一种取代哌啶基丁酸乙酯季铵盐及其制备方法与流程
本发明涉及一种新化合物:4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐,本发明还涉及该化合物的制备方法。
背景技术:
:
控制药品中的杂质已经成为药品质量控制中的重要问题,杂质研究有助于对原料药合成工艺的进行有效监控,从而保证原料药生产的质量稳定性,同时,杂质研究为原料药进一步进行毒理学安全评价,药理学研究和药剂学研究提供数据参考。
本发明人发现,在合成抗过敏药物苯磺贝他斯汀的中间体时会产生一种杂质:4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐,其分子结构如式Ⅰ。
式Ⅰ是苯磺贝他斯汀杂质谱中的重要杂质,研究式I化合物的合成方法对于原料药杂质研究和药品质量控制有着非常重要的意义。
技术实现要素:
本发明的目的是研究式I化合物的合成方法,特别是研究式I化合物的产生与合成贝他斯汀时的反应条件的区别。
发明人在合成苯磺贝他斯汀的关键中间体时,监测到一个杂质,其使用LC-MS对该杂质进行质谱检测(仪器:AB Sciex API3000质谱仪,ESI电离方式,正离子扫描),谱图显示[M-Br]=531.4,与推测的季铵盐阳离子分子量符合。证实其结构为式I。
根据反应机理推断,胺与卤化物可以形成季铵盐,发明人通过加大卤化物比例,延长反应时间,柱层析纯化等手段,开发了一种合成式I化合物的方法。其合成路线如下所示:
以上反应式中,原料式II是4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]哌啶;原料式III是4-溴丁酸乙酯。
上述式II是手性化合物,原料可选用消旋体或单一构型的光学异构体;
当使用消旋体为起始原料时,产物为消旋体(S与R异构体各50%);
当使用S-构型的光学异构体为起始原料时,产物为主要含有S-构型的光学异构体或消旋体;
当使用R-构型的光学异构体为起始原料时,产物为主要含有R-构型的光学异构体或消旋体。
当需要制备高光学纯度的式I化合物,可以使用高光学纯度的(S)-构型的式II作为原料。
式II与式III两种原料的摩尔比例为式II:式III=1:0.5~10;
优选两种原料的摩尔比例为式II:式III=1:3~4
采用碱作为催化剂,原料式II与催化剂的摩尔比例为1:0.5~10,优选比例为1:3~4
所述碱选自碳酸钠,碳酸钾,三乙胺,二异丙胺中的一种或几种,优选碳酸钾和/或三乙胺作为催化剂。
本发明发现,若选用强碱如氢氧化钠、氢氧化钾作为催化剂时,收率大幅度降低(如对比例1、2)。这是由于产物是季铵盐,在较强的碱性条件下容易发生水解反应,生成4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸。
采用丙酮、无水乙醇、乙酸乙酯、乙腈作溶剂,优选丙酮;
溶剂与原料式II的重量比为5~10:1,优选比例为5~6:1
反应温度为0~100℃,优选40~60℃。
反应时间48~56小时左右,可以达到80%的收率。
反应完成后的后处理,可选用本领域常用的各种分离纯化方法。在一个合成实例中,发明人将上述两种原料混合溶于溶剂中,再加入碳酸钾和三乙胺催化,40~60℃反应48~56小时,反应完成后再通过柱层析得到产品。
柱层析所用的流动相体系为二氯甲烷:甲醇,溶剂比例为二氯甲烷:甲醇=5~50:1,优选比例是二氯甲烷:甲醇=30:1;
或者用石油醚:乙酸乙酯体系,溶剂比例为石油醚:乙酸乙酯=5~50:1。
本发明合成方法的优点是:原料易得,稳定,反应条件相对简单温和,合成成本低。
附图说明
图1是实施例1中4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐HPLC图;
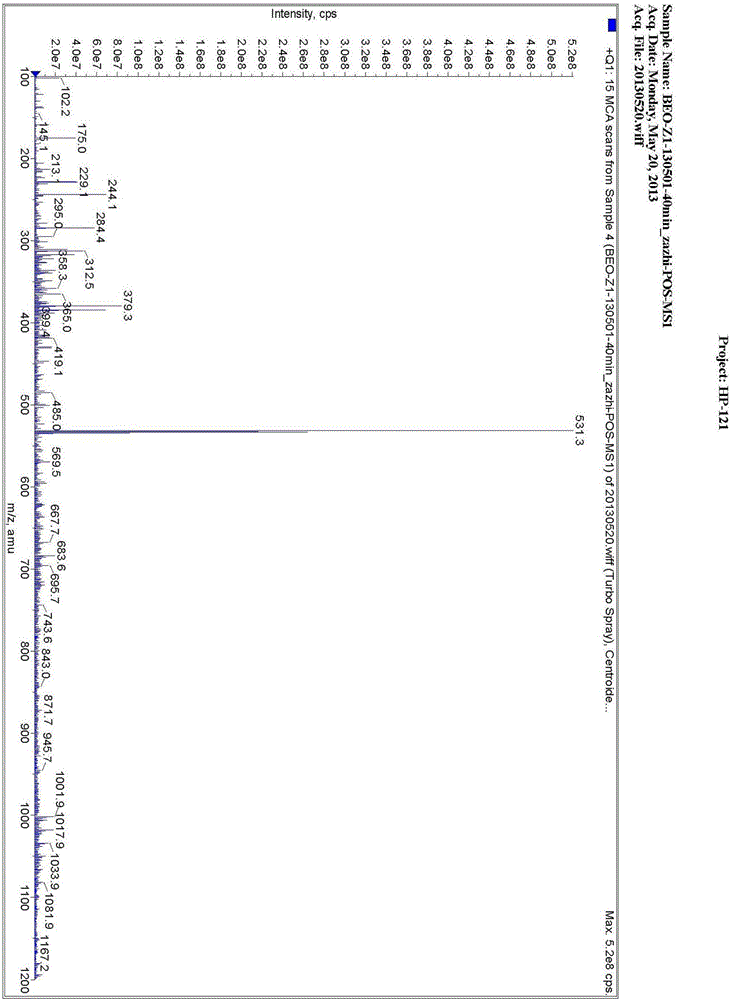
图2是实施例1中4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐一级质谱图;
图3是实施例1中4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐二级质谱图。
具体实施方式
以下实施例仅用于举例说明本发明的方法和装置,并不限定本发明的范围。实施例中所述HPLC分析方法的条件统一如下:
色谱条件:Agilent Eclipse XDB-C8(150×4.6mm,5μm)
检测波长:240nm 流速:1.0ml/min
柱温:30℃ 进样量:20μl
流动相:乙腈:磷酸盐缓冲溶液(取磷酸二氢钾6.8g,加水1000ml使溶解,每700ml缓冲盐中加入戊烷磺酸钠1.0g,用磷酸调节pH至4.0)=40:60
稀释剂:乙腈-水(20:80)
实施例1
在500ml三口反应瓶中,加入4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶30g,然后在搅拌条件下,加入180g丙酮,58g 4-溴丁酸乙酯,30g三乙胺,41g碳酸钾,52℃反应50小时
后处理:停止反应,过滤,60℃减压浓缩干,冰浴下用稀盐酸调节pH约1~2,乙酸乙酯提取,水层用氢氧化钠调至pH值约8~10,乙酯乙酸提取,然后无水硫酸镁干燥,过滤后减压浓缩,所得产物用二氯甲烷:甲醇=30:1柱层析得到产物4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐49.7g,收率82%
HPLC含量:96.519%;(详见附图1);
质谱:ESI显示[M+1]=531.3,与其理论分子量相符(详见附图2);二级质谱显示,碎片峰与其理论分子量相符(详见附图3)
实施例2
在500ml三口反应瓶中,加入4-[(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶(消旋体)15g,然后在搅拌条件下,加入150g丙酮,38.6g 4-溴丁酸乙酯,20g三乙胺,27.4g碳酸钾,40oC反应56小时
后处理:停止反应,过滤,60oC减压浓缩干,冰浴下用稀盐酸调节pH约1~2,乙酸乙酯提取,水层用氢氧化钠调至pH值约8~10,乙酯乙酸提取,然后无水硫酸镁干燥,过滤后减压浓缩,所得产物用二氯甲烷:甲醇=40:1柱层析得到产物4-[(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐24.3g,收率80%
实施例3
在500ml三口反应瓶中,加入4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶10g,然后在搅拌条件下,加入100g无水乙醇,64.4g 4-溴丁酸乙酯,33.4g二异丙基胺,45.6g碳酸钾,60℃反应56小时
后处理:停止反应,过滤,60℃减压浓缩干,冰浴下用稀盐酸调节pH约1~2,乙酸乙酯提取,水层用氢氧化钠调至pH值约8~10,乙酯乙酸提取,然后无水硫酸镁干燥,过滤后减压浓缩,所得产物用石油醚:乙酸乙酯=50:1柱层析得到产物4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐8.5g,收率42%
实施例4
在500ml三口反应瓶中,加入4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶10g,然后在搅拌条件下,加入80g乙酸乙酯,38.6g 4-溴丁酸乙酯,21g碳酸钠,48℃反应56小时
后处理:停止反应,过滤,60℃减压浓缩干,冰浴下用稀盐酸调节pH约1~2,乙酸乙酯提取,水层用氢氧化钠调至pH值约8~10,乙酯乙酸提取,然后无水硫酸镁干燥,过滤后减压浓缩,所得产物用二氯甲烷:甲醇=50:1柱层析得到产物4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐7.1g,收率35%
实施例5
在500ml三口反应瓶中,加入4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶10g,然后在搅拌条件下,加入60g丙酮,19.5g 4-溴丁酸乙酯,10.1g三乙胺,10.6g碳酸钠,50℃反应56小时
后处理:停止反应,过滤,60℃减压浓缩干,冰浴下用稀盐酸调节pH约1~2,乙酸乙酯提取,水层用氢氧化钠调至pH值约8~10,乙酯乙酸提取,然后无水硫酸镁干燥,过滤后减压浓缩,所得产物用二氯甲烷:甲醇=30:1柱层析得到产物4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐13.1g,收率65%
以下对比实施例,都因碱性太强,造成产物水解,导致收率低。
对比例1
在500ml三口反应瓶中,加入4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶10g,然后在搅拌条件下,加入70g乙酸乙酯,25.8g 4-溴丁酸乙酯,7.4g氢氧化钾,78℃反应48小时
后处理:停止反应,过滤,60℃减压浓缩干,冰浴下用稀盐酸调节pH约1~2,乙酸乙酯提取,水层用氢氧化钠调至pH值约8~10,乙酯乙酸提取,然后无水硫酸镁干燥,过滤后减压浓缩,所得产物用二氯甲烷:甲醇=30:1柱层析得到产物4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐0.8g,收率4%
对比例2
在500ml三口反应瓶中,加入4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶10g,然后在搅拌条件下,加入70g乙腈,32.2g 4-溴丁酸乙酯,6.6g氢氧化钠,65℃反应56小时
后处理:停止反应,过滤,60℃减压浓缩干,冰浴下用稀盐酸调节pH约1~2,乙酸乙酯提取,水层用氢氧化钠调至pH值约8~10,乙酯乙酸提取,然后无水硫酸镁干燥,过滤后减压浓缩,所得产物用石油醚:乙酸乙酯=30:1柱层析得到产物4-[(S)-(4-氯苯基)-2-吡啶基甲氧基]-1-哌啶基丁酸乙酯季铵盐1.2g,收率6%。
- 还没有人留言评论。精彩留言会获得点赞!