去甲万古霉素衍生物及其制备纯化方法与流程
本发明属于药物化学领域,具体而言,本发明涉及去甲万古霉素衍生物及其制备纯化方法。
背景技术:
去甲万古霉素是一类糖肽类抗生素,在中国已经商业化应用近四十年,临床主要应用于治疗心内膜炎、骨髓炎等严重感染性疾病1。去甲万古霉素主要对革兰氏阳性菌有效,特别是耐甲氧西林金黄色葡萄球菌、耐甲氧西林表皮葡萄球菌和耐青霉素的肠杆菌、肺炎链球菌以及梭状芽孢杆菌。去甲万古霉素的产生菌为1959年李群等2从贵州土壤中分离出的放线菌万-23号,后来命名为东方拟无枝酸菌(Amycolatopsis orientalis)。去甲万古霉素有着与另一个糖肽类化合物万古霉素(VCM)基本类似的化学结构和生物活性3,它们均由一个三环组成的七肽母体和一个由葡萄糖-氨基糖组成的双糖取代基,不同之处在于去甲万古霉素的N-末端较万古霉素缺少一个N-甲基(图1)。文献中组成七肽的氨基酸分别以AA-1到AA-7表示,五个芳香环从A-E依次编号,另外,三个大环的编号以芳香环编号组成A-B,C-O-D和D-O-E,为了方便化合物结构解析,本申请将沿用文献的编号方法。
早期,万古霉素初始使用阶段,临床使用过程中发现其引起多种负反应,如肾毒性和耳毒性等,可能的原因是其纯度低,杂质含量高的原因。随后,人们使用高效液相色谱(HPLC)4-7,毛细管电泳(CE)8以及液相质谱-质谱连用技术(LC-MS/MS)4,9对万古霉素有关物质进行过系统深入的研究,从中发现了八个杂质分别是去氯万古霉素A和B10,去双氯万古霉素10,万古霉素结晶过程降解产物CDP-IM和CDP-Im11,去双糖万古霉素12,去氨基糖万古霉素12和去甲万古霉素。进一步药理活性实验研究发现,含有CDP-IM和CDP-Im的万古霉素药品能引起临床治疗失败13,而三个去氯万古霉素类似物的抗菌活性比万古霉素降低了2至10倍3,去氨基糖的万古霉素的体内抗菌活性也大大降低,MIC约为万古霉素的五倍。因此,《欧洲药典》14和《美国药典》15将去氨基-,去双糖基-,和去甲基万古霉素定义为万古霉素原料三个主要杂质,并且规定其单杂不得超过4%,总杂不得超过7%。
相比而言,去甲万古霉素由于其杂质成分不明确,《中国药典》16规定以峰面积积分计算,总杂不得超过12%,单杂少于4%。因此,需要更为有效的纯化及鉴定去甲万古霉素的方法,同时,为了明确去甲万古霉素原料药中有关物质成分结构,还需对去甲万古霉素原料药进行更为系统的化学成分研究,发掘其新结构的结构类似物。
参考文献:
1.Wu,X.J.,et al.Establishment of norvancomycin fluorescence polarization immunoassay for therapeutic drug monitoring.J Antibiot(Tokyo).65,35-39(2012).
2.Li Q.,S.A.L.,Liu J.R.,&Wang X.Y.Actinomycetes Van 23-vancomycin producing strain (Shanghai scientific&Technical Publishers,Shanghai,China,1962).
3.Nagarajan,R.Structure-activity relationships of vancomycin-type glycopeptide antibiotics.J Antibiot(Tokyo).46,1181-1195(1993).
4.Diana,J.,Visky,D.,Hoogmartens,J.,Van Schepdael,A.&Adams,E.Investigation of vancomycin and related substances by liquid chromatography/ion trap mass spectrometry.Rapid Commun Mass Spectrom.20,9(2006).
5.Diana,J.,Visky,D.,Roets,E.&Hoogmartens,J.Development and validation of an improved method for the analysis of vancomycin by liquid chromatography selectivity of reversed-phase columns towards vancomycin components.J Chromatogr A.996,115-131(2003).
6.Inman,E.L.Determination of vancomycin related substances by gradient high-performance liquid chromatography.J Chromatogr.410,363-372(1987).
7.Jesus Valle,M.J.,Lopez,F.G.&Navarro,A.S.Development and validation of an HPLC method for vancomycin and its application to a pharmacokinetic study.J Pharm Biomed Anal.48,835-839(2008).
8.Kang,J.W.,Van Schepdael,A.,Roets,E.&Hoogmartens,J.Analysis of vancomycin and related impurities by micellar electrokinetic capillary chromatography.Method development and validation.Electrophoresis.22,4(2001).
9.Hadwiger,M.E.,Sommers,C.D.,Mans,D.J.,Patel,V.&Boyne,M.T.2nd.Quality assessment of U.S.marketplace vancomycin for injection products using high-resolution liquid chromatography-mass spectrometry and potency assays.Antimicrob Agents Chemother.56,7(2012).
10.Harris,C.M.,Kannan,R.,Kopecka,H.&Harris,T.M.The role of the chlorine substituents in the antibiotic vancomycin:preparation and characterization of mono-and didechlorovancomycin.Journal of the American Chemical Society.107,6652-6658(1985).
11.Harris,C.M.,Kopecka,H.&Harris,T.M.Vancomycin:structure and transformation to CDP-I.Journal of the American Chemical Society.105,6915-6922(1983).
12.Nagarajan,R.&Schabel,A.A.Selective cleavage of vancosamine,glucose,and N-methyl-leucine from vancomycin and related antibiotics.Journal of the Chemical Society,Chemical Communications.1306-1307(1988).
13.Somerville,A.L.,Wright,D.H.&Rotschafer,J.C.Implications of vancomycin degradation products on therapeutic drug monitoring in patients with end-stage renal disease.Pharmacotherapy.19,702-707(1999).
14.The European Pharmacopoeia Commission.The European Pharmacopoeia 8th edn.(Council of Europe:Strasbourg,2008).
15.The United States Pharmacopeia Commission(The United States Pharmacopeia Convention,Inc,Rockville,MD,USA,2012).
16.Chinese Pharmacopoeia Commission,Pharmacopoeia of the People's Republic of China(in Chinese)2015edn(China Medical Science and Technology Press,Beijing,China,2015).
技术实现要素:
本发明从去甲万古霉素原料药中分离得到三个糖肽类化合物,借助于1D,2D NMR和HRESIMS等方法确定三个化合物结构,均为去甲万古霉素类似物,包括一个D-O-E扩环和两个去糖基衍生物。
本发明首先涉及一种去甲万古霉素及其衍生物的HPLC制备分离方法,所述方法为:
(1)流动相:甲醇:0.05~0.2%甲酸水溶液,
(2)梯度洗脱步骤:30~50min内甲醇从10%逐渐上升至70%,
(3)将去甲万古霉素原料药水溶液过色谱柱,流速为1.0~5.0ml/min,检测波长280nm,所述的去甲万古霉素原料药优选为去甲万古霉素盐酸盐原料药,
(4)收集主峰产物即得去甲万古霉素纯品,收集次峰产物即得去甲万古霉素衍生物。
所述方法中,色谱柱优选为C18色谱柱或苯基柱。
本发明还涉及由所述方法分离获得的去甲万古霉素衍生物,所述的去甲万古霉素衍生物为:化合物1(结构如下式1所示)、化合物2或3(结构如下式2、3所示),
本发明还涉及所述化合物1-3在制备药物中的应用,所述药物为抗革兰氏阳性菌的药物,所述的革兰氏阳性菌优选为:耐甲氧西林金黄色葡萄球菌、耐甲氧西林表皮葡萄球菌、耐青霉素的肠杆菌、肺炎链球菌以及梭状芽孢杆菌。
附图说明
图1化合物1-3,去甲万古霉素和万古霉素的结构,化合物1的HMBC相关用虚线弯箭头表示。
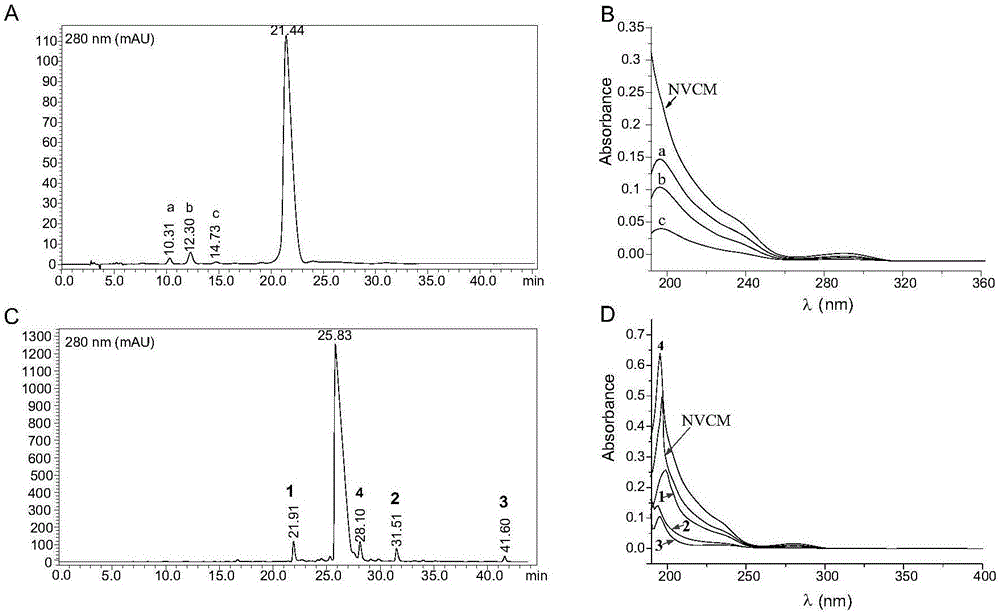
图2去甲万古霉素原料药的HPLC色谱分析图。
2A,使用《中国药典》方法的HPLC图谱;
2B,A图中a,b,c和去甲万古霉素的紫外图谱叠加图;
2C,方法优化后HPLC图谱;
2D,C图中杂质1,2,3,4和去甲万古霉素的紫外图谱叠加图。
图3去甲万古霉素(母离子为[M+H]+m/z 1434,图3A)和化合物1(母离子为[M+2H]2+m/z 724.5,图3C)的ESIMS/MS图和两者的裂解途径(3B为去甲万古霉素,3D为化合物1)。
具体实施方式
去甲万古霉素盐酸盐由华北制药(中国河北石家庄市)提供;
万古霉素标准品购于中国食品药品检定研究院;
色谱级乙腈和甲醇购于Adamas公司(中国上海);
DMSO-d6由Cambridge Isotope公司生产(Cambridge,MA,USA.);
所有其它试剂(甲酸、磷酸、盐酸、三氟乙酸、氨水(含氨气质量分数为25~28%)、三乙胺、四氢呋喃)均为化学级。
使用的仪器,包括P-2000旋光仪(JASCO,Tokyo,Japan),JASCOP-650紫外分光光度计(JASCO),SYS 600MHz核磁仪(Palo Alto,CA,USA),Finnigan LTQ XT离子阱质谱仪(Finnigan,San Jose,CA),Finnigan LTQ轨道离子阱谱仪(Finnigan),岛津LC-20液相色谱仪(Shimadzu,Japan)。
核磁测定方法:
所有样品的1H和13C NMR均在带低温探头的SYS 600MHz核磁仪上测定,溶剂为氘代DMSO。1H和13C NMR化学位移均使用TMS(δ=0.00ppm)和DMSO-d6(δ=39.5ppm)溶剂做内标。
实施例1化合物1~3的制备与分离纯化
首先,我们使用了《中国药典》中去甲万古霉素原料药HPLC方法来分析样品(见图2A和2B),发现在保留时间为21.44min的一个主峰,积分面积占所有峰面积总和的95.5%,根据药典中去甲万古霉素出峰位置为18~22min,推测该物质就是去甲万古霉素,另外,如图2A中所示,保留时间在该主峰前后还存在至少三个积分面积较小的杂质峰,保留时间分别为10.31(a),12.30(b),14.73(c)min,它们的在线紫外吸收峰型也与去甲万古霉素相类似,如图2B所示。然而,由于该方法中使用了磷酸盐,并且因为制备过程或者制备后样品中磷酸盐的残留会严重干扰质谱检测,因此,这方法不适合对样品进行液相色谱-质谱联用(LC-MS)。
我们为了借助于LC-MS初步获得这些衍生物杂质分子量等相应信息,建立了一套流动相不含盐的HPLC方法。当我们使用甲醇-0.1%甲酸水溶液进行HPLC分析时候,发现相应的主峰和杂质峰都能获得很好的峰型,并且杂质与杂质之间,杂质与主峰之间也具有较好的分离度,如图2C所示,衍生物杂质1,2,3,4峰面积分别占总面积的3.4%,1.91%,0.96%,和2.02%,主峰为87.97%。
(1)优化的HPLC分析方法如下:
Agilent C18色谱柱(150×4.6mm i.d.,5μm),
流动相:甲醇:0.1%甲酸水溶液,
梯度洗脱步骤:(1)40min内甲醇从10%逐渐上升至70%。
1.0ml min-1流速,280nm波长检测,柱温和检测池温度均为室温(25℃),
3.0mg ml-1去甲万古霉素原料药水溶液,进样量为20μl。
(2)优化的HPLC制备方法如下:
YMC-Pack苯基柱(150×20mm i.d.,20μm,YMC Co.,Ltd,Kyoto,Japan),
流动相配置与梯度洗脱方法与分析实验一致。
流速为5.0ml min-1,单次洗脱时间为120min。
100mg ml-1去甲万古霉素原料药水溶液,单次进样量为300μl。
利用优化的无盐HPLC方法对各杂质进行了系统分离制备,经减压蒸馏回收溶剂后,从5.0g去甲万古霉素原料药中分离得到了化合物1(5.0mg),化合物2(5.0mg),化合物3(1.5mg)以及4.5g去甲万古霉素纯品(纯度大于95%)。
实施例2化合物1~3结构解析
化合物1,2,3的紫外吸收与去甲万古霉素相似(见图2D),提示它们可能为去甲万古霉素结构类似物。
化合物1,无色油状物,[α]20D-44.5(c 0.22,MeOH),高分辨电喷雾电离质谱(HRESIMS)给出其准分子离子峰[M+2H]2+m/z 724.7505,分子量为1447Da,结合NMR数据推测其分子式为C66H75O24N9Cl2,不饱和度为33,提示其分子中含有多个芳环及其它不饱和结构。将化合物1的分子式与去甲万古霉素(C65H73O24N9Cl2)相比较,发现仅有一个亚甲基片段CH2的差异,因此,推测化合物1可能是去甲万古霉素的甲基化产物。将化合物1电喷雾二级质谱(ESI-MS/MS)图谱与去甲万古霉素相应图谱进行比较(图3),发现其碎片离子峰如m/z1305,1143与去甲万古霉素的碎片离子峰(m/z 1291,1129)也具有相同差值。进一步对两个裂解途径进行分析(图3),发现化合物1与去甲万古霉素具有相同的双糖结构片段,因此,甲基化位点只可能发生在七肽的母体骨架上。
利用核磁异核单量子相关(HSQC)图谱将化合物1的1H NMR和13C NMR的相应信号一一对应,并根据二维同核质子相关谱(HHCOSY),核磁异核多量子远程相关谱(HMBC)等数据解析,将化合物1结构中组成七肽的相应氨基酸归属出来,包括三个苯基甘氨酸,一个亮氨酸,两个3,4-二取代的苯丙氨酸和一个天冬酰胺。由于化合物1可能是去甲万古霉素的甲基化产物,因此将化合物1与去甲万古霉素在相同溶剂中(DMSO-d6)的NMR数据进行比较(表1),发现具有化学位移变化最大的位置集中在D-O-E环的天冬酰胺和苯丙氨酸片段上。比如化合物1中H-(AA-3)-2和H-(AA-3)-3a与去甲万古霉素相应位置化学位移比较分别向高场位移了ΔδH-0.37和-0.12ppm,而H-(AA-3)-3a向低场位移了ΔδH+0.4ppm。这些集中于天冬氨酸残基上的核磁数据的变化与CDP-I(包括CDP-IM和CDP-Im,差别仅在E环上的C-Cl键取向)11和万古霉素差异是一致的。CDP-I是万古霉素D-O-E扩环重排的产物,所以推测化合物1也可能是D-O-E环扩环重排的类似物。
尤其在2D-NOESY图谱中,H2-(AA-3)-3与H-(AA-2)-5,H-(AA-3)-2,H-(AA-2)-6相关,和H-(AA-2)-5与H-(AA-4)-2相关表明这些质子处于环平面一侧;H-(AA-2)-2与H-(AA-2)-7,H-(AA-2)-8,H-(AA-4)-6相关,H-(AA-4)-6与H-(AA-4)-7相关,表明这些质子处于平面的另一侧,而且E环上的C-Cl键的取向与去甲万古霉素、万古霉素以及CDP-IM相同。而其余部分NMR数据与去甲万古霉素相应部分差别不大,表明化合物1具有与去甲万古霉素相同的A-B,C-O-D环组成部分。
在化合物的1H和13C NMR中处于δH 3.17(3H,s)和δC 48.5ppm的信号说明化合物中含有一个N-甲基结构片段。通过分析化合物1与去甲万古霉素以及万古霉素相同条件下水解产物的ESI-MS数据确认,这个N-甲基位于结构中亮氨酸片段的N-末端。在ESI-MS中,化合物1具有与万古霉素水解产物相同的N-甲基亮氨酸离子峰(m/z 146[M+H]+)。因此,化合物1的结构被确定为D-O-E扩环的N-甲基去甲万古霉素。
化合物1和去甲万古霉素1H和13C NMR数据见下表1。
表1
注:化合物1中NH-(AA-1)-1,NH-(AA-2)-9,NH-(AA-4)-8,NH-(AA-5)-8和NH-(AA-6)-9的化学位移分别为δH 8.59,8.43,8.88,8.50和8.50ppm;而N-CH3的NMR数值分别为δH3.17ppm,δC 48.5ppm.
化合物2的光谱数据显示其也是去甲万古霉素一个类似物,将化合物2的NMR数据(表2)与去甲万古霉素进行比较,发现去甲万古霉素的双糖部分被化合物2的一个葡萄糖基取代,而其它位置数据没有大的改变。特别是1H NMR中属于去甲万古霉素中vancosaminyl片段的甲基信号δH 1.29(s)和1.06(d,J=6.6Hz),以及该片段的糖基端基信号δ5.23(m),在化合物2中消失。而且13C NMR数据显示化合物2的葡萄糖基的C-2位的化学位移较去甲万古霉素相应位置向高场位移(ΔδC–4.4ppm),这些数据结合HRESIMS中化合物2分子量较去甲万古霉素低143,说明化合物2就是去甲万古霉素的去氨基糖化合物。因此,化合物2被定为去氨基糖去甲万古霉素。
化合物3的紫外和核磁数据显示其为化合物2的类似物,仔细比较两者NMR数据,特别是1H NMR数据,显示化合物3就是2的去葡萄糖基类似物,另外,化合物3的HRESIMS谱图给出了一个[M+H]+的准分子离子峰在m/z 1129.2780,计算得出其分子式为C52H50O17N8Cl2,并且该分子量比化合物2少了162,进一步验证了化合物3就是化合物2的去葡萄糖基化合物,所以化合物3被确定为去双糖去甲万古霉素。
化合物2的1H和13C NMR数据和化合物3的1H NMR数据见下表2。
表2
实施例3化合物1~3活性测定
化合物的最低抑菌浓度(MIC)使用由美国临床和实验室标准协会提供的平板稀释法,使用万古霉素作为阳性对照,所有的测试菌为ATCC标准菌株或者临床分离菌株。Muller-Hinton(M-H)牛肉汤被用作为检定培养基,接种量为10,000cfu/spot,每一个样品的浓度范围为0.5至128μg ml-1,在35℃培养条件下培养18小时,以肉眼不可见菌体生长为最低抑菌浓度MIC。
化合物1~3和万古霉素(VCM)、去甲万古霉素(NVCM)原料药及纯品(纯度>95%)的抗菌活性比较见下表3。
表3
a:纯度>95%的去甲万古霉素样品。ATCC,美国菌种保藏中心;MRSA,耐甲氧西林金黄色葡萄球菌;MSSA,甲氧西林敏感金黄色葡萄球菌;MRSE,耐甲氧西林表皮葡萄球菌;MSSE,甲氧西林敏感表皮葡萄球菌;VISA,万古霉素中介金黄色葡萄球菌;VRE,耐万古霉素肠球菌;VSE,万古霉素敏感肠球菌。
结果显示,去甲万古霉素纯品的抗菌活性与去甲万古霉素原料药以及万古霉素相当,所获新化合物均对革兰氏阳性菌尤其是耐药菌MRSA和MRSE有活性。化合物1的抗菌活性较去甲万古霉素明显减弱;化合物2显示出与去甲万古霉素相当或略低的抗MRSA和MRSE能力;而化合物3显示出与去甲万古霉素相当的抗菌能力,在部分菌株上略优于去甲万古霉素。此外,包括阳性对照在内的所有测试化合物均无抗大肠杆菌(Escherichia coli)等革兰氏阴性菌和耐万古霉素肠球菌的能力。
最后需要说明的是,以上实施例仅用作帮助本领域技术人员理解本发明的技术方案的实质,并不用作对本发明保护范围的限定。
- 还没有人留言评论。精彩留言会获得点赞!