新型糖衍生物凝胶化剂的制作方法
技术领域:
本发明涉及含有糖衍生物的新型凝胶化剂。
背景技术:
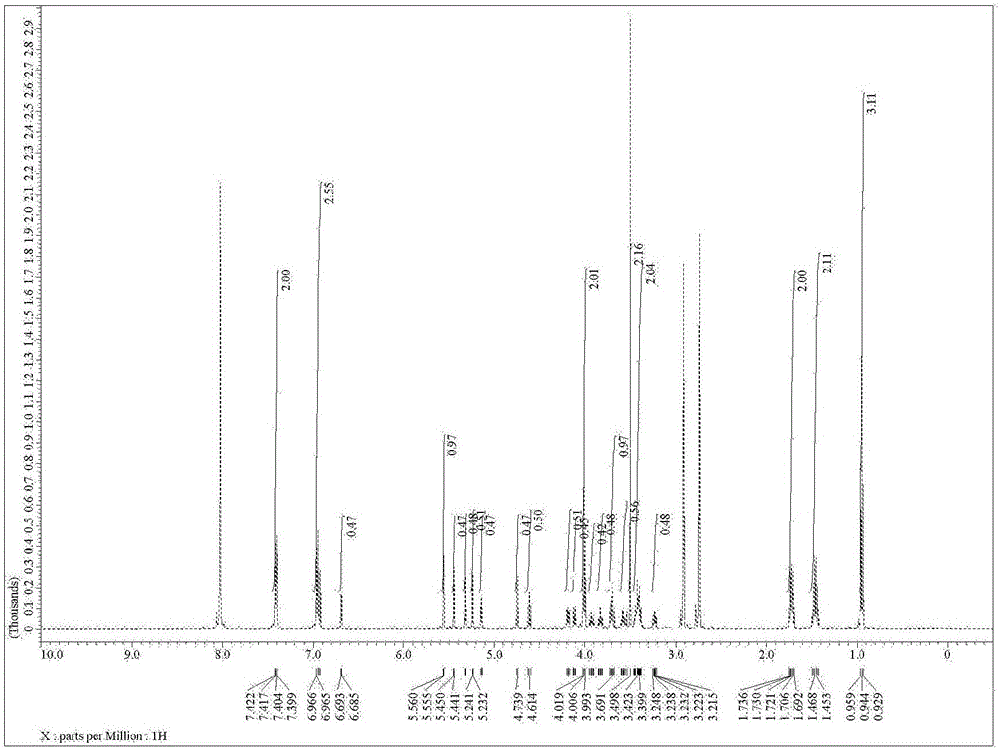
:由具有凝胶形成能力的物质(以下称凝胶化剂)形成的三维网孔结构中含有流体的结构体被称为凝胶,通常流体为水时被称为水凝胶(ハイドロ凝胶),为水以外的有机液体(有机溶剂或油等)时被称为有机凝胶或油凝胶。油凝胶(有机凝胶)在化妆品、医药品、农药、食品、粘接剂、涂料、树脂等领域中被用于化妆品及涂料的流动性调节。另外,还在例如,将废油凝胶化而形成固形物来防止水质污染等环境保护领域中得到了广泛的应用。关于凝胶化剂的研究主要是对高分子化合物来进行,但是近年来正在针对与高分子化合物相比,容易引入多种多样功能的低分子化合物进行研究开发。如上所述,油凝胶(有机凝胶)已在广阔的领域中得到的应用,且被期待今后的应用领域继续扩大。因此,对于低分子化合物凝胶化剂(以下有时称为低分子凝胶化剂),面对油凝胶的用途扩展,要求其对广泛种类的有机溶剂都有凝胶形成能力。对于这种课题,到目前为止已公开了作为对于各种有机溶剂仅以少量添加量即可形成稳定性优异的凝胶的低分子凝胶化剂的尿素化合物(例如,专利文献1、2)。另外,还公开了α-氨基内酰胺衍生物对于角鲨烷及液体石蜡等具有凝胶化能力(例如专利文献3)。另一方面,已报导了由于衍生自各种单糖类的糖衍生物相互间容易形成氢键的结构,使各种有机溶剂发生凝胶化(非专利文献1)。现有技术文献专利文献专利文献1:日本特开2000-256303号公报专利文献2:日本特开2004-359643号公报专利文献3:日本特开平10-265761号公报非专利文献非专利文献1:S.Shinkaietal.,Chem.Eur.J.2001,7,No20,4327-4334技术实现要素:发明所要解决的问题到目前为止,虽然已提出了面向有机溶剂等非水性介质的由低分子化合物组成的油凝胶化剂,但存在着能够进行凝胶化的介质受限等问题,由此,在具有新用途、功能性的新型油凝胶的创制中,正在摸索能够使各种介质凝胶化的新的低分子油凝胶化剂的方案。另外在糖衍生物的凝胶化剂方面虽然可以通过改变糖的种类来使各种溶剂凝胶化,但反过来用一种糖衍生物使广泛的溶剂凝胶化就相当困难。另外由非专利文献1记载的糖衍生物得到的凝胶不具备贮存稳定性。另外,就本发明人所知还没有能够使水和油(亲水性有机溶剂及疏水性有机溶剂)两方面的溶剂都可进行凝胶化的凝胶化剂的报道例,人们期望开发表现出这样的凝胶化性能的凝胶化剂。特别是即使不使用表面活性剂也能够使水和油(疏水性有机溶剂)的混合溶剂进行凝胶化的凝胶化剂,可用于生物体安全性要求高的化妆品及医药产品基材方面,尽管如此至今还未提出这种划时代的凝胶化剂方案。本发明是基于上述情况做出的,其所要解决的课题是提供具有至今未提出的结构的新型凝胶化剂。特别是本发明者发现了不但能够使水或油(亲水性有机溶剂及疏水性有机溶剂)各自单独地进行凝胶化,而且即使对于水和油(亲水性有机溶剂及疏水性有机溶剂)的混合溶剂,特别是水和油(疏水性有机溶剂)的混合溶剂也能进行凝胶化的葡萄糖型凝胶化剂,从而完成了本发明。解决问题的手段为了解决上述问题,本发明人进行了锐意研究,结果发现对糖衍生物上的取代基进行变换,不但适合作为水或有机溶剂等的凝胶化剂,令人惊奇的是不改变糖衍生物中使用的糖的种类也能使无论有机溶剂还是水形成凝胶,通过控制取代基的种类就改变了凝胶化溶剂的种类,从而完成了本发明。即,本发明的第1方面涉及凝胶化剂,包含下述式(1)或式(2)表示的化合物。(式中,R1及R3各自独立地表示碳原子数1至20的直链状或支化链状的烷基、碳原子数3至20的环状烷基、或碳原子数2至20的直链状或支化链状的烯基,n表示0或1~4的整数,R2表示氢原子、碳原子数1至10的直链状或支化链状的烷基、或可以具有取代基的芳基,R4及R5表示羟基)作为第2方面,涉及第1方面所述的凝胶化剂,前述式(1)表示的化合物为式(3)表示的化合物。[式中,R1、R2、R3及n与前述式(1)中记载的定义含义相同。]作为第3方面,涉及第1方面所述的凝胶化剂,前述式(1)表示的化合物为式(4)表示的化合物。[式中,R1、R2、R3及n与前述式(1)中记载的定义含义相同。]作为第4方面,涉及凝胶,由第1方面至第3方面中任一项所述的凝胶化剂和疏水性有机溶剂、亲水性有机溶剂、水、亲水性有机溶液、疏水性有机溶液或水溶液形成。作为第5方面,涉及第4方面所述的凝胶,前述疏水性有机溶剂是选自植物油、酯类、硅油及烃类中的至少一种。作为第6方面,涉及第4方面所述的凝胶,前述亲水性有机溶剂是选自甲醇、乙醇、2-丙醇、异丁醇、戊醇、己醇、1-辛醇、异辛醇、丙酮、环己酮、乙腈、二氧杂环己烷、甘油、丙二醇、乙二醇及二甲基亚砜中的至少一种。作为第7方面,涉及第4方面所述的凝胶,前述亲水性有机溶液为第6方面所述的亲水性有机溶剂和水的混合溶剂。作为第8方面,涉及第5方面所述的凝胶,前述疏水性有机溶液为第5方面所述的疏水性有机溶剂和水的混合溶剂。作为第9方面,涉及第4方面所述的凝胶,前述水溶液是将有机酸、或无机酸、或选自无机碳酸盐、无机硫酸盐、无机磷酸盐及无机磷酸氢盐中的至少一种的无机盐、或选自乙酸盐、乳酸盐、柠檬酸盐、有机胺盐酸盐及有机胺乙酸盐中的至少一种的有机盐进行溶解所形成的水溶液。作为第10方面,涉及第9方面所述的凝胶,前述有机酸是选自乙酸、柠檬酸、琥珀酸、乳酸、苹果酸、马来酸、富马酸及三氟乙酸中的至少一种的有机酸,前述无机酸是选自盐酸、磷酸、碳酸、硫酸、硝酸及硼酸中的至少一种的无机酸,前述无机盐是选自碳酸钙、碳酸钠、碳酸钾、硫酸钠、硫酸钾、硫酸镁、磷酸钾、磷酸钠、磷酸氢二钠及磷酸二氢钠中的至少一种的无机盐,前述有机盐是选自乙酸钠、乙酸钾、乳酸钠、乳酸钾、柠檬酸钠、柠檬酸钾、乙二胺盐酸盐、乙二胺四乙酸盐及三羟基甲基氨基甲烷盐酸盐中的至少一种的有机盐。作为第11方面,涉及化妆品基材或医疗用基材,包含第1方面至第3方面中任一项所述的凝胶化剂。作为第12方面,涉及化妆品基材或医疗用基材,包含第1方面至第3方面中任一项所述的凝胶化剂及至少一种高分子化合物。作为第13方面,涉及凝胶电解质,包含第1方面至第3方面中任一项所述的凝胶化剂。作为第14方面,涉及细胞培养基材,包含第1方面至第3方面中任一项所述的凝胶化剂。作为第15方面,涉及生体分子贮存用基材,包含第1方面至第3方面中任一项所述的凝胶化剂。作为第16方面,涉及外用基材,包含第1方面至第3方面中任一项所述的凝胶化剂。作为第17方面,涉及生物化学用基材,包含第1方面至第3方面中任一项所述的凝胶化剂。作为第18方面,涉及食品用基材,包含第1方面至第3方面中任一项所述的凝胶化剂。作为第19方面,涉及干燥地农业用基材,包含第1方面至第3方面中任一项所述的凝胶化剂。作为第20方面,涉及制造第一方面中所述的式(1)或式(2)表示的化合物的方法,其特征在于在乙醇和对甲苯磺酸的存在下通过一锅法进行下述制造前述式(1)或式(2)表示的化合物的工序,所述工序是使下述式[A]表示的化合物与缩醛化剂反应,接着使得到的缩醛衍生物与葡萄糖或甘露糖或它们的衍生物进行缩环反应,从而制造前述式(1)或式(2)表示的化合物的工序,(式中,R1及R3各自独立地表示碳原子数1至20的直链状或支化链状的烷基、碳原子数3至20的环状烷基、或碳原子数2至20的直链状或支化链状的烯基,n表示0或1~4的整数)。发明效果本发明的凝胶化剂无论在水系或有机溶剂系或双系中都可形成凝胶。另外,本发明的凝胶化剂,通过改变糖衍生物上的取代基的种类,可以改变要进行凝胶化的溶剂的种类。而且,本发明的凝胶化剂的贮存稳定性良好。另外,由于本发明的凝胶化剂是用葡萄糖、甘露糖或被称为它们的衍生物的单糖类作为原料形成的,可以形成生物体安全性优异的凝胶。特别是具有葡萄糖部分的上述式(3)表示的化合物(以下称其为葡萄糖型凝胶化剂),作为原料的葡萄糖在单糖类中被普遍地使用,不仅在凝胶化剂的制造中这种原料成本可控制地极低,而且凝胶化剂合成的整个过程的总收率高,而且不容易产生副反应,因此与以前的凝胶化剂相比制造容易且制造成本可控制地绝对低。另外,葡萄糖型凝胶化剂对于水和油(疏水性有机溶剂)均具有凝胶形成能力,可以形成水/油分散型凝胶。另外还可以实现醇系溶剂的凝胶化。因此,成为可以使具有宽广特性的介质进行透明且低浓度地凝胶化的通用性高的凝胶化剂。特别是对于水,可以形成透明性高的凝胶。另一方面,具有甘露糖部分的上述式(4)表示的化合物(以下称其为甘露糖型凝胶化剂),对于油(亲水性有机溶剂及疏水性有机溶剂)可以形成透明性高的凝胶,另外还可以得到触变性优异的凝胶。由于可以形成可自立(具有自支撑性的)的透明凝胶,可用作棒用基材。另外,根据本发明的制造方法,可以由葡萄糖及甘露糖的衍生物一锅法制造上述的凝胶化剂,能够便宜而简便地制造作为凝胶化剂的化合物。而且制造时不需要甲醇及金属催化剂,因此特别可以制造特别是针对化妆品基材及医疗用基材,以及食品用基材等要求高安全性的基材可以作为凝胶化剂使用的化合物。附图的简要说明图1显示了通过实施例制备的精制后的化合物〔G3”〕的HPLC图。图2显示了通过实施例制备的精制后的化合物〔G3”〕的1HNMR(500MHz,重DMF中)谱图。图3显示了甘露糖型凝胶化剂:用M1、M3以及作为公知物质的化合物A(均为0.05wt%)和SH245制备的SH245凝胶在室温下放置7天后的稳定性。图4显示了使用葡萄糖型凝胶化剂:G4(1wt%)的肉豆蔻酸异丙酯(IPM)和水中的水/油分散凝胶的显微镜照片。图5显示了使用葡萄糖型凝胶化剂:G3(0.25~1wt%)的SH245和水中的水/油分散凝胶的外观。图6显示了使用葡萄糖型凝胶化剂:G3(0.25wt%)的SH245和水中的水/油分散凝胶(搅拌手段:涡旋混合器)的共聚焦激光扫描显微镜照片。图7显示了使用葡萄糖型凝胶化剂:G3(0.25wt%)的SH245和水中的水/油分散凝胶(搅拌手段:均质器)的共聚焦激光扫描显微镜照片。图8显示了由使用葡萄糖型凝胶化剂:G4的预混料制备的凝胶的外观(图8(A))及显微镜照片(图8(B))。图9显示了由使用甘露糖型凝胶化剂:M3(0.5wt%)的角鲨烯凝胶的样品管中取出的凝胶的外观。图9(A)显示了刚取出后的图,图9(B)显示了将取出的凝胶用玻璃盖片(厚度:0.12-0.17mm)切断后的图,图9(C)显示了将切断的凝胶沿切断面相互叠合后的图。图10显示了对于使用甘露糖型凝胶化剂:M1~M6及M8及作为公知物质的化合物A制备甲苯凝胶,相对于凝胶化剂的浓度的溶胶-凝胶相转变温度(Tgel)。图11显示了使用甘露糖型凝胶化剂:M2或作为公知物质的化合物A制备的甲苯凝胶(1wt%)的外观以及它们的干凝胶的SEM图像。图12显示了使用甘露糖型凝胶化剂:M2或M6制备的甲苯凝胶的外观及它们的干凝胶的AFM图像。图13显示了使用甘露糖型凝胶化剂:M2制备的甲苯凝胶(0.5wt%)及水凝胶(0.1wt%)的外观以及它们的干凝胶的SEM图像。图14显示了使用甘露糖型凝胶化剂:M2或葡萄糖型凝胶化剂G3制备的水凝胶(0.1wt%)的外观以及它们的干凝胶的SEM图像。发明的实施方式[凝胶化剂]本发明的凝胶化剂包含下述式(1)或式(2)表示的化合物。(式中,R1及R3各自独立地表示碳原子数1至20的直链状或支化链状的烷基、碳原子数3至20的环状烷基、或碳原子数2至20的直链状或支化链状的烯基,n表示0或1~4的整数,R2表示氢原子、碳原子数1至10的直链状或支化链状的烷基、或可以具有取代基的芳基,R4及R5表示羟基)作为前述碳原子数1至20的直链状的烷基,可以列举甲基、乙基、正丙基、正丁基、正戊基、正己基、正庚基、正辛基、正壬基、正癸基、正十一烷基、正十二烷基、正十三烷基、正十四烷基、正十五烷基、正十六烷基、正十七烷基、正十八烷基、正十九烷基及正二十烷基等。作为前述碳原子数1至20的支化链状的烷基,可以列举异丙基、异丁基、仲丁基、叔丁基及2-乙基己基等。作为前述碳原子数3至20的环状烷基,可以列举具有环戊基环、环己基环结构的基团等。作为前述碳原子数2至20的直链状的烯基,可以列举乙烯基、烯丙基、丁烯基、戊烯基、己烯基、庚烯基、辛烯基、壬烯基及癸烯基等。作为前述碳原子数2至20的支化链状的烯基,可以列举2-甲基-2-丙烯基、异丙烯基、2-甲基-1-丙烯基及2-甲基烯丙基等。作为前述碳原子数1至10的直链状或支化链状的烷基,可以列举前述例示的前述碳原子数1至20的直链状的烷基及前述碳原子数1至20的支化链状的烷基中,碳原子数为1至10的品种。作为前述芳基,可以列举苯基、苄基、1-萘基、2-萘基、1-蒽基及1-菲基等。另外,该芳基可以具有取代基,作为这种取代基,可以列举可含有酯键、酰胺键及醚键的直链状、支化链状或环状烷基及卤素原子等。从使用本发明的凝胶化剂,使溶剂进行良好地凝胶化方面考虑,前述式(1)或(2)中,R2优选为氢原子或甲基。另外,从针对疏水性有机溶剂使用时能够得到不会发生脱水收缩的透明性高的凝胶方面考虑,R1优选为丁基、戊基、己基。另一方面,从针对亲水性有机溶剂使用时能够得到不会发生脱水收缩的透明性高的凝胶方面考虑,R1优选为辛基、癸基、十二烷基。另外,从能够使可以含有亲水性有机溶剂的水和油等疏水性有机溶剂的双溶剂得到各自单独的凝胶、或双溶剂混合均匀性高的凝胶方面考虑,R1优选为丙基、丁基、戊基。前述(1)或式(2)表示的化合物可以通过公知的方法,例如使苯环上带有上述取代基的苯甲醛二甲基缩醛和单糖进行反应而得到。作为可以使用的单糖,只要是显示吡喃糖环结构的单糖就没有特别的限定,可以列举阿洛糖、阿卓糖、葡萄糖、甘露糖、古洛糖、艾杜糖、半乳糖及塔罗糖等。其中,从比较便宜和特别期待生物体适合性方面考虑,作为单糖,优选葡萄糖及甘露糖。在上述式(1)或(2)表示的化合物中,特别优选具有葡萄糖部分的下述式(3)表示的化合物、或具有甘露糖部分的下述式(4)表示的化合物。[式中,R1、R2、R3及n与前述式(1)中记载的定义含义相同。][式中,R1、R2、R3及n与前述式(1)中记载的定义含义相同。]上述式(3)表示的化合物的中,优选下述式(5)表示的化合物,更优选式(7)表示的化合物。[式中,R1及R2与前述式(1)中记载的定义含义相同。][式中,R1与前述式(1)中记载的定义含义相同。]另外,上述式(4)表示的化合物的中,优选下述式(6)表示的化合物,更优选式(8)表示的化合物。[式中,R1及R2与前述式(1)中记载的定义含义相同。][式中,R1与前述式(1)中记载的定义含义相同。]还有,如上所述,对于上述式(3)表示的化合物(葡萄糖型凝胶化剂)的凝胶化能力和上述式(4)表示的化合物(甘露糖型凝胶化剂)的凝胶化能力,具有完全不同的特性,例如能够凝胶化的溶剂各不相同等。特别是上述式(3)表示的化合物(葡萄糖型凝胶化剂),最大的特征是对于水和油(疏水性有机溶剂)均具有凝胶形成能力,对于水/油混合溶剂可以形成水/油分散型凝胶。另外还能够使醇系溶剂的凝胶化成为可能,对于水形成透明性高的凝胶。另一方面,作为上述式(4)表示的化合物(甘露糖型凝胶化剂),对于油(亲水性有机溶剂及疏水性有机溶剂)可以形成透明性高的凝胶,能够得到触变性优异的凝胶。也具有能够形成可以自立(具有自支撑性)的透明凝胶的特性。这样一来,葡萄糖型凝胶化剂和甘露糖型凝胶化剂的凝胶化能力的各自特征不同,可期待在符合各自特征的宽广领域中应用。[凝胶]作为本发明的凝胶,可以通过利用上述凝胶化剂使溶剂凝胶化来得到。具体来说,可以例示将规定量的凝胶化剂加热溶解在溶剂中,并进行冷却的制造方法。通常,加热溶解时优选使其完全溶解。还有,在本说明书中,凝胶化是指具有流动性的液体达到失去流动性的状态。使溶剂凝胶化时,本发明的凝胶化剂的使用量,只要能达到本发明的效果就没有特别的限定,但相对于进行凝胶化的溶剂的质量通常为20~0.001质量%,优选为2~0.05质量%。作为前述溶剂,只要不防止凝胶化就没有特别的限定,作为优选的具体例子,可以列举疏水性有机溶剂、亲水性有机溶剂、水、水和亲水性有机溶剂的混合溶剂(在本说明书中称为亲水性有机溶液)、疏水性有机溶剂和水的混合溶剂(在本说明书,称为疏水性有机溶液)、或在水中溶解有机酸或无机酸、或在水中溶解有无机盐或有机盐而形成的水溶液(在本说明书中称为水溶液)等。作为前述疏水性有机溶剂的优选具体例子,可以列举例如,橄榄油、椰子油、蓖麻油、荷荷巴油或向日葵油等的植物油;辛酸鲸蜡酯、肉豆蔻酸异丙酯或棕榈酸异丙酯等酯类;甲苯、二甲苯、正己烷、环己烷、辛烷、矿物油、硅油或氢化聚异丁烯等烃类。在这些物质中,作为前述疏水性有机溶剂,优选橄榄油、肉豆蔻酸异丙酯、甲苯、环己烷、直链状硅酮、环状硅酮、烷基改性硅酮、苯基改性硅酮、聚二甲基硅氧烷或聚二甲基硅氧烷醇等硅油及辛烷。对于上述硅油,可以使用能够由东レ·ダウコーニング(株)获得的直链状硅酮(商品名:2-1184)、环状硅酮(商品名:SH245)、烷基改性硅酮(商品名:SS-3408)、苯基改性硅酮(商品名:PH-1555)、聚二甲基硅氧烷(商品名:BY-11-0系列)、聚二甲基硅氧烷醇(商品名:CB-1556)等。前述亲水性有机溶剂是指可以在水中以任意比例溶解的有机溶剂,可以列举醇、或丙酮、环己酮、乙腈、二氧杂环己烷、甘油及二甲基亚砜等。前述醇优选为在水中自由溶解的水溶性醇,更优选列举碳原子数1至9的醇、多元醇、高级醇、甘油酯类。具体来说,作为碳原子数1至9的醇,可列举甲醇、乙醇、2-丙醇、异丁醇、戊醇、己醇、1-辛醇、异辛醇等;作为多元醇,可列举乙二醇、丙二醇、聚丙二醇等;作为高级醇,可列举辛基十二烷醇、硬脂醇、油醇等;作为甘油酯类,可列举三辛酸甘油酯、三(辛癸酸)缩水甘油酯、硬脂酸缩水甘油酯等。在这些物质中,作为前述亲水性有机溶剂,优选甲醇、乙醇、2-丙醇、异丁醇、戊醇、己醇、1-辛醇、异辛醇、丙酮、环己酮、乙腈、二氧杂环己烷、甘油、丙二醇、乙二醇及二甲基亚砜,更优选甘油、丙二醇及乙二醇。前述有机酸或无机酸可以添加多种,作为优选有机酸的例子,可以列举乙酸、柠檬酸、琥珀酸、乳酸、苹果酸、马来酸、富马酸及三氟乙酸。更优选为乙酸、柠檬酸、琥珀酸、乳酸、苹果酸,进一步优选为乙酸、柠檬酸、乳酸。另外,作为优选的无机酸的例子,可以列举盐酸、磷酸、碳酸、硫酸、硝酸及硼酸。更优选为盐酸、磷酸、碳酸、硫酸,进一步优选为盐酸、磷酸、碳酸。前述无机盐或有机盐可以添加多种,但优选为1或2种。通过添加2种盐,还可期待溶液具有缓冲能力。作为优选的无机盐的例子,可以列举无机碳酸盐、无机硫酸盐、无机磷酸盐及无机磷酸氢盐。更优选为碳酸钙、碳酸钠、碳酸钾、硫酸钠、硫酸钾、硫酸镁、磷酸钾、磷酸钠、磷酸氢二钠或磷酸二氢钠,进一步优选为碳酸钙、硫酸镁、磷酸氢二钠或磷酸二氢钠。另外,作为优选的有机盐的例子,可以列举无机乙酸盐、无机乳酸盐、无机柠檬酸盐这样的无机有机酸盐、有机胺的盐酸盐或有机胺乙酸盐。更优选为乙酸钠、乙酸钾、乳酸钠、乳酸钾、柠檬酸钠、柠檬酸钾、乙二胺盐酸盐、乙二胺四乙酸盐、三羟基甲基氨基甲烷盐酸盐。本发明的凝胶化剂,相对于作为介质的前述的疏水性有机溶剂、亲水性有机溶剂、水、亲水性有机溶液、疏水性有机溶液或水溶液,优选按照0.001至10质量%,优选0.1至10质量%,例如0.1至5质量%的量进行使用。将本发明的凝胶化剂添加到作为介质的前述的疏水性有机溶剂、亲水性有机溶剂、水、亲水性有机溶液、疏水性有机溶液或水溶液中,必要时进行加热搅拌使其溶解后,在室温下放置,从而可以得到凝胶。凝胶强度可以利用凝胶化剂的浓度进行调节。还有,利用本发明的凝胶化剂形成的凝胶,可以在不损害凝胶化剂的凝胶化能力的范围内,根据其应用用途等,根据需要混入各种添加剂(表面活性剂、紫外线吸收剂、保湿剂、防腐剂、抗氧化剂、香料、生理活性物质(药效成分)等有机化合物、及氧化钛、滑石、云母、水等无机化合物等)。[化妆品基材或医疗用基材]本发明的化妆品基材或医疗用基材含有上述凝胶化剂。另外,对于本发明的化妆品基材或医疗用基材,除了上述凝胶化剂之外,还可以含有水、醇、多元醇、亲水性有机溶剂、疏水性有机溶剂、或它们的混合溶液。还有,作为前述醇、多元醇、亲水性有机溶剂及疏水性有机溶剂,可以列举前述醇、多元醇、亲水性有机溶剂及疏水性有机溶剂中例示的化合物。另外,对于本发明的化妆品基材或医疗用基材,必要时可含有一般在化妆品基材或医疗用基材中配入的生理活性物质及功能性物质等添加成分,作为这样的添加成分,可以列举油性基剂、保湿剂、触感提高剂、表面活性剂、高分子、增粘·凝胶化剂、溶剂、喷射剂、抗氧化剂、还原剂、氧化剂、防腐剂、抗菌剂、杀菌剂、螯合剂、pH调节剂、酸、碱、粉体、无机盐、紫外线吸收剂、美白剂、维生素类及其衍生物类、生发用药剂、血液循环促进剂、刺激剂、激素类、抗皱剂、抗老化剂、绷紧剂、冷感剂、温感剂、伤口愈合促进剂、刺激减缓剂、镇痛剂、细胞活化剂、植物·动物·微生物提取物、除痒剂、角质剥離·溶解剂、抑制出汗剂、清凉剂、收敛剂、酶、核酸、香料、色素、着色剂、染料、颜料、消炎剂、消炎剂、抗喘、抗慢性阻塞性肺病、抗过敏、免疫调节剂、抗感染剂及抗真菌剂等。另外,本发明的化妆品基材或医疗用基材含有上述凝胶化剂及至少一种高分子化合物。作为前述高分子化合物,可以列举明胶、海藻酸钠、海藻酸丙二醇酯、阿拉伯胶、聚乙烯基醇、聚丙烯酸、聚丙烯酸钠、羧甲基纤维素、结兰胶、黄原胶、角叉胶聚苯乙烯、聚甲基丙烯酸甲酯、聚乙烯基吡咯烷酮、聚氧乙烯、聚乳酸、聚苯乙烯磺酸、聚丙烯腈、聚乙烯及聚对苯二甲酸乙二酯等。[凝胶电解质]本发明的凝胶电解质是使包含有机溶剂或水的电解液(液体电解质)进行凝胶化而得到。使用的凝胶化剂和电解液没有特别的限制,根据使用进行适时选择即可。例如,在包含前述有机溶剂的电解液的情况下,是在至少一种非质子性有机溶剂中溶解电解质盐而形成。作为前述非质子性有机溶剂,可以列举乙二醇二甲醚、碳酸亚烷基酯、碳酸烷基酯、环状醚、酰胺类、腈类、酮类及酯类等,作为优选的具体例子,可以列举碳酸亚丙酯、碳酸亚乙酯、碳酸二乙酯、γ-丁内酯、1,2-二甲氧基乙烷、四氢呋喃、2-甲基四氢呋喃、二甲基亚砜、1,3-二氧戊环、甲酰胺、二甲基甲酰胺、1,4-二氧杂环己烷、乙腈、硝基甲烷、乙基单乙二醇二甲醚、磷酸三酯、三甲氧基甲烷、二氧戊环衍生物、环丁砜、3-甲基-2-噁唑烷酮、碳酸亚丙酯衍生物、四氢呋喃衍生物、二乙基醚及1,3-丙磺酸内酯等。这些有机溶剂可以单独使用或组合使用2种以上。前述电解质盐由金属阳离子和抗衡阴离子组成,作为金属阳离子,可以列举Li+、Na+、K+等,作为其抗衡阴离子,可以列举ClO4-、LiBF4-、PF6-、CF3SO3-、CF3CO2-、AsF6-、SbF6-、(CF3SO2)2N-、B10Cl102-、(1,2-二甲氧基乙烷)2ClO4-、低级脂肪族羧酸盐、AlCl4-、Cl-、Br-、I-、氯硼烷化合物及四苯基硼酸等。在这些物质中,作为优选的电解质盐,可以列举锂盐。这些电解质盐可以单独使用或组合使用2种以上。另外,本发明的凝胶化剂及由其得到的凝胶可以被用于细胞培养基材、细胞及蛋白质等生物体分子贮存用基材、外用基材、生物化学用基材、食品用基材、接触眼镜、纸尿布、人造激励器、干燥地农业基材等各种领域中的材料。另外,作为酶等生物反应器载体,可以在研究、医疗、分析、各种产业进行广泛地应用。[凝胶化剂的制造法]本发明另外还以制造作为前述本发明的凝胶化剂的式(1)或式(2)表示的化合物的方法作为对象。即,该制造方法的特征在于,在乙醇和对甲苯磺酸的存在下通过一锅法进行使下述式[A](式中,R1及R3各自独立地表示碳原子数1至20的直链状或支化链状的烷基、碳原子数3至20的环状烷基、或碳原子数2至20的直链状或支化链状的烯基,n表示0或1~4的整数)表示的化合物与缩醛化剂反应,接着使得到的缩醛衍生物与葡萄糖或甘露糖或它们的衍生物进行缩环反应,从而制造前述式(1)或式(2)表示的化合物的工序。实施例以下,为了进一步明确本发明的特征而列出实施例,但是本发明并不受这些实施例的限制。还有,下述路线A及路线1~10中显示的符号是各个路线内赋予的符号,与发明的详细说明及权利要求书中赋予的符号有区别。以下列出作为以下实施例的合成原料使用的试剂。甲基α-D-吡喃甘露糖苷、对甲氧基苯甲醛、对乙氧基苯甲醛、对(十二烷氧基)苯甲醛、4-溴-1-丁烯、1-溴-3-甲基-2-丁烯、1-溴丁烷、1-溴己烷、D(+)-葡萄糖、乙基α-D-葡糖甙从和光纯药工业(株)获得,甲基α-D-吡喃葡萄糖苷、对甲苯磺酸一水和物、4-丙氧基苯甲醛、4-正丁氧基苯甲醛、4-正戊氧基苯甲醛、4-正己氧基苯甲醛、4-正辛氧基苯甲醛、4-正癸氧基苯甲醛、原甲酸三甲酯、溴环己烷、碘化四丁基铵、原甲酸三乙酯、4-羟基苯甲醛、3-羟基苯甲醛、香草醛、对甲苯磺酸(酐)、3,4-二甲氧基苯甲醛从东京化成工业(株)获得,四氟硼酸铜(II)水合物、2-乙基己基溴化物、溴化香叶酯从Sigma-AldrichJapan(株)获得。另外,作为反应溶剂使用的N,N-二甲基甲酰胺(DMF)(脱水,有机合成用)、甲醇(脱水,有机合成用)及乙醇(超脱水,有机合成用)从和光纯药工业(株)获得。反应后处理及精制时使用的硫酸钠(特级)、氯化钠(特级)、碳酸氢钠(特级)、二乙基醚(特级)从和光纯药工业(株)获得,己烷(特级)、乙酸乙酯(特级)、氯仿(特级)、二异丙基醚(特级)从关东化学(株)获得。NMR测定时使用的重氯仿(含有0.03%TMS(四甲基硅烷))及重二甲基甲酰胺从Sigma-AldrichJapan(株)获得。以下列出凝胶化试验中使用的溶剂。辛烷(特级)、环己烷(特级)、甲苯(特级)、乙醇(特级)、橄榄油、肉豆蔻酸异丙酯(一级)、甘油(特级)、角鲨烷(一级)、角鲨烯(特级)、乙腈(特级)、甲醇(特级)、丙二醇(特级)、1,3-丁二醇(别名:亚丁基二醇)(特级)、甘油(特级)从和光纯药工业(株)获得,乙二醇(特级)从关东化学(株)获得,SH245从东レ·ダウコーニング(株)获得,戊二醇从ITO(株)获得,二甲基亚砜(DMSO)从东京化成工业(株)获得。水使用纯水。另外,凝胶化试验中使用的防腐剂、表面活性剂如下。防腐剂:对羟苯甲酸甲酯、2-苯氧基乙醇(特级)从和光纯药工业(株)获得。表面活性剂:聚氧乙烯山梨聚糖单月桂酸酯(Tween20)、十二烷基硫酸钠(SDS、一级)、硬脂基三甲基溴化铵(STAB)、3-[(3-胆固醇氨丙基)二甲基氨基]丙磺酸盐(CHAPS)从和光纯药工业(株)获得。另外还列出以下各种测定及分析中使用的装置及条件。(1)1H-NMR谱·装置:AVANCE500,BrukerBioSpin(株)制JNM-ECA600,日本电子(株)制(2)高速液相色谱(HPLC)·装置:1200系列,AgilentTechnology(株)制(3)LC-MS·装置:e2695(LC)、2489(UV)、3100(MS),日本ウォーターズ(株)制(4)光学显微镜·装置:DM2500,LeicaMicrosystems(株)制(5)共聚焦激光扫描显微镜·装置:LSM700,CarlZeiss(株)制(6)扫描型电子显微镜(SEM)·装置:SU8000,日立HighTechnologies(株)制(7)原子力显微镜(AFM)·装置:Nanocute,SIINanotechnology(株)制(8)涡旋混合器·装置:VoltexGenie2,ScientificIndustries社制(9)均质器·装置:OmniTipTMHomogenizingKit,OmniInternational公司制[实施例1:凝胶化剂的合成]形成甘露糖型凝胶化剂及葡萄糖型凝胶化剂的化合物可以按下述列出的路线A进行合成。对于作为凝胶化剂的原料的具有各种烃基的芳香族醛化合物,市售的通过购买获得(参照上述内容),未市售的按后述的路线1合成。另外,对于芳香族醛化合物和糖的缩环反应,一般经由缩醛进行缩环反应来实施,或由醛在加热条件下实施,但是作为本发明,作为特征之一可以列举,由芳香族醛化合物在室温下,而且通过一步缩环反应得到。路线A:1步反应和2步反应的合成路线[具有烃基的芳香族醛化合物的合成]按照下述路线1,合成具有各种烃基的芳香族醛化合物。路线1化合物〔1〕的合成:在氮气氛下将对羟基苯甲醛(4.9g,40mmol)、4-溴-1-丁烯(8.1g,60mmol)及碳酸钾(8.3g,60mmol)添加到80mL干燥N,N-二甲基甲酰胺(DMF)中,在80℃下加热6小时。将反应溶液放置冷却到室温后,通过过滤除去不溶物,对滤液进行减压浓缩。在残留物中加入乙酸乙酯和饱和氯化钠进行分液操作,提取出有机层。用饱和氯化钠洗涤有机层。用硫酸钠干燥有机层,过滤硫酸钠后,对滤液进行减压浓缩。通过柱色谱(二氧化硅凝胶,己烷:乙酸乙酯=9:1(v/v))精制残留物,得到目标产物。收率91%,1HNMR(500MHz,CDCl3):δ9.88(s,1H),7.85-7.81(m,2H),7.02-6.95(m,2H),5.95-5.86(m,1H),5.22-5.12(m,2H),4.15-4.05(m,2H),2.61-2.55(m,2H)。化合物〔2〕的合成:在氮气氛下将4-羟基苯甲醛(10.0g,82mmol)、溴环己烷(15.0mL,122mmol)及碳酸钾(34.0g,246mmol)、碘化四丁基铵(0.25g,0.67mmol)添加到80mL干燥N,N-二甲基甲酰胺(DMF)中,在150℃下加热12小时。将反应溶液放置冷却到室温后,通过过滤除去不溶物,对滤液进行减压浓缩。在残留物中加入乙酸乙酯和饱和氯化钠进行分液操作,提取出有机层。用饱和氯化钠洗涤有机层。用硫酸钠干燥有机层,过滤硫酸钠后,对滤液进行减压浓缩。通过柱色谱(二氧化硅凝胶,己烷:乙酸乙酯=9:1(v/v))精制残留物,得到目标产物。收率12%δ9.85(s,1H),7.83-7.79(m,2H),7.00-6.96(m,2H),4.41-4.35(m,1H),2.05-1.98(m,2H),1.87-1.79(m,2H),1.63-1.50(m,3H),1.45-1.29(m,3H)。化合物〔3〕的合成:在氮气氛下将4-羟基苯甲醛(4.9g,40mmol)、2-乙基己基溴化物(9.7g,50mmol)及碳酸钾(8.3g,60mmol)添加到80mL干燥N,N-二甲基甲酰胺(DMF)中,在80℃下加热3小时。将反应溶液放置冷却到室温后,通过过滤除去不溶物,对滤液进行减压浓缩。在残留物中加入乙酸乙酯和饱和氯化钠进行分液操作,提取出有机层。用饱和氯化钠洗涤有机层。用硫酸钠干燥有机层,过滤硫酸钠后,对滤液进行减压浓缩。通过柱色谱(二氧化硅凝胶,己烷:乙酸乙酯=9:1(v/v))精制残留物,得到目标产物。收率95%,1HNMR(500MHz,CDCl3):δ9.88(s,1H),7.85-7.81(m,2H),7.02-6.98(m,2H),3.94-3.91(m,2H),1.78-1.73(m,1H),1.53-1.30(m,8H),0.95-0.85(m,6H)。化合物〔4〕的合成:在氮气氛下将香草醛(3.9g,26mmol)、1-溴-3-甲基-2-丁烯(3.4g,23mmol)及碳酸钾(4.2g,30mmol)添加到50mL干燥N,N-二甲基甲酰胺(DMF)中,在50℃下加热3小时。将反应溶液放置冷却到室温后,通过过滤除去不溶物,对滤液进行减压浓缩。通过柱色谱(二氧化硅凝胶,己烷:乙酸乙酯=9:1~7:3(v/v))精制残留物,得到目标产物。收率75%,1HNMR(500MHz,CDCl3):δ9.85(s,1H),7.44(d,1H,J=8.2Hz),7.41(s,1H),6.98(d,1H,J=8.2Hz),5.52(t,1H,J=6.8Hz),4.68(d,2H,J=6.6Hz),3.94(s,3H),1.80(s,3H),1.77(s,3H)。化合物〔5〕的合成:在氮气氛下将香草醛(1.9g,12mmol)、溴化香叶酯(2.4g,11mmol)及碳酸钾(2.1g,15mmol)添加到20mL干燥N,N-二甲基甲酰胺(DMF)中,在80℃下加热3小时。将反应溶液放置冷却到室温后,通过过滤除去不溶物,对滤液进行减压浓缩。通过柱色谱(二氧化硅凝胶,己烷:乙酸乙酯=10:0~6:4(v/v))精制残留物,得到目标产物。收率75%,1HNMR(500MHz,CDCl3):δ9.85(s,1H),7.44(d,1H,J=8.2Hz),7.41(s,1H),6.97(d,1H,J=8.2Hz),5.51(t,1H,J=6.5Hz),5.07(t,1H,J=7.0Hz),4.72(d,2H,J=6.3kHz),3.94(s,3H),2.17-2.05(m,4H),1.76(s,3H),1.66(s,3H),1.60(s,3H)。化合物〔6〕:在氮气氛下将3-羟基苯甲醛(4.9g,40mmol)、1-溴己烷(7.3g,44mmol)及碳酸钾(6.9g,50mmol)添加到60mL干燥N,N-二甲基甲酰胺(DMF)中,在80℃下加热2小时。将反应溶液放置冷却到室温后,通过过滤除去不溶物,对滤液进行减压浓缩。在残留物中加入乙酸乙酯和饱和氯化钠进行分液操作,提取出有机层。用饱和氯化钠洗涤有机层。用硫酸钠干燥有机层,过滤硫酸钠后,对滤液进行减压浓缩。通过柱色谱(二氧化硅凝胶,己烷:乙酸乙酯=10:0至7:3(v/v))精制残留物,得到目标产物。收率97%,1HNMR(500MHz,CDCl3):δ9.97(s,1H),7.46-7.41(m,2H),7.40-6.36(m,1H),7.20-7.14(m,1H),4.01(t,2H,J=6.5Hz),1.84-1.76(m,2H),1.52-1.43(m,2H),1.40-1.30(m,4H),0.96-0.86(m,3H)。化合物〔6’〕的合成:在氮气氛下将3-羟基苯甲醛(3.7g,30mmol)、1-溴丁烷(4.5g,33mmol)及碳酸钾(5.3g,38mmol)添加到30mL干燥N,N-二甲基甲酰胺(DMF)中,在80℃下加热2小时。将反应溶液放置冷却到室温后,通过过滤除去不溶物,对滤液进行减压浓缩。通过柱色谱(二氧化硅凝胶,己烷:乙酸乙酯=10:0~9:1(v/v))精制残留物,得到目标产物。收率95%,1HNMR(500MHz,CDCl3):δ9.97(s,1H),7.46-7.37(m,3H),7.17(s,1H),4.02(t,2H,J=6.6Hz),1.83-1.75(m,2H),1.55-1.46(m,2H),0.98(t,3H,J=7.5Hz)。[缩醛衍生物的合成]<使用金属催化剂(四氟硼酸铜(II)水合物)的二甲基缩醛化(路线2)>按照下述路线2,实施芳香族醛化合物的二甲基缩醛化,得到缩醛衍生物〔17〕~〔32〕。路线2化合物〔17〕的合成:在氮气氛下将对甲氧基苯甲醛〔7〕(2.7g,20mmol)和原甲酸三甲基酯(4.4mL,40mmol)溶解在8mL干燥甲醇中,在该溶液中加入四氟硼酸铜(II)水合物(48mg)。在室温下搅拌1小时,添加饱和碳酸氢钠终止反应。在该溶液中加入乙酸乙酯进行抽提操作,用饱和氯化钠洗涤有机层。用硫酸钠干燥有机层,过滤硫酸钠后,对滤液进行减压浓缩。收率99%、1HNMR(500MHz,CDCl3):δ7.36(d,2H,J=8.8Hz),6.89(d,2H,J=8.8Hz),5.35(s,1H),3.81(s,3H),3.31(s,6H)。其它化合物〔18〕~〔32〕也按照与化合物〔17〕相同的方法合成。对它们进行分析的数据列于下面。化合物〔18〕:收率94%1HNMR(500MHz,CDCl3):δ7.35(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.35(s,1H),4.04(q,2H,J=6.9Hz),3.31(s,6H),1.41(t,3H,J=7.1Hz)。化合物〔19〕:收率95%1HNMR(500MHz,CDCl3):δ7.37(d,2H,J=8.2Hz),6.91(d,2H,J=8.8Hz),5.37(s,1H),3.95(t,2H,J=6.5Hz),3.34(s,6H),1.91-1.79(m,2H),1.06(t,3H,J=7.5Hz)。化合物〔20〕:收率99%,1HNMR(500MHz,CDCl3):δ7.35(d,2H,J=8.8Hz),6.89(d,2H,J=8.8Hz),5.35(s,1H),3.96(t,2H,J=6.6Hz),3.31(s,6H),1.80-1.73(m,2H),1.54-1.44(m,2H),0.97(t,3H,J=7.6Hz)。化合物〔21〕:收率95%1HNMR(500MHz,CDCl3):δ7.35(d,2H,J=8.8Hz),6.88(d,2H,J=8.5Hz),5.35(s,1H),3.96(t,2H,J=6.6Hz),3.31(s,6H),1.82-1.75(m,2H),1.48-1.33(m,4H),0.93(t,3H,J=7.1Hz)。化合物〔22〕:收率99%,1HNMR(500MHz,CDCl3):δ7.35(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.35(s,1H),3.95(t,2H,J=6.6Hz),3.31(s,6H),1.81-1.74(m,2H),1.51-1.41(m,2H),1.39-1.29(m,4H),0.94-0.87(m,3H)。化合物〔23〕:收率99%,1HNMR(500MHz,CDCl3):δ7.35(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.35(s,1H),3.95(t,2H,J=6.6Hz),3.31(s,6H),1.82-1.73(m,2H),1.50-1.40(m,2H),1.39-1.22(m,8H),0.92-0.85(m,3H)。化合物〔24〕:收率98%,1HNMR(500MHz,CDCl3):δ7.34(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.34(s,1H),3.95(t,2H,J=6.6Hz),3.31(s,6H),1.82-1.73(m,2H),1.50-1.40(m,2H),1.39-1.20(m,12H),0.92-0.85(m,3H)。化合物〔25〕:收率99%,1HNMR(500MHz,CDCl3):δ7.35(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.35(s,1H),3.95(t,2H,J=6.6Hz),3.31(s,6H),1.81-1.73(m,2H),1.49-1.40(m,2H),1.39-1.20(m,16H),0.88(t,3H,J=6.9Hz)。化合物〔26〕:收率97%,1HNMR(500MHz,CDCl3):δ7.02-6.97(m,2H),6.88-6.84(m,1H),5.33(s,1H),3.90(s,3H),3.89(s,3H),3.33(s,6H)。化合物〔27〕:收率75%,1HNMR(500MHz,CDCl3):δ7.37-7.33(m,2H),6.92-6.87(m,2H),5.96-5.85(m,1H),5.35(s,1H),5.21-5.08(m,2H),4.02(t,2H,J=6.6Hz),3.31(s,6H),2.58-2.51(m,2H)。化合物〔28〕:收率98%、1HNMR(500MHz,CDCl3):δ7.33(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.33(s,1H),4.31-4.23(m,1H),3.32(s,6H),2.03-1.92(m,2H),1.86-1.73(m,2H),1.64-1.23(m,6H)。化合物〔29〕:收率97%,1HNMR(500MHz,CDCl3):δ7.37-7.33(m,2H),6.92-6.87(m,2H),5.35(s,1H),3.87-3.80(m,2H),3.31(s,6H),1.77-1.67(m,1H),1.55-1.25(m,8H),0.96-0.86(m,6H)。化合物〔30〕:收率96%,1HNMR(500MHz,CDCl3):δ6.98(s,1H),6.96(d,1H,J=8.2Hz),6.86(d,1H,J=8.2Hz),5.52(t,1H,J=6.6Hz),5.33(s,1H),4.59(d,2H,J=6.6Hz),3.88(s,3H),3.33(s,6H),1.77(s,3H),1.73(s,3H)。化合物〔31〕:收率98%,1HNMR(500MHz,CDCl3):δ6.98(s,1H),6.95(d,1H,J=7.9Hz),6.85(d,1H,J=7.9Hz),5.51(s,1H),5.31(s,1H),5.08(s,1H),4.62(d,2H,J=6.0Hz),3.88(s,3H),3.33(s,6H),2.15-2.02(m,4H),1.72(s,3H),1.67(s,1H),1.59(s,3H),1.80-1.52(m,9H)。化合物〔32〕:收率97%,1HNMR(500MHz,CDCl3):δ7.32-7.26(m,1H),7.06-7.02(m,2H),6.91-6.86(m,1H),5.38(s,1H),3.99(t,2H,J=6.6Hz),3.36(s,6H),1.85-1.76(m,2H),1.54-1.44(m,2H),1.42-1.32(m,4H),0.98-0.91(m,3H)。<使用对甲苯磺酸作为催化剂的二甲基缩醛化(路线3)>按照下述路线3,实施芳香族醛化合物的二甲基缩醛化,得到缩醛衍生物〔21〕。路线3化合物〔21〕的合成:在氮气氛下将4-正戊氧基苯甲醛〔11〕(1.9g,10mmol)和原甲酸三甲基(2.2mL,20mmol)溶解在4mL干燥甲醇中,在该溶液中加入对甲苯磺酸一水合物(8mg)。在室温下搅拌1小时,添加饱和碳酸氢钠终止反应。在该溶液中加入乙酸乙酯进行提取操作,用饱和氯化钠洗涤有机层。用硫酸钠干燥有机层,过滤硫酸钠后,对滤液进行减压浓缩。收率94%。<使用对甲苯磺酸作为催化剂的二乙基缩醛化(路线4)>按照下述路线4,实施芳香族醛化合物的二乙基缩醛化,得到缩醛衍生物〔20’〕及〔21’〕。路线4化合物〔20’〕的合成:在氮气氛下将4-正丁氧基苯甲醛〔10〕(2.5g,14mmol)和原甲酸三乙酯(5.0mL,38mmol)溶解在10mL干燥乙醇中,在该溶液中加入对甲苯磺酸一水合物(7mg)。在室温下搅拌1小时,添加饱和碳酸氢钠终止反应。在该溶液中加入乙酸乙酯进行提取操作,用饱和氯化钠洗涤有机层。用硫酸钠干燥有机层,过滤硫酸钠后,对滤液进行减压浓缩。收率78%、1HNMR(500MHz,CDCl3):δ7.37(d,2H,J=8.8Hz),6.88((d,2H,J=8.8Hz),3.96(t,2H,J=6.5Hz),3.63-3.57(m,2H),3.54-3.48(m,2H),1.80-1.72(m,2H),1.54-1.44(m,2H),1.23(t,6H,J=7.1Hz),0.97(t,3H,J=7.5Hz)。化合物〔21’〕的合成:在氮气氛下将4-正戊氧基苯甲醛〔11〕(1.9g,10mmol)和原甲酸三乙酯(3.4mL,20mmol)溶解在4mL干燥乙醇中,在该溶液中加入对甲苯磺酸一水合物(8mg)。在室温下搅拌1小时,添加饱和碳酸氢钠终止反应。在该溶液中加入乙酸乙酯进行提取操作,用饱和氯化钠洗涤有机层。用硫酸钠干燥有机层,过滤硫酸钠后,对滤液进行减压浓缩。收率95%、1HNMR(500MHz,CDCl3):δ7.37(d,2H,J=8.5Hz),6.87(d,2H,J=8.5Hz),5.46(s,1H),3.95(t,2H,J=6.7Hz),3.64-3.57(m,2H),3.55-3.47(m,2H),1.81-1.74(m,2H),1.47-1.33(m,4H),1.23(t,6H,J=7.1Hz),0.93(t,3H,J=7.1Hz)。[由缩醛衍生物合成凝胶化剂化合物]<由二甲基缩醛衍生物合成甘露糖型凝胶化剂>按照下述所示路线5,使甲基α-D-吡喃甘露糖苷与二甲基缩醛衍生物反应,合成作为甘露糖型凝胶化剂的化合物〔M1〕~〔M10〕。路线5化合物〔M1〕的合成:在氮气氛下,在甲基α-D-吡喃甘露糖苷(4.3g,22mmol)的DMF(20mL)溶液中加入对甲苯磺酸(190mg,0.5mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-甲氧基苯甲醛二甲基缩醛〔17〕(2.7g,20mmol)的DMF(10mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌2小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率55%,1HNMR(600MHz,CDCl3):δ7.44-7.39(m,2H),6.92-6.87(m,2H),5.52(s,1H),4.75(s,1H),4.30-4.22(m,1H),4.07-4.00(m,2H),3.93-3.86(m,1H),3.85-3.76(m,5H),3.40(s,3H),2.66(br,2H)。也按照相同的方法合成其它化合物〔M2〕~〔M10〕。对它们进行分析的数据列于下面。化合物〔M2〕:收率45%,1HNMR(600MHz,CDCl3):δ7.42-7.37(m,2H),6.91-6.86(m,2H),5.52(s,1H),4.76(d,1H,J=2.4Hz),4.30-4.23(m,1H),4.09-4.02(m,2H),3.96(t,2H,J=6.6Hz),3.93-3.86(m,1H),3.85-3.76(m,2H),3.40(s,3H),2.64-2.56(m,2H),1.79-1.72(m,2H),1.52-1.44(m,2H),0.97(t,3H,J=7.6Hz)。化合物〔M3〕:收率49%,1HNMR(600MHz,CDCl3):δ7.39(d,2H,J=8.8Hz),6.89(d,2H,J=8.8Hz),5.52(s,1H),4.76(s,1H),4.30-4.22(m,1H),4.08-4.02(m,2H),3.98-3.93(m,2H),3.92-3.86(m,1H),3.84-3.76(m,2H),3.40(s,3H),2.64-2.59(br,2H),1.81-1.73(m,2H),1.48-1.40(m,2H),1.38-1.29(m,4H),0.93-0.86(m,3H)。化合物〔M4〕:收率48%,1HNMR(600MHz,CDCl3):δ7.39(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.52(s,1H),4.76(s,1H),4.30-4.23(m,1H),4.08-4.02(m,2H),3.98-3.86(m,3H),3.85-3.76(m,2H),3.40(s,3H),2.65-2.59(br,2H),1.80-1.73(m,2H),1.47-1.40(m,2H),1.37-1.22(m,8H),0.92-0.85(m,3H)。化合物〔M5〕:收率43%,1HNMR(600MHz,CDCl3):δ7.39(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.52(s,1H),4.76(d,1H,J=2.4Hz),4.30-4.22(m,1H),4.08-4.01(m,2H),3.97-3.86(m,3H),3.84-3.76(m,2H),3.40(s,3H),2.65-2.56(br,2H),1.80-1.73(m,2H),1.47-1.39(m,2H),1.37-1.21(m,12H),0.88(t,3H,J=6.9Hz)。化合物〔M6〕:收率41%,1HNMR(600MHz,CDCl3):δ7.42-7.36(m,2H),6.91-6.85(m,2H),5.51(s,1H),4.75(s,1H),4.29-4.22(m,1H),4.08-4.01(m,2H),3.97-3.85(m,3H),3.84-3.76(m,2H),3.40(s,3H),2.68-2.63(br,2H),1.80-1.72(m,2H),1.57-1.39(m,2H),1.38-1.20(m,16H),0.88(t,3H,J=6.9Hz)。化合物〔M7〕:收率32%,1HNMR(600MHz,CDCl3):δ7.05-7.01(m,2H),6.85(d,1H,J=7.9Hz),5.52(s,1H),4.75(s,1H),4.32-4.24(m,1H),4.08-4.01(m,2H),3.91-3.85(m,7H),3.83-3.76(m,2H)3.40(s,3H),2.89-2.71(m,2H)。化合物〔M8〕:收率45%,1HNMR(600MHz,CDCl3):δ7.40(d,2H,J=8.6Hz),6.89(d,2H,J=8.8Hz),5.93-5.85(m,1H),5.51(s,1H),5.18-5.13(m,1H),5.12-5.08(m,1H),4.73(s,1H),4.29-4.21(m,1H),4.06-3.98(m,4H),3.91-3.85(m,1H),3.83-3.75(m,2H),3.39(s,3H),2.81-2.78(m,1H),2.77-2.75(m,1H),2.56-2.50(m,2H)。化合物〔M9〕:收率31%,1HNMR(600MHz,CDCl3):δ7.38(d,2H,J=8.6Hz),6.88(d,2H,J=8.8Hz),5.50(s,1H),4.74(s,1H),4.28-4.21(m,2H),4.06-3.99(m,2H),3.91-3.85(m,1H),3.83-3.75(m,2H),3.39(s,3H),2.76(d,1H,J=3.4Hz),2.73(d,1H,J=2.2Hz),2.01-1.90(m,2H),1.82-1.74(m,2H),1.60-1.45(m,3H),1.40-1.25(m,3H)。化合物〔M10〕:收率40%,1HNMR(600MHz,CDCl3):δ7.39(d,2H,J=8.6Hz),6.88(d,2H,J=8.6Hz),5.50(s,1H),4.69(s,1H),4.27-4.20(m,1H),4.03-3.94(m,2H),3.89-3.74(m,5H),3.37(s,3H),3.05(d,1H,J=2.9Hz),3.00(s,1H),1.74-1.67(m,1H),1.53-1.24(m,8H),0.94-0.87(m,6H)。<由二甲基缩醛衍生物合成葡萄糖型凝胶化剂>按照下述所示的路线6,使甲基α-D-吡喃葡萄糖苷与二甲基缩醛衍生物反应,合成作为葡萄糖型凝胶化剂的化合物〔G1〕~〔G9〕及〔G11〕~〔G13〕。路线6化合物〔G1〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.5g,13mmol)的DMF(10mL)溶液中加入对甲苯磺酸(59mg,0.3mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-乙氧基苯甲醛二甲基缩醛〔18〕(2.3g,12mmol)的DMF(5mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率50%,1HNMR(500MHz,CDCl3):δ7.40(d,2H,J=8.8Hz),6.88(d,2H,J=8.5Hz),5.49(s,1H),4.81(s,1H),4.28(d,1H,10.0Hz),4.03(q,2H,J=6.9Hz),3.93(t,1H,J=9.2Hz),3.80(t,1H,J=9.8Hz),3.73(t,1H,10.1Hz),3.64(t,1H,J=9.3Hz),3.51-3.46(m,4H),2.63(s,1H),2.21(d,1H,J=9.4Hz),1.40(t,3H,J=6.9Hz)。化合物〔G2〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.9g,15mmol)的DMF(15mL)溶液中加入对甲苯磺酸(63mg,0.3mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-丙氧基苯甲醛二甲基缩醛〔19〕(2.9g,14mmol)的DMF(8mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率66%,1HNMR(500MHz,CDCl3):δ7.40(d,2H,J=8.5Hz),6.88(d,2H,J=8.9Hz),5.49(s,1H),4.81(s,1H),4.28(d,1H,J=9.9Hz),3.96-3.89(m,3H),3.80(t,1H,J=9.8Hz),3.73(t,1H,J=10Hz),3.64(t,1H,J=9.4Hz),3.51-3.46(m,4H),2.65(s,1H),2.22(d,1H,J=9.4Hz),1.84-1.75(m,2H),1.02(t,3H,J=7.5Hz)。化合物〔G3〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(4.3g,22mmol)的DMF(20mL)溶液中加入对甲苯磺酸(190mg,0.5mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-丁氧基苯甲醛二甲基缩醛〔20〕(4.7g,21mmol)的DMF(10mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率85%,1HNMR(500MHz,CDCl3):δ7.40(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.49(s,1H),4.81(d,1H,J=3.8Hz),4.28(dd,1H,J=9.8,4.4Hz),3.97-3.91(m,3H),3.83-3.70(m,2H),3.64(dt,1H,J=9.5,4.1Hz),3.50-3.47(m,4H),2.67(s,1H),2.24(d,1H,J=9.5Hz),1.75(m,2H),1.48(sext,2H,J=7.6Hz),0.96(t,3H,J=7.6Hz)。化合物〔G4〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.7g,14mmol)的DMF(12mL)溶液中加入对甲苯磺酸(59mg,0.3mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-戊氧基苯甲醛二甲基缩醛〔21〕(3.0g,13mmol)的DMF(6mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率60%,1HNMR(500MHz,CDCl3):δ7.40(d,2H,J=8.8Hz),6.88(d,2H,J=8.9Hz),5.49(s,1H),4.81(s,1H),4.28(d,1H,J=10.0Hz),3.97-3.90(m,3H),3.80(t,1H,J=9.9Hz),3.73(t,1H,J=10.3Hz),3.64(t,1H,J=9.4Hz),3.52-3.45(m,4H),2.66(s,1H),2.23(d,1H,J=9.7Hz),1.81-1.73(m,2H),1.47-1.32(m,4H),0.92(t,3H,J=7.3Hz)。化合物〔G5〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.1g,11mmol)的DMF(10mL)溶液中加入对甲苯磺酸(98mg,0.5mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-己氧基苯甲醛二甲基缩醛〔22〕(2.5g,10mmol)的DMF(5mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率72%,1HNMR(500MHz,CDCl3):δ7.40(d,2H,J=8.8Hz),6.88(d,2H,J=8.9Hz),5.49(s,1H),4.81(s,1H),4.28(d,1H,J=9.9Hz),3.97-3.90(m,3H),3.80(t,1H,J=9.9Hz),3.73(t,1H,J=10.3Hz),3.64(t,1H,J=9.4Hz),3.51-3.45(m,4H),2.61(s,1H),2.20(d,1H,J=9.5Hz),1.81-1.72(m,2H),1.48-1.40(m,2H),1.38-1.30(m,4H),0.90(t,3H,J=7.1Hz)。化合物〔G6〕:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(4.3g,22mmol)的DMF(20mL)溶液中加入对甲苯磺酸(190mg,1.0mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-辛氧基苯甲醛二甲基缩醛〔23〕(5.6g,20mmol)的DMF(10mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率63%,1HNMR(500MHz,CDCl3):δ7.40(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.49(s,1H),4.80(d,1H,J=3.5Hz),4.28(dd,1H,J=10.1,4.8Hz),3.97-3.89(m,3H),3.83-3.70(m,2H),3.67-3.61(m,1H),3.51-3.45(m,4H),2.64(d,1H,J=2.2Hz),2.22(d,1H,J=9.4Hz),1.80-1.72(m,2H),1.48-1.22(m,10H),0.93-0.85(m,3H)。化合物〔G7〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(0.9g,4.6mmol)的DMF(4mL)溶液中加入对甲苯磺酸(40mg,0.2mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-癸氧基苯甲醛二甲基缩醛〔24〕(1.3g,4.2mmol)的DMF(2mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率54%,1HNMR(500MHz,CDCl3):δ7.39(d,2H,J=8.9Hz),6.88(d,2H,J=8.8Hz),5.49(s,1H),4.80(s,1H),4.28(d,1H,J=9.9Hz),3.97-3.89(m,3H),3.79(t,1H,J=9.8Hz),3.73(t,1H,J=10.1Hz),3.63(t,1H,J=9.4Hz),3.51-3.45(m,4H),2.64(s,1H),2.22(d,1H,J=9.7Hz),1.81-1.72(m,2H),1.48-1.39(m,2H),1.38-1.21(m,12H),0.88(t,3H,J=6.9Hz)。化合物〔G8〕:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(4.3g,22mmol)的DMF(20mL)溶液中加入对甲苯磺酸(190mg,1.0mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-十二烷氧基苯甲醛二甲基缩醛〔25〕(6.8g,20mmol)的DMF(10mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率33%,1HNMR(500MHz,CDCl3):δ7.39(d,2H,J=8.8Hz),6.88(d,2H,J=8.8Hz),5.49(s,1H),4.81(d,1H,J=4.1Hz),4.28(dd,1H,J=9.8,4.4Hz),3.97-3.90(m,3H),3.83-3.77(m,1H),3.73(d,1H,J=10.1Hz),3.64(dt,1H,J=9.2,4.1Hz),3.51-3.45(m,4H),2.66(s,1H),2.23(d,1H,J=9.5Hz),1.80-1.72(m,2H),1.47-1.39(m,2H),1.37-1.20(m,16H),0.88(t,3H,J=6.9Hz)。化合物〔G9〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.4g,13mmol)的DMF(10mL)溶液中加入对甲苯磺酸(55mg,0.3mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-(3-丁烯氧基)苯甲醛二甲基缩醛〔27〕(2.5g,11mmol)的DMF(5mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率81%,1HNMR(500MHz,CDCl3):δ7.40(d,2H,J=8.5Hz),6.89(d,2H,J=8.9H),5.94-5.84(m,1H),5.49(s,1H),5.16(d,1H,J=17.0Hz),5.10(d,1H,J=10.0Hz),4.81(s,1H),4.28(d,1H,J=9.9Hz),4.01(t,2H,J=6.6Hz),3.93(t,1H,J=9.2Hz),3.80(t,1H,J=9.8Hz),3.73(t,1H,J=10.2Hz),3.64(t,1H,J=9.3Hz),3.52-3.45(m,4H),2.63(s,1H),2.53(q,2H,J=6.7Hz),2.21(d,1H,J-9.5Hz)。化合物〔G11〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.5g,13mmol)的DMF(13mL)溶液中加入对甲苯磺酸(58mg,0.3mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加3-甲氧基-4-二甲基烯丙氧基苯甲醛二甲基缩醛〔30〕(3.1g,12mmol)的DMF(6mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率58%,1HNMR(500MHz,CDCl3):δ7.03(s,1H),7.00(d,1H,J=8.2Hz),6.86(d,1H,J=8.2Hz),5.52-5.47(m,2H),4.82(d,1H,3.8Hz),4.58(d,2H,J=6.6Hz),4.29(q,1H,J=5.0Hz),3.95(t,1H,J=9.2Hz),3.89(s,3H),3.82(t,1H,J=10.0Hz),3.74(t,1H,J=10.3Hz),3.65(t,1H,J=9.4Hz),3.50-3.47(m,4H),2.68(s,1H),2.24(d,1H,J=9.5Hz),1.76(s,3H),1.72(s,3H)。化合物〔G12〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(1.9g,10mmol)的DMF(10mL)溶液中加入对甲苯磺酸(45mg,0.2mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加3-甲氧基-4-香叶氧基苯甲醛二甲基缩醛〔31〕(3.0g,9mmol)的DMF(5mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率58%,1HNMR(500MHz,CDCl3):δ7.03(s,1H),7.00(d,1H,J=8.2Hz),6.85(d,1H,J=8.2Hz),5.48(s,1H),5.08(t,1H,J=6.8Hz),4.81(d,1H,J=3.7Hz),4.61(d,2H,J=6.3Hz),4.29(q,1H,J=4.9Hz),3.94(t,1H,J=9.3Hz),3.89(s,3H),3.87-3.79(m,1H),3.74(t,1H,J=10.3Hz),3.65(t,1H,J=9.4Hz),3.52-3.46(m,4H),2.71(s,1H),2.26(d,1H,J=9.4Hz),2.14-2.01(m,4H),1.72(s,3H),1.67(s,3H),1.60(s,4H)。化合物〔G13〕:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.1g,11mmol)的DMF(10mL)溶液中加入对甲苯磺酸(95mg,0.5mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加3-己氧基苯甲醛二甲基缩醛〔32〕(2.5g,10mmol)的DMF(5mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌2小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二乙基醚洗涤得到的固体,用柱色谱(二氧化硅凝胶,氯仿:乙酸乙酯=10:0~8:2(v/v))精制残留物,得到作为白色固体的目标产物。收率13%,7.30-7.24(m,1H),7.06-7.02(m,2H),6.91-6.86(m,1H),5.49(s,1H),4.80(d,1H,J=6.6Hz),4.29(dd,1H,J=9.8,4.4Hz),3.98-3.90(m,3H),3.85-3.70(m,2H),3.68-3.60(m,1H),3.51-3.42(m,4H),2.73(br,1H),2.29(br,1H),1.81-1.72(m,2H),1.50-1.41(m,2H),1.39-1.30(m,4H),0.93-0.88(m,3H)。<由二乙基缩醛衍生物合成葡萄糖型凝胶化剂>按照下述所示路线7,使甲基α-D-吡喃葡萄糖苷与二乙基缩醛衍生物反应,合成作为葡萄糖型凝胶化剂的化合物〔G3〕及〔G4〕。路线7化合物〔G3〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.5g,13mmol)的DMF(6mL)溶液中加入对甲苯磺酸(46mg,0.3mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-丁氧基苯甲醛二乙基缩醛〔20’〕(2.9g,12mmol)的DMF(6mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率85%。化合物〔G4〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(2.0g,10mmol)的DMF(9mL)溶液中加入对甲苯磺酸(52mg,0.3mmol),在室温下搅拌5分钟。在室温下向该混合液中滴加4-戊氧基苯甲醛二乙基缩醛〔21’〕(2.5g,9mmol)的DMF(5mL)溶液。滴加后,在室温下搅拌10分钟,减压搅拌1小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,减压浓缩滤液,用二异丙基醚洗涤得到的固体,得到作为白色固体的目标产物。收率72%。[由芳香族醛化合物一步法合成葡萄糖型凝胶化剂]<使用甲基-α-D-吡喃葡萄糖苷合成葡萄糖型凝胶化剂>按照下述所示的路线8,由芳香族醛化合物一步法合成作为葡萄糖型凝胶化剂的化合物〔G3〕及〔G13’〕。路线8化合物〔G3〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(1.9g,10mmol)的DMF(7mL)溶液中加入4-丁氧基苯甲醛〔10〕(1.8g,10mmol)和对甲苯磺酸(52mg,0.27mmol),在室温下搅拌10分钟。在室温下向该混合液中滴加原甲酸三乙酯(1.5g,10mmol)。滴加后,在室温下搅拌20分钟,减压搅拌2小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,对滤液进行减压浓缩,用己烷洗涤得到的固体,得到作为白色固体的目标产物。收率78%。化合物〔G13’〕的合成:在氮气氛下,在甲基α-D-吡喃葡萄糖苷(3.9g,20mmol)的DMF(13mL)溶液中加入3-丁氧基苯甲醛〔G6’〕(3.6g,20mmol)和对甲苯磺酸(92mg,0.5mmol),在室温下搅拌10分钟。在室温下向该混合液中滴加原甲酸三乙酯(3.8g,26mmol)。滴加后,在室温下搅拌20钟,在室温下减压搅拌1小时,在40℃下搅拌2小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,对滤液进行减压浓缩,用己烷洗涤得到的固体,得到作为白色固体的目标产物。收率65%1HNMR(500MHz,CDCl3):δ7.30-7.25(m,1H),7.06-7.03(m,2H),6.89(d,1H,J=9.2Hz),5.50(s,1H),4.81(d,1H,J=3.8Hz),4.29(q,1H,J=4.8Hz),3.99-3.91(m,3H),3.84-3.78(m,1H),3.74(t,1H,J=10.3Hz),3.65(t,1H,J=9,5Hz),3.52-3.42(m,4H),2.63(s,1H),2.21(d,1H,J=9.4Hz),1.79-1.72(m,2H),1.53-1.44(m,2H),0.97(t,3H,J=7.5Hz)。<使用乙基-α-D-吡喃葡萄糖苷合成葡萄糖型凝胶化剂>按照下述所示的路线9,由芳香族醛化合物通过一步法合成作为葡萄糖型凝胶化剂的化合物〔G3’〕。路线9化合物〔G3’〕的合成:在氮气氛下,在乙基α-D-葡糖甙(1.1g,5mmol)的DMF(3mL)溶液中加入4-丁氧基苯甲醛〔G10〕(0.9g,5mmol)和对甲苯磺酸(22mg,0.1mmol),在室温下搅拌10分钟。在室温下向该混合液中滴加原甲酸三乙酯(0.8g,5mmol)。滴加后,在室温下搅拌1小时,在室温下减压搅拌2小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,对滤液进行减压浓缩,用己烷洗涤得到的固体,得到作为白色固体的目标产物。收率66%1HNMR(500MHz,CDCl3):δ7.40(d,2H,J=8.5Hz),6.88(d,2H,J=8.8Hz),5.49(s,1H),4.91(d,1H,J=4.1Hz),4.26(q,1H,J=4.8Hz),3.98-3.91(m,3H),3.86-3.79(m,2H),3.72(t,1H,J=10.3Hz),3.65-3.52(m,2H),3.48(t,1H,J=9.5Hz),2.63(s,1H),2.19(d,1H,J=10.1Hz),1.79-1.72(m,2H),1.51-1.44(m,2H),1.27(t,3H,J=7.1Hz),0.97(t,3H,J=7.5Hz)。<使用D(+)-葡萄糖合成葡萄糖型凝胶化剂>按照下述所示的路线10,由芳香族醛化合物通过一步法合成作为葡萄糖型凝胶化剂的化合物〔G3”〕。路线10化合物〔G3”〕的合成:在氮气氛下,在D(+)-葡萄糖(1.8g,10mmol)的DMF(7mL)溶液中加入4-丁氧基苯甲醛〔G10〕(1.9g,10mmol)和对甲苯磺酸(54mg,0.3mmol),在室温下搅拌10分钟。在室温下向该混合液中滴加原甲酸三乙酯(1.5g,10mmol)。滴加后,在室温下搅拌20分钟,在室温下减压搅拌5小时。在反应液中加入饱和碳酸氢钠水溶液并用乙酸乙酯提取。用饱和食盐水洗涤后,用无水硫酸钠干燥。过滤后,对滤液进行减压浓缩,用己烷洗涤得到的固体并进行过滤。用柱色谱(二氧化硅凝胶,氯仿:甲醇=10:0至9:1(v/v))进行精制,得到白色固体。将精制后的HPLC图及1HNMR(500MHz)分别示于图1及图2中。在HPLC中观测到2种成分(保持时间2.16分钟、2.31分钟(溶剂:乙腈/水=50/50)),从1HNMR也可推定2种成分的存在,而从LC-MS观测到目标产物的分子离子峰(ESI-MScaldforC17H25O7[M+H]+341,found341)可确定得到了作为α体和β体的混合物的目标产物。收率32%。[实施例2~实施例17:凝胶化剂的凝胶形成能力及得到的凝胶的各种评价]将实施例1中合成的化合物M1-M10、G1-G9、G11-G13作为凝胶化剂,进行针对各种溶剂的凝胶形成能力(凝胶化能力)以及得到的凝胶的各种评价。同时,分别与作为公知物质(非专利文献1)已知的化合物A:甲基α-D-4,6-亚苄基甘露糖及化合物B:甲基α-D-4,6-亚苄基葡萄糖的凝胶化能力进行比较。[实施例2:与公知物质的凝胶化能力的比较]按以下方式进行凝胶化试验。在4mL的螺丝口样品管中加入凝胶化剂及各种溶剂,对于环己烷、乙腈、甲醇、乙醇、乙醇/水混合溶剂在90℃下,对于甲苯、水、DMSO/水混合溶剂在100℃下,对于辛烷、SH245、橄榄油、肉豆蔻酸异丙酯、乙二醇、甘油在120℃下,分别加热溶解30分钟。将得到的溶液冷却到室温并放置1小时,观察凝胶的形成。还有,放冷后,将溶液失去流动性,即使倒置样品管溶液也不流下的状态判定为“凝胶化”。对于凝胶化试验,近凝胶化剂的浓度:2.0、1.0、0.5、0.25、0.1wt%进行实施,将凝胶化所需要的凝胶化剂的最低浓度(wt%)作为最低凝胶化浓度。此处,所示的浓度单位wt%是指wt/vol×100。得到的结果示于表1及表2中。表中的数字表示最低凝胶化浓度(wt%),表中的符号表示形成的凝胶的状态,透明凝胶记为“G”,半透明凝胶记为“#G”,混浊凝胶记为“*G”,结晶化时记为“Cr”,部分凝胶化时记为“PG”。另外,即使在凝胶化剂:2wt%的浓度下也不凝胶时记为“-”。表1中分别列出了作为甘露糖型凝胶化剂的化合物M1~M10及上述A化合物的凝胶化试验的结果、表2中作为葡萄糖型凝胶化剂的化合物G3及G8以及上述B化合物的凝胶化试验的结果。表1甘露糖型凝胶化剂(M1~M10)和公知物质(A)的凝胶化试验结果※表中的数值表示各溶剂的凝胶化所需要的凝胶化剂(化合物)的最低浓度(wt%)。※G:透明凝胶#G:半透明凝胶*G:混浊凝胶Cr:结晶化物PG:部分凝胶化—:不凝胶化表2葡萄糖型凝胶化剂(G3及G8)和公知物质(B)的凝胶化试验结果※表中的数值表示各溶剂的凝胶化所需要的凝胶化剂(化合物)的最低浓度(wt%)。※G:透明凝胶#G:半透明凝胶*G:混浊凝胶Cr:结晶化物PG:部分凝胶化-:不凝胶化如表1及表2所示,本发明的凝胶化剂与使用作为公知物质的化合物A或化合物B的凝胶化剂相比,对于各种溶剂都可形成透明性高的凝胶,而且表现出最低凝胶化浓度低。令人感兴趣的是,作为本发明的凝胶化剂,尽管是在公知物质的化合物A或化合物B上引入了作为疏水性官能团的烃基的化合物,对于非极性溶剂(辛烷、甲苯、SH245)并不降低凝胶化能力,对于极性高的极性溶剂等(水、乙醇、乙二醇)可以得到凝胶形成能力变高的结果。特别是作为葡萄糖型凝胶化剂的G3,对于水形成了透明性比较高的凝胶。接着,对于乙醇/水混合系溶剂及DMSO/水混合系溶剂,按上述顺序实现实施例1中合成的甘露糖型凝胶化剂M2、M4、M6和作为公知物质的化合物A的凝胶化试验,评价它们的凝胶化能力。得到的结果示于表3及表4中。表3乙醇/水混合系溶剂中的凝胶化试验的结果※表中的数值表示各混合溶剂的凝胶化所需要的凝胶化剂(化合物)的最低浓度(wt%)。※G:透明凝胶#G:半透明凝胶*G:混浊凝胶Cr:结晶化物PG:部分凝胶化—:不凝胶化表4DMSO/水混合系中的凝胶化试验的结果※表中的数值表示各混合溶剂的凝胶化所需要的凝胶化剂(化合物)的最低浓度(wt%)。※G:透明凝胶#G:半透明凝胶*G:混浊凝胶Cr:结晶化物PG:部分凝胶化—:不凝胶化如表3及表4所示,在乙醇/水混合系溶剂及DMSO/水混合系溶剂的所有体系中作为公知物质的化合物A不进行凝胶化。另一方面,对于实施例1中合成的甘露糖型凝胶化剂M2、M4及M6,得到了在乙醇/水混合系及DMSO/水混合系中可以进行凝胶化的结果。另外,还得到了凝胶化剂的化合物的苯环上结合的烃基的烷基链长度越短,就可以使水的比率越多的混合溶剂发生凝胶化的结果。[实施例3:触变性试验]对于由甘露糖型凝胶化剂组成的各种凝胶的触变性进行评价。按以下方式进行触变性试验。与实施例2一样制备最低凝胶化浓度下的凝胶。然后,使用涡旋混合器进行振荡直到由凝胶变成溶胶状态,从而使凝胶崩解,在室温下静置1小时。1小时后,将加入了溶液的样品管倒置,将溶液呈不流下的状态时判定为“有触变性”。得到的结果示于表5中。还有,在表中,判定为有触变性时记为“○”,判定为“无触变性”时记为“×”,不形成凝胶时记为“-”。表5甘露糖型凝胶化剂的触变性评价AM1M2M3M4M5M6M8M10甲苯×○○○○○○○-SH245×××××○○××如表5所示,与公知化合物A相比,引入了烃基的本发明的凝胶化剂获得了形成具有触变性的凝胶的结果。[实施例4:贮存稳定性]评价将加入了0.05wt%的实施例1中制作的甘露糖型凝胶化剂:M1及M3以及作为公知物质的化合物A的SH245的凝胶在室温下放置7天时的凝胶稳定性。得到的结果示于图3中。如图3所示,对于本发明的凝胶化剂,与使用作为公知物质的化合物A的凝胶化剂相比,得到了脱水收缩量少、贮存稳定性优异的结果。另外,随着苯环上结合的烃基的烷基链的链长变长,脱水收缩量也变少,因此得到了暗示凝胶的贮存稳定性与烷基链长非常相关的结果。[实施例5:葡萄糖型凝胶化剂的凝胶化试验]如实施例2至实施例4所示,与作为公知物质的化合物A及化合物B相比,作为本发明的凝胶化剂的甘露糖型凝胶化剂和葡萄糖凝胶化剂均得到了具有良好的凝胶化能力的结果。另外,还得到了暗示通过引入烷基链,可形成触变性及贮存稳定性优异的凝胶的结果。根据这些结果,关于被暗示对于极性溶剂具有更高凝胶化能力的葡萄糖型凝胶化剂,进行对于在化妆品中配合频率高的油剂(肉豆蔻酸异丙酯、橄榄油、SH245)及醇(乙醇)配合水溶液的凝胶化试验。结果示于表6。凝胶化试验的条件与实施例2一样进行。表6葡萄糖型凝胶化剂的凝胶化试验结果※表中的数值表示溶剂的凝胶化所需要的凝胶化剂(化合物)的最低浓度(wt%)。※G:透明凝胶#G:半透明凝胶*G:混浊凝胶Cr:结晶化物PG:部分凝胶化*PG:部分混浊的凝胶化—:不凝胶化如表6所示,关于上述试验中使用的油剂大部分凝胶化剂形成了凝胶。特别是G1~G4、G9、G13得到了对于水形成了透明性比较高的凝胶的结果。由G1~G8的乙醇配合试验的结果可看出,得到了通过使凝胶化剂的化合物的苯环上结合的烃基的烷基链长变长,即使醇配合水溶液中醇的比例增加时也倾向于发生凝胶化的结果。[实施例6:醇配合水溶液凝胶的触变性试验]由实施例5中的醇配合试验,明确了各葡萄糖型凝胶化剂的可配合醇量。因此,与实施例3一样,进行醇配合水溶液凝胶的触变性试验。按以下方式进行触变性试验。首先,凝胶制备与实施例5一样进行,而触变性试验与实施例3一样进行。此时,对于凝胶化剂的浓度,从最低凝胶化浓度进行制备,慢慢升高浓度进行制备直到表现出触变性,观察触变性的出现。结果示于表7。表的框内(下部)的数字表示表现触变性的凝胶化剂的浓度(wt%),()内(上部)的数字及记号表示实施例5中所示最低凝胶化浓度及凝胶的状态。还有,作为公知物质的化合物B再现了实施例2所示结果。设定的凝胶化剂的浓度为0.1、0.25、0.5、1.0、2.0wt%。但是在设定的浓度范围内未表现出触变性时标记为“×”。表7醇配合水溶液凝胶的触变性评价※()内的数值表示凝胶化所需要的凝胶化剂(化合物)的最低浓度(wt%)。※G:透明凝胶#G:半透明凝胶*G:混浊凝胶Cr:结晶化物PG:部分凝胶化—:不凝胶化※下部的数值表示表现出触变性的凝胶化剂(化合物)的浓度(wt%)。(×:未表现出触变性)如表7所示,可以确认随着凝胶化剂化合物的苯环上结合的烃基的烷基链的链长变长,表现出了触变性。关于作为公知化合物的化合物B,确认未表现出触变性。其中,关于形成用G及#G标记的透明凝胶或半透明凝胶的凝胶,得到了在最低凝胶化浓度附近倾向于表现出触变性的结果。[实施例7:添加剂配合试验]通常化妆品及准医药品中配合有防腐剂及表面活性剂。对于本发明的凝胶化剂,研究配入防腐剂及表面活性剂时的凝胶可能性。<防腐剂配合试验>防腐剂配合试验按以下方式进行。将防腐剂溶解在水中达到规定的浓度来配制防腐剂配合水溶液。然后,在4mL的螺纹试管中添加凝胶化剂(葡萄糖型凝胶化剂:G3)达到规定浓度,加入预先配制的防腐剂配合水溶液,在100℃加热溶解30分钟。将得到的溶液冷却至室温并放置1小时,通过倒置螺纹试管来确认凝胶的形成。结果示于表8。使用的防腐剂是对羟基苯甲酸甲酯及苯氧基乙醇各自单独物质和它们的混合物。表8防腐剂配合试验如表8所示,本发明的凝胶化剂配入防腐剂时也得到了形成凝胶的结果。<表面活性剂配合试验>表面活性剂配合试验按以下方式进行。将表面活性剂溶解在水中达到规定的浓度来制备表面活性剂配合水溶液。然后,在4mL的螺纹试管中添加凝胶化剂(葡萄糖型凝胶化剂:G3)达到规定浓度,加入预先制备的表面活性剂配合水溶液,在100℃下加热溶解30分钟。将得到的溶液冷却至室温并放置1小时,通过倒置螺纹试管来确认凝胶的形成。结果示于表9。使用的表面活性剂是作为非离子性表面活性剂的聚氧乙烯山梨聚糖单月桂酸酯(Tween20)、作为阴离子性表面活性剂的十二烷基硫酸钠(SDS)、作为阳离子性表面活性剂的硬脂基三甲基铵溴化物(STAB)及作为两性表面活性剂的3-[(3-胆固醇氨丙基)二甲基氨基]丙磺酸盐(CHAPS)。表9表面活性剂配合试验如表9所示,本发明的凝胶化剂在配入表面活性剂的情况下也可得到能形成凝胶的结果。[实施例8:使用葡萄糖型凝胶化剂的水-油分散试验]对于化妆品的乳霜及清洁剂,为了使油剂和水均匀分散,并且使其稳定化,一般要使用表面活性剂。但是,对于表面活性剂的配合,由于对皮肤的刺激及皮肤变粗糙的问题,优选减少表面活性剂的配合量。在实施例7中,本发明的凝胶化剂即使配入表面活性剂配合时也能得到形成良好凝胶的结果。另一方面,如实施例2及实施例5所示,本发明的凝胶化剂,特别是葡萄糖型凝胶化剂:G1~G4,对于水和油系溶剂均可得到能形成凝胶的结果。该结果暗示,本发明的凝胶化剂即使不使用表面活性剂也能使水和油均匀分散,并且可达到稳定化。因此,在实施例8中,为了确认本发明的凝胶化剂是否能使水和油剂的双溶剂进行均匀分散,进行水-油分散试验。按以下方式进行水-油分散试验。在4mL的螺丝口样品管中加入凝胶化剂(葡萄糖型凝胶化剂:G4或G5)及各种溶剂达到规定的浓度,在100℃加热溶解30分钟。然后,用涡旋混合器剪切2分钟后,在室温下静置1小时,观察分散状态。还有,将放冷后,溶液失去流动性,即使倒置样品管溶液也不流下的状态,并且使水和油均匀分散的状态判定为“水/油分散凝胶”。此时,将水/油分散凝胶所需要的凝胶化剂的最低浓度(wt%)作为最低凝胶化浓度。得到的结果示于表10。表中的数字表示最低凝胶化浓度,将水/油分散凝胶记为“G”,将水/油分散得显示流动性时记为“W-sol”,将部分凝胶化时记为“PG”。在水-油分散试验中,使用对水和油剂双方具有凝胶化能力的G4和仅对油剂具有凝胶化能力的G5。这些凝胶化剂的浓度按2.0、1.0、0.5、0.25、0.1wt%进行实施。表10使用葡萄糖型凝胶化剂的水-油分散试验※数值表示水/油分散凝胶化所需要的凝胶化剂(化合物)的最低浓度。※G:形成水/油分散凝胶PG:部分凝胶化W-sol:水/油分散但显示流动性如表10所示,G4和G5均可以得到形成水/油分散凝胶的结果。特别是对于水和油剂的双溶剂具有凝胶化能力的G4,与仅使油剂凝胶化的G5相比,得到了最低凝胶化浓度低,并且水和油可分散的配合比例范围宽的结果。令人感兴趣的是,已明确对于水/油分散凝胶(G)及显示W-sol体系,能够在水和油不分离的情况下在室温下稳定保持其形状(凝胶或流动性的形状)4个月。图4显示了使用G4作为凝胶化剂的肉豆蔻酸异丙酯(IPM)和水中水/油分散凝胶的光学显微镜观察结果。通过光学显微镜观察观测到的液滴约为10~50μm。另外,对于与G4一样可以使水和油的两者进行凝胶化的G3,也使用SH245作为油剂,在同样的条件下进行水-油分散试验。图5显示了得到的水/油分散凝胶的外观。另外,图6显示了水/油分散凝胶的共聚焦激光扫描显微镜观察结果。如图5所示,对于G3,也与G4一样,得到了最低凝胶化浓度低,并且可使水和油分散的配合比例的范围宽,形成了良好的水/油分散凝胶。而且得到了由图6所示的共聚焦激光扫描显微镜观察结果所观测到的液滴为约10~50μm程度,具有比较均匀的尺寸的结果。[实施例9:使用均质器的水-油分散试验]对于实施例8中的水-油分散试验,在使凝胶化剂加热溶解在溶剂中后实施的搅拌中使用涡旋混合器。在本实施例中,使用作为更高剪切的搅拌手段的均质器实施水-油分散试验。可推测通过均质器提供高剪切,使得形成的液滴更小。使用均质器的水-油分散试验按以下方式进行。在试验管中加入葡萄糖型凝胶化剂:G3(0.25wt%)及溶剂(SH245和水的混合系:30/70~70/30(vol/vol)),在干浴槽中于100℃下加热溶解30分钟。然后,使用均质器剪切5分钟后,在室温下静置1小时,用共聚焦激光扫描显微镜观察分散状态。图7列出了结果。与使用涡旋混合器的情况一样,通过使用均质器的搅拌法也形成了均匀的水/油分散凝胶。另外,由共聚焦激光扫描显微镜看出,得到了使用均质器形成的液滴的尺寸最大约为30μm(图7),与使用涡旋混合器时的液滴的尺寸(10~50μm:图6)相比形成了较小液滴的结果。即,得到了以下结果:暗示着通过控制凝胶化剂加热溶解后的剪切速度,可以控制得到的水/油分散液中的液滴直径。[实施例10:预混料试验]对于实施例2~实施例9中的凝胶化试验以及水-油分散试验,将凝胶化剂(化合物)粉末添加到各种溶剂中后,使其加热溶解实施各试验。在该方法中,凝胶化剂粉末溶解在各种溶剂中之前需要时间,除此之外对各种溶剂的溶解时间不同,难说是通用手段。假如事先配制使粉末的凝胶化剂进行高浓度溶解的分散液,并将该分散液添加到各种溶剂中,从而能够迅速地制备凝胶或水/油分散凝胶,则从凝胶制备的工艺角度来看可以认为是迅速且简便的方法。象这样形成凝胶化剂的高浓度分散液后添加到各种溶剂中,形成凝胶或水/油分散凝胶的手段,在本说明书中定义为“预混料”。为了实施用于判断本发明的凝胶化剂对预混料是否适用的预混料试验,首先研究能够使凝胶化剂高浓度溶解的分散液。<预混料用分散液的溶剂研究>分散液中使用的溶剂的研究按以下方式实施。将凝胶化剂(葡萄糖型凝胶化剂:G4)添加到各种溶剂中达到规定的浓度(10wt%),在干浴槽中于80~100℃加热30分钟后,确认凝胶化剂是否溶解,放冷1小时返回室温。然后,为了确认是否能作为预混料用分散液使用,实施80℃下的再溶解试验。分散液中使用的溶剂选择乙醇(EtOH)、丙二醇(PG)、丁二醇(BG)、戊二醇(PenG)、甘油(Gly)的醇系溶剂。结果示于表11。表11G4分散液的研究结果PG:丙二醇,BG:丁二醇,PenG:戊二醇,Gly:甘油如表11所示,对于将G4按10wt%的浓度添加到各种醇系溶剂中所配制的分散液,可确认乙醇、丙二醇、丁二醇、戊二醇加热时均匀溶解,放冷后在80℃下再溶解。另一方面,可确认甘油在加热时和再加热时均有不溶物,判定为形成10wt%G4分散液时溶解度不足,对于预混料用分散液是不适合的。基于以上结果,作为分散液的溶剂,选择可再溶解的丙二醇,按以下顺序实施预混料试验。<使用分散液的预混料试验>如下进行预混料试验。首先,在螺纹试管加入肉豆蔻酸异丙酯(21g),在80℃下加热。然后,添加加热到80℃的10wt%G4丙二醇分散液(6.6g),形成均匀溶液后,添加到加热到70℃的水(9g)。用均质器搅拌10分钟,制备水/肉豆蔻酸异丙酯分散凝胶。预混料试验后的最终组成示于表12。另外图中列出了水/肉豆蔻酸异丙酯分散凝胶的外观(图8(A))及显微镜观察的结果(图8(B))。表12预混料试验后的凝胶最终组成质量/g质量%肉豆寇酸异丙酯21.057.4水9.0024.6丙二醇5.9416.2凝胶化剂:G40.661.8合计36.6100如图8所示,得到的结果是即使在预混料的情况下也形成了良好的水/油分散凝胶(图8(A))。另外,得到的结果是如显微镜观察(图8(B))所示,液滴的尺寸也与由粉末制备时处于同等程度(最大为大约30μm)。[实施例11:凝胶取出试验]由本发明的凝胶化剂制备的凝胶是具有可以自立的固态凝胶,另一方面,对剪切来说非常弱,显示液体性,具有剪切后立即凝固的特征。由于这些特征,制备的凝胶可以容易地从容器中取出,另外也容易用锋利的刀具切断,切断后能够不脱水收缩而保持凝胶的形状。<使用甘露糖型凝胶化剂(M3)的凝胶取出试验>使用甘露糖型凝胶化剂(M3)的凝胶取出试验按以下方式进行。在样品管中添加M3及角鲨烯(2.5mL)达到0.5wt%后,在130℃下加热溶解30分钟,放冷1小时至室温,从而制成凝胶。然后,使用刮铲慢慢取出凝胶。图9中,显示了从样品管取出的凝胶的外观照片。图9(A)是刚取出后的照片,图9(B)是取出后的凝胶用玻璃盖片(厚度:0.12-0.17mm)切断后的照片,图9(C)是将切断的凝胶沿切断面相互叠合后的照片。如图9所示,得到的结果是取出的凝胶可以自立(自支撑性),另外在剪切后不脱水收缩而保持了凝胶的形状,切断面相互叠合时还可以重新粘接,即能够具有自己修复性的凝胶。<使用葡萄糖型凝胶化剂(G4)的凝胶取出试验>与甘露糖型凝胶化剂(M3)一样,使用作为葡萄糖型凝胶化剂的G4,进行凝胶取出试验。在样品管中添加葡萄糖型凝胶化剂(G4)及各种溶剂达到规定的浓度后,在120~130℃下加热溶解30分钟。然后,将该溶液(约2mL)灌入直立的圆筒状筒中,放冷1小时至室温,从而制成凝胶。从圆筒状筒中缓慢挤出凝胶,从而进行凝胶的取出试验。结果就于表13。对于表中的记号,取出后可保持凝胶形状时判定为“○”,稍微软化时判定为“△“,不能保持形状时判定为“×”。另外,未实施时用斜线表示。表13G4凝胶的取出试验结果※○:取出后保持凝胶形状,△:取出后凝胶形状稍微软化×:取出后不能保持凝胶形状如表13所示,得到的结果是与甘露糖型凝胶化剂(M3)一样,对于使用葡萄糖型凝胶化剂(G4)的凝胶,也可以进行凝胶取出。[实施例12:喷射试验]如实施例11的结果所示,由本发明的凝胶化剂制备的凝胶是具有可自立的固态性的凝胶,另一方面,对剪切来说非常弱,显示出液体性,具有剪切后立即固态化的特征。由于这些特征,本发明的凝胶在化妆品等中用于通常使用的喷射等喷雾器时,可以期待能均匀地喷雾,另外还喷雾后立即成为凝胶状,可期待具有没有滴流的良好涂布特性。喷射试验按以下方式进行。在样品管中加入葡萄糖型凝胶化剂(G3、G6)和水/乙醇混合溶剂(75/25或50/50(vol/vol)(4mL)达到0.25wt%,在80℃下加热溶解30分钟。然后,将该溶液转移到喷射瓶管((株)マルエム制,No.3L)中,放冷1小时至室温,制成喷射瓶管盛放凝胶。然后,将制备的喷射瓶管盛放凝胶从相距4cm的位置向玻璃板(10×7.5cm)上喷雾,评价是否可喷雾。另外确认被喷雾的凝胶的广度(长和宽的长度),及1分钟后的滴流。喷射试验的结果示于表14。表14喷射试验的结果如表14所示,得到的结果是使用G3或G6得到的凝胶均在玻璃板上达到了均匀分散,并且未产生滴流,即可以进行喷射喷雾。作为该结果,如实施例6所示,就是因为由G3及G6制备的凝胶具有触变性而造成的特征。另外,凝胶在倒置状态下也能稳定地保持其形状暗示着即使倒置喷射容器也能发挥喷射功能。[实施例13:溶胶ー凝胶相转变温度]测定甲苯的溶胶ー凝胶相转变温度(Tgel)。测定时,将放入了用甘露糖型凝胶化剂(M1~M6、M8)或公知物质的化合物A和甲苯制备的凝胶的样品管,放入温度得到控制的干浴槽中并在25℃下静置5分钟后,从干浴槽中取出样品管,倒置并确认凝胶是否变成溶液从从下方落下。保持了凝胶的状态时将干浴槽设定为30℃,放入加入了凝胶的样品管并静置5分钟后,进行同样的操作。重复进行以上的操作,每次将干浴槽升高5℃,将倒置样品管而凝胶变成溶液从下方落下的温度温度作为Tgel。图10中显示了各凝胶化剂的甲苯凝胶的相对于凝胶化剂的浓度的溶胶ー凝胶相转变温度(Tgel)。通过同样的试验,将溶剂变换为SH245,按最低凝胶化浓度制备的SH245凝胶的凝胶化剂的浓度和溶胶ー凝胶相转变温度(Tgel)示于表15。表15SH245凝胶的溶胶ー凝胶相转变温度AM1M2M3M4M5M6M8M10浓度/wt%0.0250.050.050.050.250.250.50.050.1Tgel/℃5585707095951009040如图10所示,可看出对于所有甲苯凝胶随着凝胶化剂的浓度提高溶胶-凝胶相转变温度Tgel变高的趋势。另外得到的结果是凝胶化剂的苯环上结合的烃基的烷基链长越长,溶胶ー凝胶转移温度越低。可以认为这是因为凝胶化剂本身对溶剂的亲和性在提高。另外,如果将烷基链长相同的凝胶化剂M2(CH3(CH2)3O-)和M8(3-丁烯基-O-)进行比较,则得到的结果是通过在烷基末端引入烯烃结构,使得溶胶ー凝胶转移温度上升。另外,如表15所示,得到的结果是与作为公知物质的已知凝胶化剂化合物A相比,使用凝胶化剂M1~M6及M8得到的SH245凝胶的溶胶-凝胶相转变温度更高。这表明与已知的化合物A相比,本发明的凝胶化剂可制备热稳定的凝胶。[实施例14:甲苯凝胶(干凝胶)SEM观察]制备加入了1wt%实施例1中制备的甘露糖型凝胶化剂:M2或公知物质化合物A的甲苯凝胶后,将凝胶真空干燥24小时,用扫描型显微镜(SEM)观察由此得到的干凝胶。得到的结果示于图11。从图11中所示的SEM图像看出,相对于在凝胶化剂A的甲苯凝胶(干凝胶)情况下得到宽度为大约500nm至800nm的纤维状图像,在凝胶化剂M2的甲苯凝胶(干凝胶)情况下得到了板状图像。[实施例15:使用凝胶化剂M2或M6的甲苯凝胶(干凝胶)的AFM观察]为了确认由实施例14的SEM图像中确认的本发明的凝胶化剂得到的甲苯凝胶(干凝胶)中板状体的详细信息,进行AFM图像观察。在100℃下加热制备添加了实施例1中合成的甘露糖型凝胶化剂:M2(0.02wt%)或M6(0.02wt%)的甲苯溶液,将其浇铸在石墨基板(HOPG)上后,静置1小时后真空干燥24小时,用原子力显微镜(AFM)观察HOPG。得到的结果示于图12。还有,图12中还一并显示了按最低凝胶化浓度(M2:0.5wt%、M6:2wt%)凝胶化的凝胶的外观照片。得到的结果是,由图12中所示的AFM图像看出由凝胶化剂M2的甲苯溶液制备的凝胶(干凝胶)具有从HOPG开始高度约为3.5nm的纤维结构。由该结果可看出,在实施例13的SEM图像中看到的凝胶化剂M2的板状体是宽度约3.5nm的纤维结构集束化而形成了板状。另外,如果利用AFM比较观察由凝胶化剂M2和M6的甲苯溶液制备的凝胶(干凝胶),则得到的结果是可看出由M2的甲苯溶液制备的凝胶(干凝胶)形成了高度约为3.5nm的纤维结构,与此相对,由M6的甲苯溶液制备的凝胶(干凝胶)中高度为3.1nm至3.8nm的纤维发生集束化而形成了带状结构。[实施例16:使用凝胶化剂M2的甲苯凝胶及水凝胶(干凝胶)的SEMM观察]制备加入了0.5wt%实施例1中制作的甘露糖型凝胶化剂:M2的甲苯凝胶及加入了0.1wt%的M2的水凝胶,分别冻结干燥24小时,通过SEM观察由此得到的干凝胶。得到的结果示于图13。由图13所示的SEM图像看出,得到的结果是甲苯凝胶(干凝胶)为纤维集束化形成的板状体,与此相对,水凝胶(干凝胶)中形成了宽度约为200nm的纤维结构。[实施例17:水凝胶(干凝胶)的SEM观察]制备加入了0.1wt%实施例1中制作的甘露糖型凝胶化剂:M2或葡萄糖型凝胶化剂:G3的水凝胶,冻结干燥24小时,通过SEM观察由此得到的干凝胶。得到的结果示于图14。由图14所示的SEM图像看出,得到的结果是使用M2的水凝胶(干凝胶)形成了宽度约为200nm的纤维结构,与此相对,使用G3的水凝胶(干凝胶)形成了宽度约为40至50nm的纤维结构。如此得到的结果是,对于水凝胶,通过将凝胶化剂的糖部分的结构从甘露糖变换为葡萄糖,凝胶的透明性就提高了,而由各种凝胶化剂组成的纤维结构的宽度倾向于变细。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1