一种高效快速分离T‑DNA插入位点侧翼序列的方法及其用途与流程
本发明属于生物技术领域,具体涉及一种高效快速分离t-dna插入位点侧翼序列的方法及其用途。
背景技术:
随着植物基因工程的飞速发展,农杆菌介导的植物转化技术已经成为植物转基因育种和植物基因功能研究过程中一项重要的方法和技术。t-dna插入位点侧翼序列的分离和鉴定对转基因植株的遗传分析起着关键作用。目前分离侧翼分离的方法主要有以下几种:
一、tail-pcr,其主要原理是基于一端的特异巢式引物和另一端随机简并引物,经过多轮扩增可以分离出侧翼序列;二、反向pcr,这种方法主要是对基因组dna进行酶切,然后进行自连,通过载体上的特异引物进行侧翼序列分离;三、接头pcr,其主要原理与反向pcr类似,不同的是酶切基因组dna以后加接头连接代替自连,在扩增过程中用接头的一端引物和载体一端引物配合进行pcr扩增;四、质粒拯救,基本原理是在载体设计的时候在载体t-dna区加入了质粒复制相关元件,对转基因植株的dna进行酶切,再进行自连,然后自连dna转化大肠杆菌,对克隆进行测序验证。以上方法均存在效率低,随机性大和成功率低的特点,如在使用tail-pcr分离过程中存在大量的假阳性;在使用反向pcr和接头pcr分离过程中,插入位点附近无合适酶切位点。而且以上方法在涉及到多拷贝t-dna插入突变体时往往很难将大部分位点分离出来,经常出现不能得到与突变性状共分离的插入位点。因此,如何高效、快速地分离出转化事件中的所有插入位点信息在基因功能分析过程中对t-dna突变体进行共分离检测和在转基因新品种培育过程中进行后代筛选和遗传稳定性分析均具有重要意义。
随着第二代测序技术的飞速发展,理论上讲,我们可以通过深入重测序的方法,无限加大测序深度从而分析出插入位点信息。但是随着测序深度的加大,数据量过大,不仅会造成数据量的浪费;而且会加大计算资源的消耗。
技术实现要素:
本发明通过利用dna捕获技术,专门针对t-dna边界序列进行测序,具有高效,经济和分析简单的特点。即本发明的目的在于克服现有侧翼序列分离技术通量低,效率低和成功率低的不足,结合dna捕获和第二代测序技术,特异性对t-dna边界序列进行测序并通过简单分析就能获得t-dna插入位点的侧翼序列,可以将其运用于植物的基因功能验证和转基因育种与品种改良。
即本发明的第一目的在于提供一种高效快速分离t-dna插入位点侧翼序列的方法,包括以下步骤:
1)提取农杆菌介导的转基因植株的基因组dna;
2)将基因组dna片段化,dna修复、加a和连接后,利用磁珠进行片段选择后进行文库构建,构建的文库与t-dna边界序列进行杂交捕获以后进行pcr富集;
3)对文库进行高通量测序,对测序数据进行生物信息学分析,获取t-dna插入位点。
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述步骤1)中提取基因组dna的方法为ctab法。
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述步骤2)中基因组dna片段化的方法为超声破碎法。
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述步骤2)中dna片段大小为300bp~500bp。
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述步骤2)中dna修复、加a和连接的方法分别为将dna双链的片段修复成平末端,并且将5’末端磷酸化;加a:在末端修复后的3’末端加一个腺苷酸;连接:与接头进行连接。
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述步骤2)中使用探针进行杂交捕获。
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述探针为seqidno1所示序列.
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述步骤2)中pcr富集的循环数为15~20个循环。
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述步骤3)中测序数据进行生物信息学分析为原始序列经ngsqctoolkit进行质量控制,获得高质量cleanreads,对cleanreads采用dna测序处理,以t-dna边界序列为参考基因组,提取一端匹配到t-dna,另一端未匹配的reads序列。后者序列以水稻基因组序列,采用blast软件进行比对分析,根据比对结果设计引物进行后续验证。
优选地,本发明所述的高效快速分离t-dna插入位点侧翼序列的方法中,所述t-dna边界序列为seqidno2所示序列。
与现有技术相比,本发明至少有以下优点:
1)本发明利用靶标dna捕获,专门针对t-dna边界序列进行低通量测序(每个样品测序数据量低),快速分析t-dna插入位点序列,本发明的方法克服了现有侧翼序列分离技术通量低,效率低和成功率低的不足,结合dna捕获和第二代测序技术,特异性对t-dna边界序列进行测序并通过简单分析就能获得t-dna插入位点的侧翼序列,具有高效,经济和分析简单的特点。
2)本发明的技术可以运用于植物的基因功能验证和转基因育种与品种改良。
附图说明
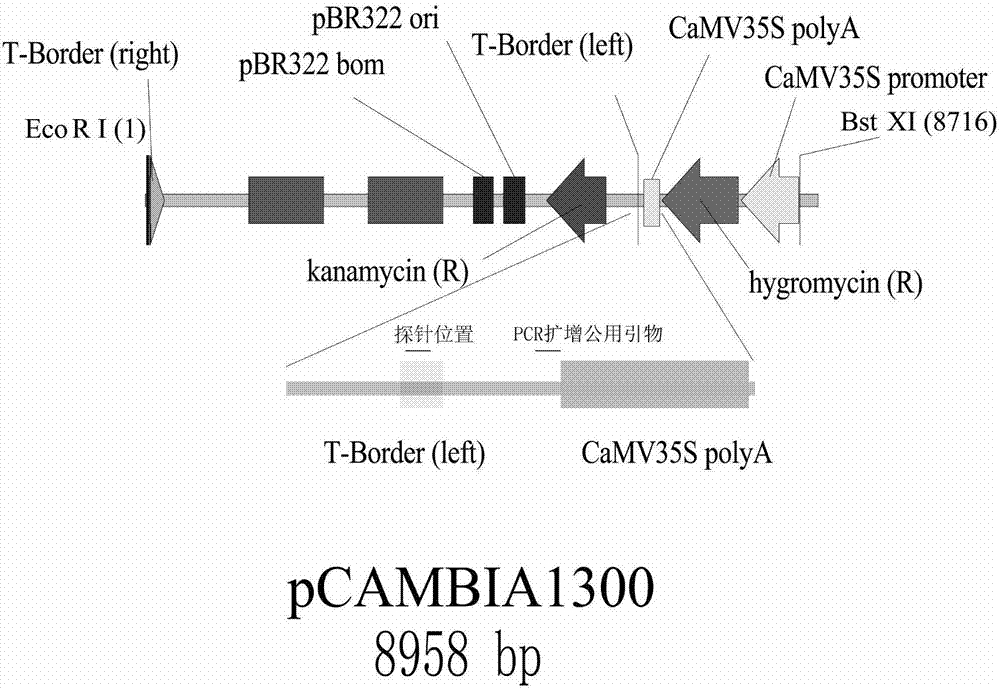
图1为本发明的一个实施例中pc1300载体图(图1a)与探针在载体中的大概位置示意图(图1b);
图2为本发明的一个实施例中dna文库电泳图;
图3为本发明的一个实施例中dna文库构建和序列分析流程图;
图4为本发明的一个实施例中pcr扩增产物电泳检测图;
图5为本发明的一个实施例中southernblotting图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
在本发明的一个实施例中,提供了一种高效快速分离t-dna插入位点侧翼序列的方法,包括以下步骤:
1)提取农杆菌介导的转基因植株的基因组dna;
2)后将基因组dna片段化,dna修复、加a和连接后,利用磁珠进行片段选择后进行文库构建,构建的文库与t-dna边界序列进行杂交捕获以后进行pcr富集;
3)对文库进行高通量测序,对测序数据进行生物信息学分析,获取t-dna插入位点。
优选地,在本发明的一个实施例中,所述步骤1)中提取基因组dna的方法为ctab法。
优选地,在本发明的一个实施例中,所述步骤2)中基因组dna片段化的方法为超声破碎法。
优选地,在本发明的一个实施例中,所述步骤2)中dna片段大小为300bp~500bp。
优选地,在本发明的一个实施例中,所述步骤2)中dna修复、加a和连接的方法分别为将dna双链的片段修复成平末端,并且将5’末端磷酸化;加a:在末端修复后的3’末端加一个腺苷酸;连接:与接头进行连接。
优选地,在本发明的一个实施例中,所述步骤2)中使用探针进行杂交捕获。
优选地,在本发明的一个实施例中,所述探针为seqidno1所示序列.
优选地,在本发明的一个实施例中,所述步骤2)中pcr富集的循环数为15~20个循环。
优选地,在本发明的一个实施例中,所述步骤3)中测序数据进行生物信息学分析为原始序列经ngsqctoolkit进行质量控制,获得高质量cleanreads,对cleanreads采用dna测序处理,以t-dna边界序列为参考基因组,提取一端匹配到t-dna,另一端未匹配的reads序列。后者序列以水稻基因组序列,采用blast软件进行比对分析,根据比对结果设计引物进行后续验证。
优选地,在本发明的一个实施例中,所述t-dna边界序列为seqidno2所示序列。
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图和具体实施方式对本发明作进一步详细的说明。
为了便于研究利用,我们通过农杆菌介导的遗传转化获得转基因植株,随机选择4株转化植株进行实验技术流程研发,然后利用捕获测序数据对插入事件进行分析,最后通过southern杂交的方法验证转化植株的拷贝数。
在本发明的一个实施例中,首先利用农杆菌介导的遗传转化方法将pc1300导入野生型中花11。在获得转基因植株以后,选择4株转化植株取分蘖期叶片,采用ctab法抽提基因组dna。样品溶于100μlddh2o备用。基因组dna经超声波打断后利用磁珠进行片段选择后进行文库构建,文库与t-dna边界序列进行杂交捕获以后进行pcr富集。对文库进行高通量测序,对测序数据进行生物信息学分析,获取t-dna插入位点。通过分析出的序列设计引物扩增基因组dna,pcr产物再经过双末端终止法测序,比较序列差异和分析结果完全一致。对选择的4株转化植株进行southern杂交检测其拷贝数,结果显示与分析高通量测序信息获取的t-dna插入数量相同。
实施例1:农杆菌介导的遗传转化
农杆菌介导的遗传转化方法主要参照华中农业大学作物遗传改良国家重点实验室发表的“农杆菌介导的遗传转化操作手册”所示的方法(林拥军等,农杆菌介导的牡丹江8号高效转基因体系的建立,作物学报,2002,28(3):294-300)。
如图1所示,转化载体为pc1300,外源基因为潮霉素筛选标记基因,pcambia1300(genbank号af234296.1),使用pc1300电转化至农杆菌菌株eha105(购自华中农业大学)备用,转化受体为水稻品种中花11成熟种子所诱导产生的胚性愈伤组织。胚性愈伤组织经过预培养3天、用含有pc1300的农杆菌侵染30min、共培养2天、水洗和2次筛选(约35天)后得到具有潮霉素抗性的愈伤组织,再经过分化(40天)、生根(10天)、练苗(4天)和移栽,得到t0代转基因植株。
实施例2:基因组dna的提取
取实施例1随机选择4株t0代转基因水稻,每株幼嫩叶片2g,经液氮碾磨后,加入2ml80℃1.5×ctab(15gctab;75ml1mtris-hcl,ph8.0;30ml0.5medta,ph8.0;61.4gnacl;定容1l,搅拌子搅拌溶解2h);65℃水浴30min;加入2ml氯仿/异戊醇(24:1)缓慢上下颠倒15min,下层液相呈深绿色为止;室温下3000r/min离心15min;取上清于一新10ml离心管,加入200μl10%ctab和2ml氯仿/异戊醇(24:1)缓慢上下颠倒约15min;室温下3000r/min离心15min;取1.5ml上清,加入4ml1%ctab,混匀后将沉淀用枪头挑出溶于1ml1mnacl(加有1μlrnase)中。加入3ml冰冻无水乙醇沉淀dna,絮状dna用枪头挑至1.5ml离心管中,加入500μl75%乙醇洗涤dna。完成后弃上清,晾干后加入200μlddh2o溶解,备用。dna采用qubit(invitrogen,http://www.thermofisher.com/cn/zh/home.html)测定浓度。
实施例3:dna文库构建
dna纯化:取1μgdna(实施例2制备)溶于50μlddh2o中后采用超声波破碎仪破碎至300-500bp大小。加入0.5x磁珠(贝克曼,http://www.beckmancoulter.cn/),混匀后室温放置15min;用磁力架(invitrogen,http://www.thermofisher.com/cn/zh/home.html)吸附磁珠,将上清转移至新的离心管中,重新加入0.5x磁珠,混匀后室温放置15min;用磁力架吸附磁珠,弃上清,加入200μl80%乙醇,洗涤2遍。晾干后加入10μlddh2o溶解dna,备用。
dna修复、加a和连接:dna修复和加a采用neb(http://www.neb-china.com/)公司试剂盒(e7546s),操作步骤参见说明书。反应完成后采用neb公司连接试剂盒(e7595s),接头采用illumina公司接头(每个样品采用不同的index接头),操作步骤参见说明书。
链霉亲和素标记磁珠准备:取20μl链霉亲和素标记磁珠,用2xwashingbuffer洗涤3遍后备用。
探针杂交与dna捕获:文库连接产物经98℃变性10min后混合一起,加入1μl(10μm)探针(seqidno1:ccag
pcr富集:参考illumina公司truseqdna建库流程进行pcr富集,pcr循环数为15-20个循环。文库用1x磁珠(贝克曼,http://www.beckmancoulter.cn/)纯化(纯化步骤见上)后送公司测序。
实施例4:数据分析
实施例3原始序列经ngsqctoolkit(http://www.nipgr.res.in/ngsqctoolkit.html)进行质量控制,高质量reads采用hisat2(http://ccb.jhu.edu/software/hisat2/index.shtml),以t-dna边界序列(seqidno2)为参考序列。
提取一端匹配到t-dna,另一端未匹配的reads序列。后者序列以水稻基因组序列(http://rice.plantbiology.msu.edu/),采用blast软件进行比对分析。对测序数据进行生物信息学分析,获取t-dna插入位点,具体过程如下:
首先,建立一个t-dna序列的文本文件,命名为target.txt。其文件格式为fasata格式,具体为如下:
>target(seqidno.2pc1300载体部分序列)
gccggtcggggagctgttggctggctggtggcaggatatattgtggtgtaaacaaattgacgcttagacaacttaataacacattgcggacgtttttaatgtactgaattaacgccgaattaattcgggggatctggattttagtactggattttggttttaggaattagaaattttattgatagaagtattttacaaatacaaatacatactaagggtttcttatatgctcaacacatgagcgaaaccctataggaaccctaattcccttatctgggaactactcacacattattatggagaaactcgagcttgtcgatcgacagatccggtcggcatctactctatttctttgccctcggacgagtgctggggcgtcggtttccactatcggcgagtacttctacacagccatcggtccagacggccgcgcttctgcgggcgatttgtgtacgcccgacagtcccggctccggatcggacgattgcgtcgcatcgacc
1.采用hisat2-build建立reference数据库具体命令为:
hisat2-buildtarget.txttarget
2.采用hisat2进行序列比对,以1号样品为例(其两端测序结果文件分别命名为1_r1.fastq和1_r1.fastq)。其比对命令如下:
hisat2-q-k2-p8--very-sensitive-xtarget-11_r1.fastq-21_r1.fastq-s1.sam
3.绝大部分的reads都不能比对上t-dna(因为非特异性杂交);有部分reads两端成对比对上t-dna,这部分不能提供t-dna侧翼的基因组序列信息,无效。真正有用的是一端在t-dna,一端在基因组上的reads。提取这类型reads的命令:
grep“target”1.sam|grep“*”|cut–f1,10>extract1.txt
得到的文件数据如下所示,数据有两列,其中第一列为第二代测序过程中生成的序列编号,第二列为该编号对应的具体序列
第1列:st-j00123:44:hcc5vbbxx:1:1210:12053:8348
第2列:
ctaccgctcgcagttcgtcgcctaaaccaccactgcccaagaaatcatgtccgatctcttcattgctttggttggtttgatcccgtggctctcctttgctgattttgcttggaaaatattgtggtgtaaacaaattgacgcttagacaact
(seqidno.3)
以上面的序列与水稻基因组序列(http://rice.plantbiology.msu.edu/)进行blast分析(可采用本地化批量进行)
根据比对结果设计引物进行后续验证。
t-dna插入位点结果见下表1:
表1
注:1.家系为t0代的转基因植株,也就是实施例2中选取的4个单株,这里的家系编号1,2,3,4与实施例6的southernblot检测结果附图5的家系编号1,2,3,4是相对应的。2.染色体定位结果为t-dna的定位结果,结束位置为t-dna左边界插入到的染色体位置。
实施例5:pcr验证
选取t-dna序列上一条引物和根据位点设计的引物进行pcr扩增验证。其中,其中引物设计如下:
表2
注:引物从上到下序列表编号为:seqidno.4~no14
pcr体系为:模板dna:10ng,10xbuffer2μl,dntp(10mm)0.4μl,左右引物(10μm)各0.3μl,rtaq0.2μl,补水至20μl,加矿物油后进行扩增反应。反应程序为95℃,30s;30个循环(95℃,10s,65℃,30s,72℃,30s);72℃,5min。取10μl反应产物采用0.8%琼脂糖凝胶电泳检测,电泳检测结果见附图4(泳道1为全式金公司2kplusdnamarker,泳道2至泳道11依次对应表3中测序样品编号9至18),剩余10μl送公司测序,测序引物采用pcr过程中的t-dna引物fls-f,测序样品编号对应的扩增引物见下表3。
表3测序结果的编号说明
测序结果如下:测序结果显示与表1中定位结果一致。
实施例6:southernblot检测
southernblot的具体步骤过程参考southern所示的方法进行(southern,e.m.1975,detectionofspecificsequencesamongdnafragmentsseparatedbygelelectrophoresis,jmolbiol98,503-517)。取8μg基因组总dna,采用hindiii进行dna酶切,酶切产物经过琼脂糖凝胶电泳,转膜,烘膜固定。然后预杂交,杂交以及洗膜与显影,获得转化植株的拷贝数。southern结果详见附图5,t0代转基因家系1至家系4的拷贝数分别为1,1,3,5个拷贝,与预期或测序结果一致。其中,m:dnamarker;1:家系1;2:家系2;3:家系3;4:家系4。不同的t0代(直接从抗性愈伤组织分化的转基因植株称为t0代)转基因水稻植株称为家系,家系1至家系4表示4株t0代的转基因植株。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
sequencelisting
<110>武汉天问生物科技有限公司
<120>一种高效快速分离t-dna插入位点侧翼序列的方法及其用途
<160>14
<170>patentinversion3.5
<210>1
<211>38
<212>dna
<213>人工序列
<400>1
ccagtactaaaatccagatcccccgaattaattcggcg38
<210>2
<211>500
<212>dna
<213>人工序列(pc1300载体)
<400>2
gccggtcggggagctgttggctggctggtggcaggatatattgtggtgtaaacaaattga60
cgcttagacaacttaataacacattgcggacgtttttaatgtactgaattaacgccgaat120
taattcgggggatctggattttagtactggattttggttttaggaattagaaattttatt180
gatagaagtattttacaaatacaaatacatactaagggtttcttatatgctcaacacatg240
agcgaaaccctataggaaccctaattcccttatctgggaactactcacacattattatgg300
agaaactcgagcttgtcgatcgacagatccggtcggcatctactctatttctttgccctc360
ggacgagtgctggggcgtcggtttccactatcggcgagtacttctacacagccatcggtc420
cagacggccgcgcttctgcgggcgatttgtgtacgcccgacagtcccggctccggatcgg480
acgattgcgtcgcatcgacc500
<210>3
<211>151
<212>dna
<213>水稻
<400>3
actaccgctcgcagttcgtcgcctaaaccaccactgcccaagaaatcatgtccgatctctt60
cattgctttggttggtttgatcccgtggctctcctttgctgattttgcttggaaaatatt120
gtggtgtaaacaaattgacgcttagacaact151
<210>4
<211>26
<212>dna
<213>人工序列
<400>4
cccgcagaagcgcggccgtctggacc26
<210>5
<211>27
<212>dna
<213>人工序列
<400>5
cgcaggagcagcccaccccttccgccg27
<210>6
<211>29
<212>dna
<213>人工序列
<400>6
gagaaatcagaacctttgcgatttgcatg29
<210>7
<211>25
<212>dna
<213>人工序列
<400>7
gccgcagcgccagctgcacccggtg25
<210>8
<211>25
<212>dna
<213>人工序列
<400>8
cttgtgcggctactacgtgtgcgag25
<210>9
<211>26
<212>dna
<213>人工序列
<400>9
ctttcacccaaaatttgaaaacattc26
<210>10
<211>24
<212>dna
<213>人工序列
<400>10
ggcgcgctcgccatgctcgagcag24
<210>11
<211>24
<212>dna
<213>人工序列
<400>11
acttccacagaacagcctagcagc24
<210>12
<211>28
<212>dna
<213>人工序列
<400>12
ctccaaatacagtctctctagggtgagg28
<210>13
<211>26
<212>dna
<213>人工序列
<400>13
ctatgtgccattcctatactaaccac26
<210>14
<211>25
<212>dna
<213>人工序列
<400>14
ctaggggtccacctctccaaatctg25
- 还没有人留言评论。精彩留言会获得点赞!