物。本领域技术人员会理解常规纯化方法耗时并经常引起产率损失来交换较高纯度。然而,中间体的纯化经常是必需的,因为中间体中所含杂质经常干扰中间体随后的反应(使随后的反应不能进行或产率低)。如本申请所公开的设计良好的反应方案,有利地规避了纯化多种中间体的需要,由此得到有高产率的高效方法。
如上面阐述的,方案3中粗制式3的烯胺,粗制式12的化合物,和粗制式14的化合物均可以通过简单操作程序(如反应混合物的萃取,清洗,过滤或蒸发)来获得,并随后分别直接用于下一反应步骤。同样地,方案1中粗制式3的烯胺,粗制式10的化合物,和其他化合物也可以不纯化分别直接用于下一反应步骤。应注意操作程序不被认为是纯化,因为操作程序后的粗制产物仍然含有有机杂质,并也可能有某些无机杂质。
本发明的方法也适合制备卡洛芬衍生物。例如,上面方法中使用的重氮盐化合物衍生自取代苯基胺。取决于要制备的卡洛芬衍生物类型,可以简单地选择不同的取代苯基胺以制备重氮盐。在某些实施方式中,取代苯基胺可以为苯胺,4-氯苯胺,4-溴苯胺,或3-氯苯胺。作为另一个例子,可以使用不同的α-取代羧酸酯与烯胺反应以得到不同的卡洛芬类似物。作为又另一个例子,也可通过以不同的酮作为起始材料来获得不同类型的烯胺。
图3显示了本方法中所用试剂的某些变化。X可以为氯或溴;而碱可以为氢化钠,NaOR1,DIEA,三乙胺,或DBU;且R1可以为氢,甲基,乙基,或C3-C6烷基。
另一方面,本发明提供了通过上述方法制备的化学品,中间体,及其衍生物。化学品,中间体,及其衍生物可用于制备卡洛芬及其衍生物。这样的化学品或中间体包括,不限于,有下列结构的式:
式10’的化合物:
当R2=4-Cl,R3=Me,n=1时,上述结构代表式10的化合物。
式11’的化合物:
当R2=4-Cl,R3=Me,n=1时,上述结构代表式11的化合物。
式14’的化合物:
当R2=4-Cl,R3=Me,n=1时,上述结构代表式14的化合物。
式15’的化合物:
当R2=4-Cl,R3=Me,n=1时,上述结构代表式15的化合物。
实施例
实施例1:1-(环己-1-烯-1-基)吡咯烷
向装有冷凝器和Dean-stark头的10L反应器中加入环己酮(1449g),吡咯烷(1kg),大孔磺酸树脂(50g)和甲苯(1.5L)。加热回流反应混合物并通过GC监测。回流7h后,GC显示反应完成。随后通过蒸发去除甲苯,并真空蒸馏残余物以提供1-(环己-1-烯-1-基)吡咯烷,1673g(78.6%),1HNMR(CDCl3):δ4.26(s,1H),2.97(s,4H),2.16(s,2H),2.07(s,2H),1.80(s,4H),1.66(m,2H),1.53(m,2H)。
实施例2:2-(2-氧环己基)丙酸甲酯
向含有1-(环己-1-烯-1-基)吡咯烷(50g)和甲醇(500mL)的1L圆底瓶中加入2-溴丙酸乙酯(60g)。搅拌的同时加热回流反应混合物29小时。随后向反应混合物中加入水(60mL)。再继续回流1小时。随后冷却混合物至室温,之后以水(500mL)稀释。以甲基叔丁基醚(MTBE)(2×300mL)萃取所得混合物。合并有机萃取物,以盐水清洗,硫酸钠上干燥并过滤。浓缩滤液。真空蒸馏粗制产品以得到纯产品,7g(11.5%)。1HNMR(CDCl3):δ3.66(d,3H),2.71-2.59(mm,2H),2.32(m,2H),2.03(m,2H),1.86(m,1H),1.62(m,2H),1.32(m,1H),1.12-1.05(dd,3H)。MS(ESI)m/z 185.2[M+1]+。
实施例3:2(3-甲酰基-2-氧环己基)丙酸甲酯
在氮气中向250ml圆底瓶中装入氢化钠(2.2g,矿物油中60%),随后室温下搅拌同时滴加THF(50mL)。随后以使得内部温度控制在25℃以下的速度向混合物中缓慢加入THF(10mL)中的2-(2-氧环己基)丙酸甲酯(5g),甲酸甲基酯(1.6g),和无水甲醇(0.1mL)溶液。加入后,反应混合物在室温下再搅拌5小时,随后以水(100mL)处理。以甲基叔丁基醚(MTBE)(2×50mL)萃取所得混合物2次。合并有机萃取物,以盐水清洗,硫酸钠上干燥,过滤并浓缩。随后通过柱层析纯化粗制产品(石油醚∶乙酸乙酯=10∶1)以得到1g(17%)纯产品。1HNMR(CDCl3):δ8.26(dd,1H),3.65(d,3H),3.10(m,1H),2.90-2.75(mm,1H),2.26(m,2H),1.86(m,2H),1.72(m,1H),1.47(m,2H),1.18-1.05(dd,3H)。MS(ESI)m/z 213.1[M+1]+。
实施例4:(E)-2-(3-(2-(4-氯苯基)腙)-2-氧环己基)丙酸甲酯
向100mL瓶中装入盐酸(6N,1.5mL)和4-氯苯胺(0.9g)。在室温下搅拌混合物直至形成澄清的溶液,随后在冰水浴中冷却至约0℃的内部温度。随后滴加亚硝酸钠(0.5g)在水(3mL)中的溶液。滴加完成后在0℃再搅拌混合物30分钟。
在含2-(3-甲酸基-2-氧环己基)丙酸甲酯(1.3g)在甲醇(7mL)中的溶液的另一个瓶中,搅拌的同时以乙酸钠(1.3g)在水(6mL)中的溶液处理。该混合物随后冷却至0℃,之后缓慢加入上述4-氯苯基重氮氯混合物。随后在0℃再搅拌混合物2.5小时。随后以水处理得到的混合物。随后去除水层,油状半固体不做进一步纯化直接用于下一步骤。MS(ESI)m/z 323.1[M+1]+。
实施例5:2-(6-氯-1-氧代-2,3,4,9-四氢-1H-咔唑-2-基)丙酸
加热回流实施例4中的粗制产品,乙酸(10mL),和浓盐酸(2mL)的混合物1小时。随后在搅拌的同时将反应混合物倒入冰-水(50mL)中。通过过滤收集黄色固体。随后随搅拌以丙酮(2mL)处理黄色固体产品直至混合物变成均一的悬液。随后通过过滤收集固体悬浮物并以最少的冷丙酮洗两次以得到固体产品,0.4g(从(E)-2-(3-(2-(4-氯苯基)腙)-2-氧环己基)丙酸甲酯计22.4%)。_1HNMR(DMSO-d6):δ12.18(S,1H),11.73(s,1H),7.72(d,1H),7.37(d,1H),7.26(d,1H),3.04(m,3H),2.86(m,1H),2.10(m,1H),2.00(m,1H),1.01(d,3H)。MS(ESI)m/z 292.1[M+1]+。
实施例6:从2-(6-氯-1-氧代-2,3,4,9-四氢-1H-咔唑-2-基)丙酸制备卡洛芬
将2-(6-氯-1-氧代-2,3,4,9-四氢-1H-咔唑-2-基)丙酸(0.38g,实施例5)和盐酸吡啶(0.36g)的混合物在氮气中加热到230℃持续3小时。随后在仍然热时以冰水处理混合物,之后在室温下搅拌20分钟。以甲基叔丁基醚(MTBE)(2×50mL)萃取所得混合物2次。在硫酸钠上干燥合并的MTBE萃取物,过滤,并蒸发以得到标题产品。1HNMR(DMSO-d6):δ12.26(s,1H),11.32(s,1H),8.14(d,1H),8.05(d,1H),7.45(d,1H),7.37(s,1H),7.32(d,1H),7.07(d,1H),3.79(t,1H),1.40(d,3H)。MS(ESI)m/z 274.1[M+1]+。
实施例7:2-(2-(吡咯-1-基)环己-2-烯-1-基)丙酸甲酯
向1L圆底瓶中加入1-(环己-1-烯-1-基)吡咯烷(100g,实施例1),乙腈(800mL),2-溴丙酸甲酯(116.3g),和碘化钠(9.9g)。随后在搅拌的同时加热回流混合物20小时。将混合物冷却至室温,加入DBU(100.3g),并搅拌30分钟。随后将得到的混合物浓缩以去除大部分乙腈,之后加入甲苯(500mL)。在室温下搅拌悬液1小时随后过滤。以小量甲苯洗滤饼2次。真空蒸馏合并的甲苯溶液以得到液体产品,143.4g(91.4%)。1HNMR(CDCI3):δ4.64(t,1H),3.60(s,3H),2.99(q,2H),2.73(m,2H),2.56(m,2H),2.05(m,1H),1.97(m,1H),1.73(m,5H),1.54(m,3H),1.01(d,3H)。
实施例7:4-氯苯基重氮四氟硼酸盐
向250mL圆底瓶中加入四氟硼酸溶液(34mL,50%)和水(40mL)。搅拌混合物并以冷水浴冷却至约10℃,随后加入4-氯苯胺(12.7g)。在相同的温度下搅拌混合物直至形成澄清的溶液。随后在冰-水浴中将得到的溶液冷却至约0℃。以使内部温度低于10℃的速度滴加亚硝酸钠(6.9g)在水(15mL)中的溶液。加入后,在冰-水浴中再搅拌混合物30分钟。通过过滤收集白色固体。将粗制固体产品溶于丙酮(50mL),随后以冷乙醚(100mL)处理。再次通过过滤收集固体,并以冷丙酮(2×50mL)洗两次以得到白色纯固体产品,19g(84.1%)。1HNMR(DMSO-d6):δ8.62(d,2H),8.04(d,2H)。MS(ESI)m/z 139.0[M+1]+。
实施例8:(E)-1-(2-(2-(4-氯苯基)腙)-6-(1-甲氧基-1-氧代丙-2-基)亚环己基)吡咯-1-鎓四氟硼酸盐
二氯甲烷(165mL)中的2-(2-(吡咯-1-基)环己-2-烯-1-基)丙酸甲酯(5g,实施例7)溶液在干冰-丙酮浴中冷却至约-78℃,随后在搅拌的同时分批加入4-氯苯基重氮四氟硼酸盐(4.8g)。加入后,通过去掉干冰-丙酮浴将混合物在2小时内温暖至室温。随搅拌以稀盐酸(1M,100mL)处理混合物。以二氯甲烷(2×100mL)萃取所得溶液2次。浓缩合并的二氯甲烷相以得到粗制油状产品。通过柱层析来纯化粗制产品,使用4∶1,1∶1,1∶2比率的石油醚-乙酸乙酯作为洗脱液和纯乙酸乙酮顺序洗脱硅胶柱以得到纯产品,2.2g(22.6%)。1HNMR(CDCl3):δ9.44(s,1H),7.21(s,4H),4.24(m,2H),4.12(m,2H),3.62(s,3H),3.32(m,1H),2.81(m,2H),2.64(m,1H),2.11(m,6H),1.80(m,2H),1.15(d,3H)。MS(ESI)m/z 376.2[M]+。
实施例9:1-(6-氯-2-(1-甲氧基-1-氧代丙-2-基)-2,3,4,9-四氢-1H-咔唑-1-亚基)吡咯-1-鎓四氟硼酸盐
向装有温度计,机械搅拌器,和带冷凝器的Dean-Stark头的500mL三颈圆底瓶中装入多磷酸(PPA,300mL)。随后在搅拌的同时将PPA加热至120℃。缓慢加入(E)-1-(2-(2-(4-氯苯基)腙)-6-(1-甲氧基-1-氧代丙-2-基)亚环己基)吡咯-1-鎓四氟硼酸盐(34.6g)在二氯甲烷(100mL)中的溶液。加入期间,二氯甲烷被蒸发并通过带冷凝器的Dean-Stark头收集。加入后,继续加热到约120℃持续3小时。以热水(800mL)处理热液体混合物以形成溶液。随后以二氯甲烷(5×300mL)萃取所得溶液。以水(500mL)洗合并的有机相,在硫酸钠上干燥,过滤并蒸发以提供9g粗制产品(27.1%),其通过柱层析纯化(石油醚/乙酸乙酯(1∶1)随后乙酸乙酯顺序洗脱)以得到纯产品。1HNMR(CDCl3):δ10.20(s,1H),7.56(dd,1H),7.43(d,1H),7.30(d,1H),4.60(m,1H),4.40(m,1H),4.20(m,1H),4.15(m,1H),4.00(m,1H),3.69-3.61(d,3H),3.05(m,1H),2.73(m,2H),2.40(m,2H),2.16(m,4H),1.36-1.22(dd,3H)。MS(ESI)m/z 359.1[M]+。
实施例10:从1-(6-氯-2-(1-甲氧基-1-氧代丙-2-基)-2,3,4,9-四氢-1H-咔唑-1-亚基)吡咯-1-鎓四氟硼酸盐(实施例9)制备卡洛芬
1-(6-氯-2-(1-甲氧基-1-氧丙-2-基)-2,3,4,9-四氢-1H-咔唑-1-亚基)吡咯-1-鎓四氟硼酸盐(1g,实施例9)和盐酸吡啶的混合物在氮气中加热至200℃持续1小时。当热时将混合物溶于甲醇(50mL)中,随后以水(50mL)处理。通过过滤收集固体以提供粗制产品。将粗制产品溶于乙酸乙酯(100mL)中,随后以水(100mL)洗。通过过滤去除不溶悬浮物。随后分离有机相,以盐水洗,在硫酸钠上干燥并蒸发以得到标题化合物,0.5g(81.8%)。MS(ESI)m/z 274.10[M]+。
实施例11:不纯化中间体直接从1-(环己-1-烯-1-基)吡咯烷(实施例1)制备卡洛芬
向250mL反应器中加入1-(环己-1-烯-1-基)吡咯烷(50g,实施例1),2-溴丙酸甲酯(55.2g),2.5g碘化钠和100mL乙腈。随后加热回流混合物16.5小时,之后在冰-水浴中冷却至0℃。随后分批加入19.6g甲醇钠使得内部温度保持在低于5℃。加入后,在0℃继续搅拌1小时,随后旋转蒸发去除所有乙腈。随后向残余物中加入120mL甲苯,并在室温搅拌得到的混合物20分钟。随后通过过滤去除固体并以2×15mL甲苯洗2次。合并甲苯滤液和洗液,并蒸发去除所有甲苯以提供80g粗制产品,2-(2-(吡咯-1-基)环己-2-烯-1-基)丙酸甲酯,其直接用于下一步骤。
向500mL反应器中加入上述粗制中间体(80g)和160mL THF。搅拌的同时将溶液冷却至0℃,随后以使内部温度<5℃的速度分批加入4-氯苯基重氮四氟硼酸盐(45.8g,实施例7)。加入后在0℃搅拌混合物1小时,随后在室温下搅拌1小时。蒸发反应混合物以去除所有THF以得到深色油状产品,(E)-1-(2-(2-(4-氯苯基)腙)-6-(1-甲氧基-1-氧代丙-2-基)亚环己基)吡咯-1-鎓四氟硼酸盐,其直接用于下一步骤。
向上述含(E)-1-(2-(2-(4-氯苯基)腙)-6-(1-甲氧基-1-氧代丙-2-基)亚环己基)吡咯-1-鎓四氟硼酸盐的反应器中装入224mL甲醇和56mL浓盐酸。加热回流混合物12小时。随后将反应混合物冷却到室温,之后搅拌30分钟。通过过滤收集固体并以3×15mL乙醇洗2次以得到白色固体产品,2-(6-氯-1-氧代-2,3,4,9-四氢-1H-咔唑-1-基)丙酸甲酯,15.2g(从1-(环己-1-烯-1-基)吡咯烷计15.0%)。1HNMR(DMSO-d6):δ11.75(s,1H),7.72(s,1H),7.37(d,1H),7.28(d,1H),7.61(s,3H),3.04(m,3H),2.86(m,1H),2.12(m,1H),2.01(m,1H),1.05(d,3H)。MS(ESI)m/z 306.1[M]+。
向100mL反应器中装入上述2-(6-氯-1-氧代-2,3,4,9-四氢-1H-咔唑-2-基)丙酸甲酯(10g)和盐酸吡啶(11.4g),向瓶中充入氮气以从瓶中去除氧气,随后加热到200℃持续3小时。当仍然热时将混合物倒入100mL冰水中。以30mL甲醇洗反应器,并将甲醇溶液也倒入冰水混合物中。随后在室温搅拌混合物30分钟。通过过滤收集固体并以水(2×30mL)洗两次,在真空加热烘干。
在室温下将上述粗制卡洛芬(9.5g)与50mL丙酮搅拌以制成澄清溶液,随后滴加三甲胺(7g,2当量)。加入后继续搅拌1小时。通过过滤收集固体并加热干燥以得到9.4g卡洛芬三甲胺盐。
在搅拌的同时,将100mL水中上述9.4g盐混合物加热到60℃。在搅拌的同时,加热到60℃10分钟后混合物仍为悬液。向该悬液中加入15mL 5%的HCl使pH=3。在60℃继续搅拌20分钟,并随后在室温搅拌1小时。通过过滤收集固体,以水(2×15mL)洗两次,并在真空加热到60℃烘干,得到标题产品,卡洛芬,7.4g(82.7%)。1HNMR(DMSO-d6):δ12.26(s,1H),11.32(s,1H),8.14(d,1H),8.05(d,1H),7.45(d,1H),7.37(s,1H),7.32(d,1H),7.07(d,1H),3.79(t,1H),1.40,(d,3H)。MS(ESI)m/z 274.1[M+1]+。
本发明的描述本质上仅为示例性的,并且因此,不背离本发明主旨的变化应在本发明范围内。这样的变化不认为是背离了本发明的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!