一种高流速多糖微球及其制备方法与流程
本发明属于生物技术领域,具体涉及一种高流速多糖微球的制备方法。
背景技术:
琼脂糖是一种天然多糖,系海洋植物石花菜或江蓠等红藻提取的琼脂经精制加工而成,是由1,3-连接的β-D-吡喃半乳糖和1,4-连接的3,6-脱水-α-L-吡喃半乳糖残基交替连接而成的直链线性多糖。琼脂糖在温度较高时可以溶于水,形成琼脂糖水溶液,在冷却过程中则形成凝胶,其中含有大量能允许大分子扩散的孔道。琼脂糖可用于制备凝胶微球,具有羟基密度大、亲水性好、可取代基团多、易于活化、优良的孔结构、非特异性吸附低以及稳定性好等特点,常用于层析、酶固定化和细胞培养等领域。以层析介质为例,迄今为止,琼脂糖凝胶过滤介质及在此基础上得到的各种功能性层析介质已广泛应用于生物制药、食品工业及化工等领域,可分离蛋白、核酸、肽类及糖等多种物质。但是,传统琼脂糖层析介质机械强度低、不耐压,实际应用中受到流速限制,影响分离效率。制备耐压性能好、流速快的琼脂糖微球,在其基础上制备其他功能性层析介质,以能够满足快速分离生物大分子的要求。
目前,琼脂糖微球的制备方法包括制球和交联两步。首先是制备琼脂糖微球,通常采用搅拌法(Hjertén S.The preparation of agarose spheres for chromatography of molecules and particles.Biochim.Biophys.Acta,1964,79:393-398)、喷射法(Bengtsson S,Philipson L.Chromatography of animal viruses on pearl-condensed agar.Biochim.Biophys.Acta,1964,79:399-406)和膜乳化法(CN 1304101C)完成。其次是对琼脂糖微球进行交联,传统的交联方法采用双功能团交联剂或多功能团交联剂,如环氧氯丙烷(US Patent 3,507,851)、草酰氯(US Patent 3,860,573)和季戊四醇三缩水甘油醚(US Patent 4,665,164)等,虽然在一定程度上提高了微球的机械强度,但是,由于交联过程中交联剂容易发生水解及自交联等副反应,导致交联效率大大降低且交联程度不可控,制得的琼脂糖微球耐压性能差。图1(a)是这种琼脂糖微球的结构示意图,其交联位点分布松散且随机性大,从而影响微球机械强度。
为了进一步提高琼脂糖微球的交联强度,Lindgren等以溴丙烯作为交联剂,交联琼脂糖微球(US Patent 4,973,683)。与上述双功能团或多功能团交联剂相比,溴丙烯的结构特点是一端为活性、另一端为惰性,一开始活性端基与琼脂糖反应,接下来通过控制反应条件,使另一惰性端基与琼脂糖反应。该反应的特点是交联程度可控,交联效率高。Hans Berg等在制球过程中引入交联剂,即在琼脂糖溶液中加入溴丙烯,制得琼脂糖微球上连有双键,再通过一系列反应打开双键,从而得到交联琼脂糖微球(US Patent 6,602,990)。图1(b)是以溴丙烯为交联剂制备的交联琼脂糖微球内部凝胶结构示意图,与传统的双功能团或多功能团交联剂相比,溴丙烯交联微球内部交联位点数目更多且分布更均匀,微球的强度大大提高。赵希等在该方法基础上,将一部分多糖经交联剂改性,得到修饰多糖,并与未修饰多糖混合后作为水相,成球后再经后期活化交联,得到高流速多糖微球(CN 201210413412.9)。
以溴丙烯作为交联剂,尽管对改善琼脂糖微球的交联程度以及提高微球流速起到一定效果,但是,由于天然琼脂糖具有较大的刚性,分子链较长,分子尺寸较大,溶液粘度大(几百—几千CP,甚至更高),溴丙烯与其反应时存在一定空间位阻效应,易造成交联不均匀,在很大程度上影响了琼脂糖微球的最终交联效率。
有鉴于上述的缺陷,本设计人,积极加以研究创新,以期创设一种高流速多糖微球及其制备方法,使其更具有产业上的利用价值。
技术实现要素:
为解决上述技术问题,本发明的目的是提供一种高流速多糖微球及其制备方法。以该方法制备的多糖微球具有机械强度高、交联程度可控的特点。
本发明的多糖微球的制备方法,包括降解多糖溶液得到多糖降解溶液的步骤。所得多糖降解溶液的粘度为10-1000cP。
进一步的,还包括将所述多糖溶液和多糖降解溶液改性的步骤。
进一步的,多糖微球的制备方法包括以下步骤:
(1)在60-100℃和碱性条件下,将所述多糖降解溶液与多糖溶液分别与改性剂反应,反应时间为0.5-8h,得到改性多糖降解溶液和改性多糖溶液,将改性后的两溶液混合,得到多糖混合溶液;所述多糖降解溶液与多糖溶液中[OH-]浓度为0.01-5mol/L,优选为0.1-2mol/L,且反应结束后用浓冰醋酸溶液调节体系pH至中性。
(2)将所述多糖混合溶液在60-100℃和碱性条件下与改性剂反应,反应时间为0.5-8h,得到改性多糖混合溶液,作为水相;
(3)将所述改性多糖混合溶液加入含乳化剂的油相中乳化、固化,得到改性多糖微球;其中油相可以是液体石蜡、甲苯/CCl4、环己烷体系等;乳化剂是油溶性乳化剂,如Span系列、PO500等,优选为Span85与Span60复配而成,二者复配比例为19:1-5:1,优选为19:1-9:1,乳化剂含量为油相的0.5-5%(w/v);成球过程包括搅拌法、喷射法和膜乳化法。
(4)将改性多糖微球经洗涤并与溴水反应得到溴化多糖微球,在碱性条件下,所述溴化多糖微球和交联剂在有机溶剂进行交联反应,得到多糖微球。所述有机溶剂为二甲亚砜和丙酮中一种或组合,其用量为多糖微球的1-30%(v/w),且有机溶剂中[OH-]浓度为0.01-3mol/L,优选为0.05-2mol/L;交联剂为环氧氯丙烷或1,4-丁二醇双缩水甘油醚;根据需要,交联反应可以反复多次进行,交联过程中还可补加交联剂。
进一步的,所述多糖溶液中的多糖为琼脂、琼脂糖、葡聚糖、魔芋多糖和纤维素中的一种。
进一步的,所述多糖溶液和多糖降解溶液中多糖的质量浓度均为1-15%。
进一步的,所述改性剂为一端双键、另一端可以与多糖分子上的羟基反应的双键试剂,优选双键试剂为溴丙烯或烯丙基缩水甘油醚中的一种或组合。
进一步的,所述改性剂在被改性溶液中的体积百分比为0.1-30%,优选为1-10%。
进一步的,步骤(1)中,多糖降解溶液在多糖溶液中的体积百分比为10-90%,优选为20-40%。
进一步的,采用化学降解法(如酸降解法)、生物降解法(如酶降解法)或物理降解法(如超声降解法)对所述多糖溶液进行降解。
本发明的另一方面还提供一种多糖微球,该多糖微球具有更高的机械强度和耐压性能,操作过程中可以实现高流速,在层析等领域具有更加广泛的应用前景。
借由上述方案,本发明至少具有以下优点:
本发明通过对多糖溶液进行降解和改性,将短链改性多糖降解溶液和改性多糖溶液混合,再与改性剂反应,得到改性多糖混合溶液(水相),多次改性保证能够得到更多的惰性官能团,改性多糖混合溶液经乳化、固化过程成球后,再经碱化交联反应,最终制得高流速多糖微球。
与现有的多糖微球相比,本发明制备的高流速多糖微球具有明显的优点:首先,由于多糖的分子链较长,空间位阻作用导致传统交联方法的交联效果有限,形成的网络结构较为松散,仅靠增加交联剂用量无法有效解决这一问题,反而会造成局部交联过度,而本发明首先对多糖溶液进行降解,得到一定粘度范围的短链多糖降解溶液,再对其进行改性,以这种改性短链多糖降解溶液与改性多糖溶液分别作为短、长链两种单体,在一定条件下发生共聚,形成类似“block polymer”的结构,如图1(c),与传统交联方法相比,长、短链单体形成的交联结构具有更多的交联连接点,网络结构更为紧密,多糖微球的耐压性能更高;本发明中的短链多糖降解溶液来源于对多糖溶液的降解,较结构单一且不可控的市售多糖而言,本发明得到的短链多糖降解溶液具有一定粘度范围,且通过调节降解工艺条件可以实现多糖粘度可控,从而形成更有效的凝胶网络结构;其次,传统交联方法的碱化反应在水相中进行,副反应较多,交联效率低,需要进一步进行交联反应以提高微球的强度,操作步骤繁琐。为解决该问题,本发明在有机相中进行碱化反应,同时加入双功能团交联剂,不仅有效避免了副反应的发生,大大提高了反应效率,而且减少了反应步骤,缩短了反应时间。与现有的多糖微球相比,本发明制备的多糖微球具有更高的机械强度和耐压性能,操作过程中可以实现高流速,在层析等领域具有更加广泛的应用前景。
本发明的方法步骤中:(1)降解多糖溶液,采用化学降解法、物理降解法或生物降解法对多糖溶液进行降解,得到粘度范围为10-1000cP的短链多糖降解溶液,短链多糖降解溶液的分子尺寸及其分布极大地影响多糖微球的最终交联程度;(2)多糖溶液的改性过程,采用含烯丙基改性剂分别对短链多糖降解溶液和多糖溶液进行改性,得到共聚反应的两种单体;多糖溶液的改性程度是影响多糖微球交联程度的重要因素之一;(3)改性短链多糖降解溶液和改性多糖溶液以一定比例混合,并进一步改性后,以其作为水相,与含有乳化剂的油相发生乳化反应,再固化成球,两种单体的混合比例极大地影响多糖微球的机械强度;(4)碱化过程在有机相中进行,反应同时加入双功能团交联剂,有机溶剂可以是二甲亚砜或丙酮等,从而有效减少副反应的发生,交联剂是环氧氯丙烷或1,4-丁二醇双缩水甘油醚等,能够大大提高交联效率,根据需要,交联反应可以反复多次进行,交联过程中还可补加交联剂。
上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例并配合附图详细说明如后。
附图说明
图1为现有技术的交联方法和本发明的交联方法制备的琼脂糖微球结构示意图,其中(a)环氧氯丙烷交联,(b)溴丙烯交联,(c)改性长短链单体共聚交联;
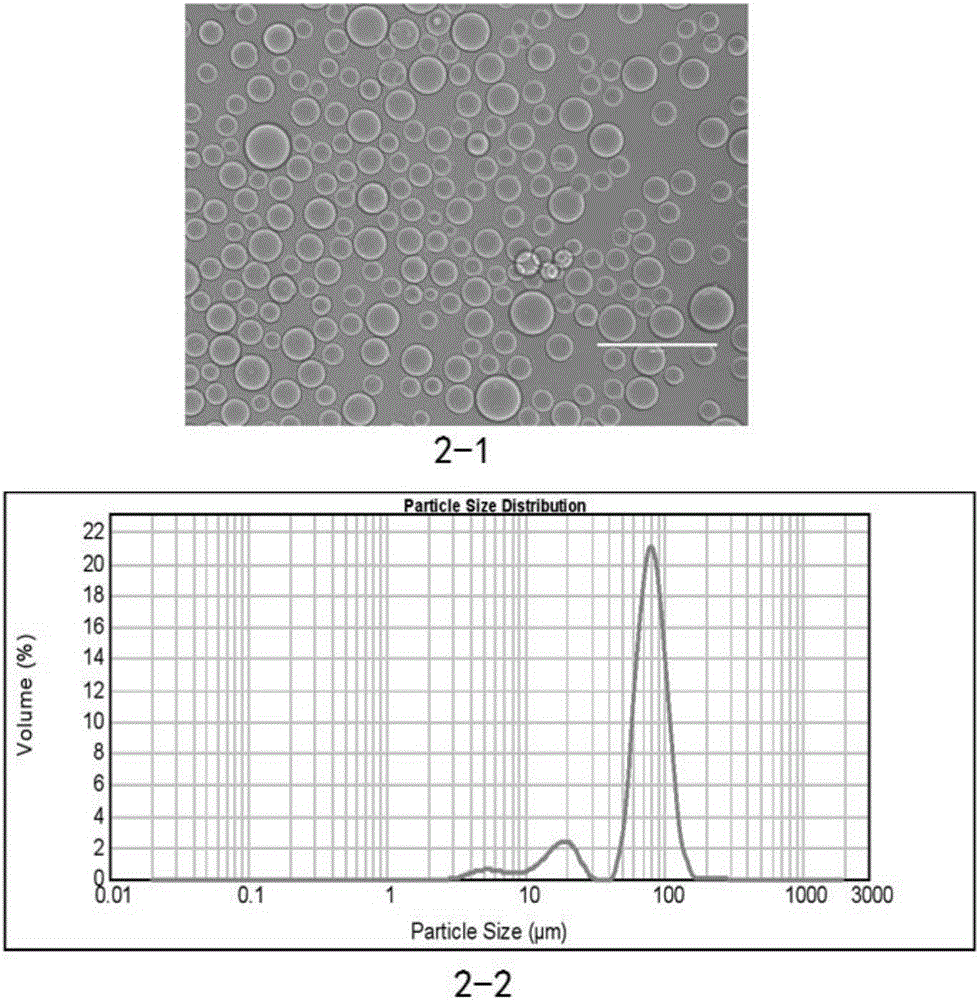
图2为本发明中实施例1制备的质量浓度为6%的高流速琼脂糖微球的光学显微镜照片(2-1)和粒径分布图(2-2);
图3为本发明中实施例1至7和对比例1至2与商品介质Sepharose 6FF的压力-流速曲线比较图;
图4为本发明中三种荧光标记蛋白分别在由实施例1制备的6%高流速琼脂糖微球(a)和商品介质Sepharose 6FF(b)中分布情况的激光共聚焦显微镜照片,图中(a)和(b)从左至右依次为核糖核酸酶、牛血清白蛋白和甲状腺球蛋白;
图5为本发明中实施例1制备的6%琼脂糖微球的凝胶过滤色谱图;
图6为本发明中实施例1、实施例5、对比例1及对比例2的凝胶过滤色谱实验各组分Kav值。
图7为本发明中实施例2制备的微球的光学显微镜照片(7-1)和粒径图(7-2);
图8为本发明中实施例3制备的微球的光学显微镜照片(8-1)和粒径图(8-2);
图9为本发明中实施例4制备的微球的光学显微镜照片(9-1)和粒径图(9-2);
图10为本发明中实施例6制备的微球的光学显微镜照片(10-1)和粒径图(10-2);
图11为本发明中实施例7制备的微球的光学显微镜照片(11-1)和粒径图(11-2)。
具体实施方式
下面结合附图和实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,但不用来限制本发明的范围。
实施例1:制备质量浓度为6%的高流速琼脂糖微球
(1)琼脂糖降解溶液的制备
采用酸降解法降解琼脂糖溶液,在100℃下加热90mL琼脂糖溶液(含琼脂糖6.7g)20min,加入浓盐酸调节体系pH 3.0,继续保持5min,得到短链琼脂糖降解溶液,用NaOH溶液调节体系至中性。
将琼脂糖降解溶液加热至100℃待其溶解后,采用Brookfiled DV-II+Pro粘度计测定溶液粘度,测得短链琼脂糖降解溶液的粘度值为800cP。
(2)改性琼脂糖降解溶液和改性琼脂糖溶液的制备
向步骤(1)中的琼脂糖降解溶液体系加入5mL溴丙烯、5mL20%NaOH溶液,于90℃反应2h。反应结束后用冰醋酸调节体系pH至中性,得到改性琼脂糖降解溶液(单体A)。
在100℃下加热90mL琼脂糖溶液(含琼脂糖6.7g)30min,加入5mL溴丙烯、5mL 20%NaOH溶液,于90℃反应2h。反应结束后用冰醋酸调节体系pH至中性,得到改性琼脂糖溶液(单体B)。
(3)改性琼脂糖混合溶液(水相溶液)的制备
在90℃下将30mL单体A与60mL和单体B混合,加入5mL溴丙烯、5mL 20%NaOH溶液,反应2h。反应结束后用冰醋酸调节体系pH至中性,得到质量浓度为6%的改性琼脂糖混合溶液(水相溶液)。
(4)改性琼脂糖微球的制备
在60℃下加热600mL液体石蜡(含1g Span60,9g Span85)30min,得到油相。将100mL上述步骤(3)得到的改性琼脂糖混合溶液加入油相,于600rpm搅拌30min,冷水浴降温固化,得到改性琼脂糖微球。
(5)溴化琼脂糖微球的制备
按照溴化钾-溴酸钾标准溶液滴定法测定改性琼脂糖微球的烯丙基密度,其中改性琼脂糖微球的烯丙基密度为200μmol/g抽干湿胶,改性琼脂糖微球经反复洗涤抽干后,加入过量饱和溴水,(室温)20-35℃下反应2h,加入甲酸钠后体系由黄色变无色,充分洗涤改性琼脂糖微球,得到溴化琼脂糖微球。
(6)交联过程
称取步骤(5)中50g溴化琼脂糖微球,加入200mL二甲亚砜、10mL环氧氯丙烷、4mL 40%NaOH溶液,于20-35℃下反应4h后,升温至40℃,继续反应4h。反应结束后充分洗涤微球,得到高流速琼脂糖微球。
采用XSZ-H3型光学显微镜观察高流速琼脂糖微球的形貌,所得的高流速琼脂糖微球形貌规整;采用Mastersizer 2000E激光粒度分析仪分析琼脂糖微球的粒径,光学显微镜照片和粒径分布图如图2中(2-1)和(2-2)所示。
高流速琼脂糖微球装柱待床层稳定后,层析柱连入PPS 100蛋白层析系统。设定流速,待压力稳定后记录此时压力值。逐渐增加柱子流速,测定柱子在不同流速下的压力值。随着流速的不断增大,柱子压力值相应增加。以压力为横坐标,流速为纵坐标,得到微球的压力-流速曲线。该曲线在一定范围内呈线性,该范围内微球未发生变形,其最高点对应的流速即为微球的最大线性流速,其值越大,说明微球的流通性能越好。测得高流速琼脂糖微球的最大线性流速为2751cm/h,如图3所示,高于传统环氧交联法和溴丙烯交联法制备的微球。
配制1mg/mL标准蛋白溶液(核糖核酸酶、牛血清白蛋白和甲状腺球蛋白),向其中加入少量荧光染料FITC,4℃、pH 8下振荡过夜。次日,离心数次,洗去未被结合的荧光染料,得到经荧光标记的标准蛋白。向其中加入30mg微球,4℃下振荡过夜,次日离心洗球。采用激光共聚焦观察荧光标记蛋白在微球孔道中的分布情况。激光共聚焦显微镜实验结果表明,采用该法制备的高流速琼脂糖微球对一系列不同分子量标准蛋白均有很好的渗透性能,蛋白都能进入微球孔道,且均匀分布在微球内部,如图4所示。
配制一系列标准蛋白溶液(核糖核酸酶(RNA),Mw 13.7kDa;牛血清白蛋白(BSA),Mw66.12kDa;甲状腺球蛋白(Thyrobolulin),Mw 660kDa),浓度为2mg/mL。介质装柱采用50mM PB-0.15M NaCl(pH7.0)缓冲液平衡柱子。蛋白依次上柱,继续采用50mM PB-0.15M NaCl(pH7.0)缓冲液洗脱蛋白,记录蛋白保留体积。分别采用Blue Dextran 2000(Mw 2,000kDa)和丙酮测定柱子的外水相体积和总流动相体积。计算各组分的保留系数(Kav)。凝胶过滤色谱实验结果表明,微球具有良好的凝胶过滤色谱性能,蛋白按照分子量由大到小的顺序先后洗脱,如图5所示,各组分Kav值如图6中的表格所示。
实施例2:制备质量浓度为6%的高流速琼脂糖微球
与实施例1中的步骤相同,区别在于:采用酶降解法降解琼脂糖溶液,在100℃下加热90mL琼脂糖溶液(含琼脂糖6.7g)20min,降温至42℃,加入100mg琼脂糖酶,继续保持2h,得到短链琼脂糖降解溶液,测定其粘度值为700cP;依次得到改性琼脂糖微球和溴化琼脂糖微球,其中改性琼脂糖微球的烯丙基密度为180μmol/g抽干湿胶,最终制备得到的高流速琼脂糖微球最大线性流速为2610cm/h,如图3所示,光学显微镜照片和粒径分布图如图7中(7-1)和(7-2)所示。
实施例3:制备质量浓度为6%的高流速琼脂糖微球
与实施例1中的步骤相同,区别在于:采用超声降解法法降解琼脂糖溶液,在100℃下加热90mL琼脂糖水溶液(含琼脂糖6.7g)20min,降温至50℃,超声继续保持2h(超声条件:35kHZ,300W/cm2)得到短链琼脂糖降解溶液,测定其粘度值为450cP;以烯丙基缩水甘油醚代替溴丙烯作为改性剂,依次得到改性琼脂糖微球和溴化琼脂糖微球,其中改性琼脂糖微球的烯丙基密度为890μmol/g抽干湿胶,光学显微镜照片和粒径分布图如图8中(8-1)和(8-2)所示;最终制备得到的高流速琼脂糖微球最大线性流速为2680cm/h,如图3所示。
实施例4:制备质量浓度为4%的高流速琼脂糖微球
与实施例1中的步骤相比,增加了补交交联剂的步骤,其区别在于:在100℃下加热90mL琼脂糖水溶液(含琼脂糖4.5g)20min,测得短链琼脂糖降解溶液粘度值为200cP;100℃下加热90mL琼脂糖水溶液(含琼脂糖4.5g)30min;依次得到改性琼脂糖微球和溴化琼脂糖微球,其中改性琼脂糖微球的烯丙基密度为140μmol/g抽干湿胶,光学显微镜照片和粒径分布图如图9中(9-1)和(9-2)所示;交联反应中,为进一步提高交联效果,采取交联过程补加交联剂的方法,称取50g溴化琼脂糖微球,加入200mL二甲亚砜,10mL环氧氯丙烷,4mL 40%NaOH溶液,于室温下反应4h后,升温至40℃,补加10mL环氧氯丙烷,继续反应4h,反应结束后充分洗涤微球,得到高流速琼脂糖微球。最终制备得到的高流速琼脂糖微球最大线性流速为1560cm/h,如图3所示。
实施例5:制备质量浓度为6%的高流速琼脂糖微球
与实施例1中的步骤相同,区别在于:采取多次交联的方法,即称取50g实施例1中制备得到的6%交联琼脂糖微球,加入200mL二甲亚砜,10mL环氧氯丙烷,4mL 40%NaOH溶液,于室温下反应4h后,升温至40℃,继续反应4h。反应结束后充分洗涤琼脂糖微球,得到高流速琼脂糖微球,其最大线性流速为3013cm/h,高于传统环氧交联法和溴丙烯交联法制备的微球,如图3所示;凝胶过滤色谱实验结果表明,微球具有良好的凝胶过滤色谱性能,各组分Kav如图6中的表格所示。
实施例6:制备质量浓度为6%的高流速琼脂微球
与实施例1中的步骤相同,区别在于:在100℃下加热90mL琼脂溶液(含琼脂6.7g)20min,测得短链琼脂溶液的粘度值为500cP;在100℃下加热90mL琼脂水溶液(含琼脂6.7g)30min;依次得到改性琼脂微球和溴化琼脂微球,其中改性琼脂微球的烯丙基密度为110μmol/g抽干湿胶,光学显微镜照片和粒径分布图如图10中(10-1)和(10-2)所示;最终制备得到的高流速琼脂微球最大线性流速为2180cm/h,如图3所示。
实施例7:制备质量浓度为12%的高流速葡聚糖微球
与实施例1中的步骤相同,区别在于:采用超声降解法降解葡聚糖溶液,在10℃下超声90mL葡聚糖溶液(含葡聚糖6.7g)30min(超声条件:35kHZ,500W/cm2),得到短链葡聚糖降解溶液,用浓NaOH溶液调节体系至中性。测定短链葡聚糖降解溶液的粘度值为100cP;依次得到改性葡聚糖微球和溴化葡聚糖微球,其中改性葡聚糖微球的烯丙基密度为520μmol/g抽干湿胶光学显微镜照片和粒径分布图如图11中(11-1)和(11-2)所示;最终制备得到的高流速葡聚糖微球最大线性流速为2570cm/h,如图3所示。
对比例1:制备质量浓度为6%的琼脂糖微球
采用环氧试剂交联法制备,配制质量浓度为6%琼脂糖溶液,按照实施例1中步骤(4)、(5)和(6)的方法制备6%琼脂糖微球。其最大线性流速为992cm/h,低于改性降解多糖参与制备的琼脂糖微球,如图3所示。
对比例2:制备质量浓度为6%的琼脂糖微球
采用溴丙烯交联法。在100℃下加热90mL琼脂糖水溶液(含琼脂糖6g)30min,加入5mL溴丙烯、5mL 20%NaOH溶液,于90℃反应2h。反应结束后用冰醋酸调节体系pH至中性,得到6%改性琼脂糖溶液。按照实施例1步骤步骤(4)、(5)的方法依次制备改性琼脂糖微球和溴化琼脂糖微球,其中改性琼脂糖微球的烯丙基密度为105μmol/g抽干湿胶。按照实施例1步骤(6)的方法对琼脂糖微球进行交联,得到的琼脂糖微球最大线性流速为1152cm/h,低于改性降解多糖参与制备的琼脂糖微球,如图3所示。
本发明中多糖溶液(如琼脂糖溶液、琼脂溶液、葡聚糖溶液等)、多糖降解溶液(如琼脂糖降解溶液、琼脂降解溶液、葡聚糖降解溶液等)指多糖水溶液(如琼脂糖水溶液、琼脂水溶液、葡聚糖水溶液等)、多糖降解水溶液(如琼脂糖降解水溶液、琼脂降解水溶液、葡聚糖降解水溶液等)。
以上所述仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!