水稻氮素吸收利用位点qNUE6及其分子标记方法与流程
本发明属于分子遗传学领域,其涉及水稻氮素吸收利用位点qNUE6及其分子标记方法。
背景技术:
水稻是世界上最重要的粮食作物之一,全世界一半以上的人口以稻米为主要粮食。氮是水稻生产中最重要的营养元素之一(Yoshida,1981)。自20世纪60年代以来,水稻品种的选育一直朝着高氮投入获得最大的籽粒产量方向发展(Tong,2006)。然而,过量的施用氮肥导致氮素利用率下降和环境污染。因此,当前水稻氮高效品种的需求变得非常迫切,选育高效吸收利用氮素水稻品种,提高水稻的氮素利用效率是全世界水稻育种中的一个重要目标。虽然水稻氮素高效吸收利用种质资源评价及氮素高效吸收利用基因研究取得了一些进展,定位了一批氮吸收利用相关QTL。但由于研究所用的材料原始或遗传基础狭窄、传统作图群体个体间遗传背景的干扰以及指标不确定等原因,使QTL定位及利用存在一些不足,因而到目前为止还没有可直接应用于育种的氮素吸收利用基因,也还没有真正用分子标记辅助选择的手段获得氮效率改良成功的报道。
技术实现要素:
针对以上技术问题,本发明公开了一种水稻氮素吸收利用位点qNUE6及其分子标记方法,可用分子标记NUE2来检测氮素高效吸收利用品种GH998及其衍生品种(系)中是否含有氮素吸收利用基因位点,可预测其氮素吸收利用水平,大大提高氮素高效吸收利用水稻的选择效率。
对此,本发明的技术方案为:
水稻氮素吸收利用位点qNUE6,其位于分子标记NUE2和NUE23之间的一个位点,所述分子标记NUE2引物:SEQ ID NO.1/ SEQ ID NO.2,存在qNUE6时的扩增条带为135bp条带。
分子标记NUE2的引物序列为:
上游引物:GTTCCACATGTTGATGGG(如SEQ ID NO.1)
下游引物:GATGAGGACGAGGAGGAG(如SEQ ID NO.2)
本发明公开了如上所述的水稻氮素吸收利用位点qNUE6的分子标记方法,用分子标记NUE2引物水稻材料的DNA,如果能够扩增出135bp扩增片段,则标志着氮素吸收利用基因位点qNUE6的存在。其中,所述水稻材料优选为氮素吸收利用品种或育种材料。
筛选所述的水稻氮素吸收利用位点qNUE6的分子标记的方法,包括以下步骤:
步骤S1:以普通野生稻材料Y11为供体,氮素高效吸收利用品种GH998为受体,通过杂交、回交和自交构建一套包含212个株系的水稻染色体片段导入系BC4F7;
步骤S2:采用在田间低氮、高氮条件下进行氮素吸收利用率鉴定;其中,所述低氮为田间氮含量为0 kg/hm2,所述高氮为田间氮含量为150 kg/hm2。
步骤3:在导入系中取氮素高效吸收和氮素低效吸收水稻各30株,利用CTAB法提取水稻植株的DNA,构建DNA氮素高效利用池、低效利用池,利用基于全基因组测序技术QTL-seq对水稻氮素吸收利用基因进行主效基因定位;其中,采用CTAB法,可以很好去除多糖,更有利于提取和纯化水稻植株的DNA。
步骤4:在水稻第6染色体上,均匀选取40对分子标记(Mccouch, 2002);通过对Y11和GH998进行比对,找出两者之间≥6 bp的插入或者缺失,进而设计InDel标记;选取在两亲本、两个池间多态性好标记,用于分析212份BC4F7群体;对多态性标记基因型和相应家系的氮素吸收利用级别进行连锁分析,并将Kosambi函数转换为遗传距离;利用Windows QTL Cart V2.5软件复合区间作图法,以2cM为步长在基因组内进行扫描,QTL的检测采用5%总体显著水平,相应LOD统计量的显著阀值用排列测验方法进行估计,共重复分析1000次。
步骤5:获得氮素吸收利用品种GH998氮素吸收利用位点qNUE6一个,位于标记NUE2附近,通过氮素吸收利用主基因的分子标记来检测氮素吸收利用品种GH998及其衍生品种(系)中是否含有该主基因位点,预测其氮素吸收利用水平。
进一步的,步骤3中,所述主效基因定位于第6染色体上。
进一步的,步骤4中,在水稻第6染色体上,均匀选取40对分子标记(Mccouch, 2002);通过BWA软件对Y11和998进行比对,找出两者之间的插入或者缺失(≥6 bp),进而使用Primer5 设计InDel标记。选取在两亲本、两个池间多态性好标记,用于分析212份BC4F7群体。利用MAPMARKER/EXP3.0软件对多态性标记基因型和相应家系的氮素吸收利用级别进行连锁分析,并将Kosambi函数转换为遗传距离;利用Windows QTL Cart V2.5软件复合区间作图法,以2cM为步长在基因组内进行扫描,QTL的检测采用5%总体显著水平,相应LOD统计量的显著阀值用排列测验方法进行估计,共重复分析1000次。
本发明所述的水稻氮素吸收利用位点qNUE6在水稻育种中的应用。
进一步的,所述水稻氮素吸收利用位点qNUE6在筛选氮素高效吸收利用品种或者品系中的应用。
本发明所述的氮素吸收利用位点qNUE6的分子标记引物在水稻分子育种中的应用。
进一步的,所述的氮素吸收利用位点qNUE6的分子标记引物在快速筛选氮素高效吸收利用品种或品系中的应用。
与现有技术相比,本发明的有益效果:
采用本发明的技术方案,利用普通野生稻材料Y11与氮素高效吸收利用品种—GH998杂交、回交和自交获得的BC4F7进行关联分析和遗传连锁分析,获得一个氮素吸收利用位点qNUE6,通过与qNUE6连锁的分子标记来检测氮素高效吸收利用品种GH998及其衍生品种(系)中是否含有氮素吸收利用位点,可以预测其氮素吸收利用率水平,大大提高氮素高效吸收利用水稻的选择效率。本发明所提供的氮素吸收利用主效基因位点的分子标记方法,具有以下优点:
(1)本发明在国际上首次采用联合基于全基因组测序的水稻QTL-seq方法和QTL遗传连锁分析定位了氮素吸收利用基因位点。
(2)通过本发明分子标记定位的氮素吸收利用基因位点位置精确,鉴定方便、快捷。通过检测这些与氮素吸收利用基因位点连锁的分子标记,即可以预测水稻植株的氮素吸收利用率水平,用于水稻品种或品系的基因型检测,以判断该品种或品系是否具有氮素高效吸收利用性,进而快速筛选氮素高效吸收利用品种或品系用于水稻育种,氮素吸收利用基因位点的检测方便快速,不易受环境影响。
(3)分子标记辅助育种选择目标明确,效率高。以往的传统育种技术,在提高氮素吸收利用率育种方法中,通常利用含氮素高效吸收利用基因的供体亲本与受体亲本进行杂交、多次回交或聚合复交,而且要对氮素吸收利用的效率进行单株选择;然而,由于水稻氮素吸收利用率的鉴定过程复杂,影响因素多,鉴定难度很大,表型鉴定的结果可靠性低;因此,氮素高效吸收利用育种不仅费时费工,而且难度大、成本高。通过利用本发明中的分子标记检测氮素吸收利用基因位点,可以在苗期就鉴定出氮素高效吸收利用的单株,淘汰其它植株,不仅节约成本,而且大大提高了氮素高效吸收利用水稻的选择效率。
附图说明
图1是本发明的氮素吸收利用基因紧密连锁分子标记的电泳图谱,
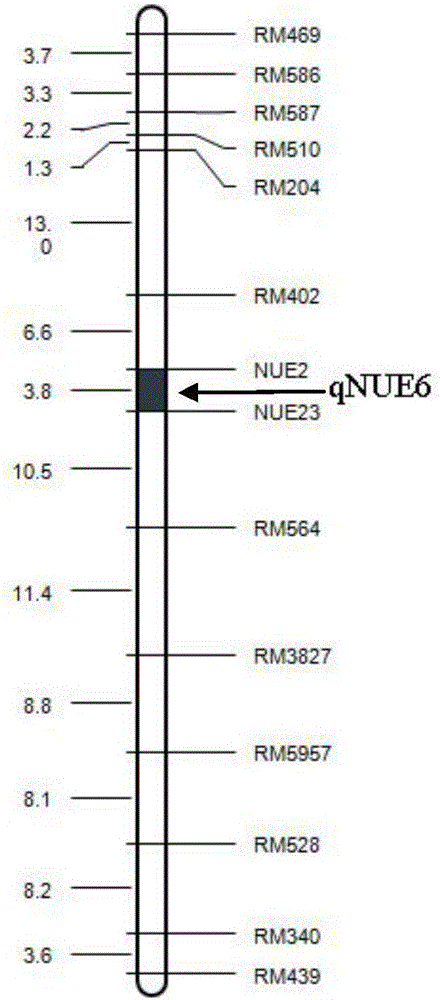
图2是本发明的GH998氮素吸收利用基因在染色体上的位置。
图1中标记包括: 1:Y11扩增带型;2~4:氮素低效吸收利用率株系扩增带型;5~10:杂合株系扩增带型;11~13:氮素高效吸收利用率株系扩增带型;14:GH998扩增带型。
具体实施方式
下面对本发明的较优的实施例作进一步的详细说明。
材料与方法:
步骤1:遗传分析分离群体的构建;
以普通野生稻材料Y11为供体,氮素高效吸收利用品种GH998为受体,通过杂交、回交和自交构建了一套包含212个株系的水稻染色体片段导入系BC4F7
以普通野生稻材料Y11为父本,氮素高效吸收利用品种GH998为母本,配制F1杂种,再以GH998为母本,998/Y11杂种为父本进行回交直到BC4F1,之后进行自交加代繁种获得BC4F7群体。
步骤2:采用田间鉴定进行氮素吸收率鉴定;
2013 年在广西农业科学院水稻研究所试验田(22 o51′N,108o14′)开展试验。试验田为水稻土,所述水稻田的营养成分为:有机质17.05 g/kg,全氮质量含量为 0.12%,全磷质量含量为 0.11%,全钾质量含量为1.78%,速效氮含量为90.50 mg/kg,速效磷含量为34.00 mg/kg ,速效钾含量为198.50 mg/kg, pH值为 6.48。
试验设不施氮(0 kg/hm2)和施氮(150 kg/hm2)2 个处理,施氮处理分别在插秧后第5天施105 kg/hm2和第25天施45 kg/hm2。钾肥和磷肥两个处理相同,钾肥施用量为100 kg/hm2,分别在插秧第5天施钾肥70 kg/hm2,在第25天施钾肥30 kg/hm2,磷100 kg/hm2全部作为基肥。小区面积为20 m2,行距设为23.1 cm,株距设为13.2 cm ,3次重复。
水稻成熟后,以小区普查结果的平均值为依据,取有代表性的植株10 穴,沿地面割取,茎叶与籽粒分开后,于 105℃烘箱杀青30 min ,75℃烘4天至恒重后称重,粉碎。使用半微量凯氏定氮法测定植株全氮。按照下式计算氮素吸收利用率:
氮素吸收利用率RE= (TNF-TN0)/N×100
其中,TN0为不施氮植株总含氮量;TNF为正常施氮植株总含氮量。
步骤3:Y11/GH998 分离群体的氮素吸收利用主基因定位;
(1)在分蘖盛期采取所有单株叶片,放入-80℃的冰箱内保存。根据2013年BC4F7群体中所有植株吸收利用率的鉴定结果,选出吸收利用率极低的30个单株和极高的30个单株,选出这60个单株和2个亲本的叶片,利用CTAB法提取植物基因组DNA。用琼脂糖凝胶电泳分析单个样品DNA的纯度和完整性,将氮素吸收利用率30个极低单株的DNA等量混合组成极低池(L-pool),30个极高的单株DNA等量混合组成极高池(H-pool)。
(2)DNA样品通过Covaris破碎机随机打断成长度为350bp的片段。采用TruSeq Library Construction Kit 进行建库。DNA片段经末端修复、加ployA尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备。构建好的文库通过illumina HiSeq 2500进行测序,测序深度亲本为20×,子代为30×。
(3) QTL-seq分析
测序得到的原始测序序列(Sequenced Reads)或者 raw reads,里面含有带接头的、低质量的reads。为了保证信息分析质量,必须对raw reads过滤,得到clean reads,后续分析都基于 clean reads。本次测序共产生Raw data 53.68 Gb,过滤后的Clean data 53.00 Gb。数据处理的步骤如下:
①去除带接头(adapter)的reads pair;
②当单端测序read中含有的N的含量超过该条read长度比例的10%时,需要去除此对paired reads;
③当单端测序read中含有的低质量(Q ≤ 5)碱基数超过该条read长度比例的 50% 时,需要去除此对paired reads。
④SNP-index的计算
SNP-index的计算是对子代池中SNP的一种统计方法,其原理是利用测序reads对每个碱基位点的碱基进行统计,以某一亲本或参考基因组为参考,统计子代池中和亲本或者参考基因组在某一个碱基位点相同或者不相同的reads条数,计算不相同reads条数占总条数的比例,即为该碱基位点的SNP -index。对于有两个子代池数据的项目,我们会过滤掉两个池中SNP-index均小于0.3的点。对于过滤后的SNP-index利用滑窗口的方式统计某窗口中所有SNP的SNP-index的平均值作为该窗口的SNP-index,一般默认参数是1Mb的窗口,10kb滑动。按照上述方法分别计算两个子代池的SNP-index,然后在计算两个子代池的SNP-index的差值即为△(SNP-index)。
⑤氮素利用率QTL定位
计算△(SNP-index),即两个子代SNP-index作差:△(SNP-index) =SNP-index(极端性状B)-SNP-index(极端性状A)。进行1000次置换检验,选取95%置信水平作为筛选的阈值。分析95%置信水平下,大于阈值的窗口作为候选区间,在该区域中挑选两个子代在SNP-index差异显著的SNP位点,即挑选子代2(极端性状B)SNP-index大于等于0.7,且子代1(极端性状A)SNP-index小于等于0.3的位点,符合这些特点的区域可能与氮素吸收利用率相关的QTL位点。
步骤4:连锁遗传作图分析
在水稻第6染色体上,均匀选取40对分子标记(Mccouch, 2002);通过BWA软件对Y11和998进行比对,找出两者之间的插入或者缺失,进而使用Primer5 设计InDel标记。选取在两亲本、两个池间多态性好标记,用于分析212份BC4F7群体。
SSR分析程序如下:
①所述12μlPCR反应体系包括:DNA(10ng/μl),2μl;Primer(4pmol/μl),1.5μl;2×Taq PCR Master Mix(中科瑞泰),6μl;ddH2O,2.5μl;
②扩增反应在东盛龙ETC811扩增仪上进行,所述PCR反应条件为:95℃预变性5min,95℃变性35s,55℃复性35s,72℃延伸1min,35个循环,最后72℃延伸7min。扩增产物用8%的非变性PAGE胶分离,通过银染显色。亲本间有多态的引物在BC4F7群体进行分析,获取样本基因型资料;
③利用MARMAKER/EXP3.0分析软件进行氮素吸收利用效率与SSR标记的分离分析,并将Kosambi函数转换为遗传距离(cM)。利用Windows QTL Cartographer V2.5软件复合区间作图法(Composite interval mapping ,CIM),以2cM为步长在基因组内进行扫描。QTL的检测采用5%总体显著水平,相应LOD统计量的显著阈值用排列测验方法进行估计,其重复分析1000次。
步骤5:结果与分析
Y11和GH998的氮素吸收利用率经田间鉴定,其值分别为7.09%和33.18.%,表明GH998具有较高的氮素吸收利用能力,而Y11的氮素吸收利用能力很低。经过对比氮素利用率低池和高池的SNP-index,分析95%置信水平下,大于阈值的窗口作为候选区间,我们发现在第6条染色体上的6,099,043-8,940,631 bp之间出现了SNP不平衡的状况,是控制水稻氮素利用率的QTL位点,我们将其命名为qNUE6。
从40对SSR标记中选取在亲本间表现多态的16对SSR标记对63个BC4F7单株进行了背景检测,结果发现他们在水稻第6染色体标记RM539和RM136间区域有差异。我们利用Y11和GH998重测序结果设计了30对插入缺失标记,其中24对具有多态性,结合上述16对SSR标记,对212个高世代群体单株进行基因型分析,利用MAPMAKER3.0和WinQTL Cart 3.0软件将基因NUE6范围缩小到8,051,882-8,990,567 bp之间。因此,水稻第6染色体区间8,051,882-8,990,567 bp可能含有控制水稻氮素利用性状的基因。如图2所示,结果在分子标记NUE2和NUE23之间检测到一个所述水稻氮素吸收利用位点qNUE6,LOD值为4.96,贡献率为16.8%,可解释表型变异的16.8%。
分子标记NUE2引物序列:
上游引物:GTTCCACATGTTGATGGG
下游引物:GATGAGGACGAGGAGGAG
用分子标记NUE2引物扩增氮素高效吸收利用品种或育种材料DNA,如图1所示,用分子标记NUE2能够扩增出135bp的扩增片段,则标志着水稻品种氮素吸收利用的位点qNUE6的存在。
通过与上述基因位点连锁的分子标记来检测氮素高效吸收利用品种GH998及其衍生品种(系)中是否含有氮素吸收利用基因位点,可预测其氮素吸收利用水平,大大提高氮素高效吸收利用水稻的选择效率。
以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
序列表(SEQUENCE LISTING)
<110> 广西壮族自治区农业科学院水稻研究所
<120> 水稻氮素吸收利用位点qNUE6及其分子标记方法
<160> 2
<170> PatentIn version 3.3
<210> 1
<211> 18
<212> DNA
<213> 人工序列
<400> 1
gttccacatg ttgatggg 18
<210> 2
<211> 18
<212> DNA
<213> 人工序列
<400> 2
gatgaggacg aggaggag 18
- 还没有人留言评论。精彩留言会获得点赞!