一种IDTT衍生物的新型合成方法与流程
本发明涉及有机太阳能电池领域,具体涉及一种idtt衍生物的新型合成方法。
背景技术:
有机太阳能电池由于具有质量轻、成本低、制备工艺简单、柔韧性好且可大面积制备等优点,成为近年来的研究热点。因而使得有机光电材料的种类层出不穷,其中由电子给体材料和电子受体材料所组成的复合体系的异质结型光电材料发展较为迅速。
idtt作为一种新型的电子受体材料,具有相当好的稳定性,其结构的对称性和刚性稠芳香体系可以提高电子的离域,分子间的作用力可以提高电荷迁移率,具有优异的光电性能。
2012年,jen等设计合成了idtt,并以其为基体,对其进行修饰,从而对其光学带隙进行微调,设计合成了给体材料pidtt-dfbt,并与受体材料pc71bm共混制备了有机太阳能电池器件,其相应的光伏器件能量转化率可达5.72%;2015年,zhan等人同样以idtt为基体,对其进行修饰,设计并合成了itic,作为一类优异的受体材料,与给体材料ptb7-th共混制备了太阳能电池器件,使pce提高到了7.52%,同时也开辟了一新的研究领域,供体材料通过修饰微调可与受体材料互变;2017年,同样是zhan等人继续将此类体系拓展,将并三噻吩替代并噻吩,合成了一类新型的idtt衍生物,并将其氟化,所得的受体材料制备成器件的pce提高到了11%。
通过研究发现,基于idtt修饰的小分子受体,在可见光区域内有着更强的吸收和更宽的吸收范围,而且制备得到的器件其热稳定性和化学稳定性都很好。具有突出的光电性能。在此体系的光电材料中,具有很好的应用前景。
在之前的文献中有报道采用直接加入硫酸与醋酸并回流3小时来制备idtt的方法,虽然此合成方法可以得到纯度相对较高的产物,但是此方法要用到浓硫酸,同时需要在醋酸中回流,因此实验操作危险系数较高,能耗较高,后处理复杂,且溶剂消耗量较大,产率不高。
本方法采用三氟化硼乙醚溶液在室温下就可以反应,且反应最终直接用甲醇淬灭,真空旋干过柱,与之前文献报道的方法相比较,后处理简便,得到的最终产物idtt的产率较高,环保压力小,能耗低,安全性高。
技术实现要素:
本发明旨在提供一种idtt衍生物的新型合成方法,该合成方法简单、易操作,有机溶剂消耗量小,后处理简单,产品收率高,纯度高。
本发明所采用的技术方案是:
一种idtt衍生物的新型合成方法,包括如下步骤:
步骤(1),在氮气环境下,将烷基溴苯溶解在除水的四氢呋喃中,得到烷基溴苯溶液;
步骤(2),在-78~-80℃下,向步骤(1)所得烷基溴苯溶液中加入正丁基锂的己烷溶液,并反应0.9-1.2h;
步骤(3),将2,5-二溴对苯二甲酸酯加入步骤(2)所得反应液中,室温下反应16-20h,使之反应完全;
步骤(4),将反应用水淬灭,乙酸乙酯萃取,无水硫酸钠干燥旋干,得到粗产物;
步骤(5),在氮气环境下将粗产物用二氯甲烷溶解,0~5℃下加入三氟化硼乙醚溶液,室温下反应1-1.5h;
步骤(6),向步骤(5)所得混合溶液中加入甲醇,反应16-20h,使其反应完全;
步骤(7),反应结束后,用水淬灭,二氯甲烷萃取,用柱色谱法分离,得到idtt衍生物。
优选地,以摩尔比计,烷基溴苯:正丁基锂:2,5-二溴对苯二甲酸酯:三氟化硼乙醚溶液=6:6:1:2。
优选地,步骤(1)中所述烷基溴苯溶液的浓度为0.020g/ml-0.030g/ml。
优选地,步骤(2)中烷基溴苯与正丁基锂反应温度为-78℃,反应时间为1h;步骤(3)中的反应温度为25℃,反应时间为16h;步骤(5)中在0℃下加入三氟化硼乙醚溶液,反应温度为25℃,反应时间为17h。
优选地,柱色谱法所用洗脱剂的体积比例为石油醚:二氯甲烷=15:1。
本发明的优点在于:
1、本方法简便易于操作,反应条件温和,总体产率和产品纯度较原有方法有所提高。文献中的方法一方面在反应中用到浓硫酸与醋酸,反应操作较为危险;另一方面,反应过程都需要加热,能耗较多。
2、本方法操作安全性高,无需加热,能耗低。与文献报道的采用浓硫酸与醋酸加热回流的方法相比较,本方法采用三氟化硼乙醚溶液在室温下就可以反应,且反应用甲醇淬灭,与文献报道的方法相比较环保压力小,能耗低,安全性高。
附图说明
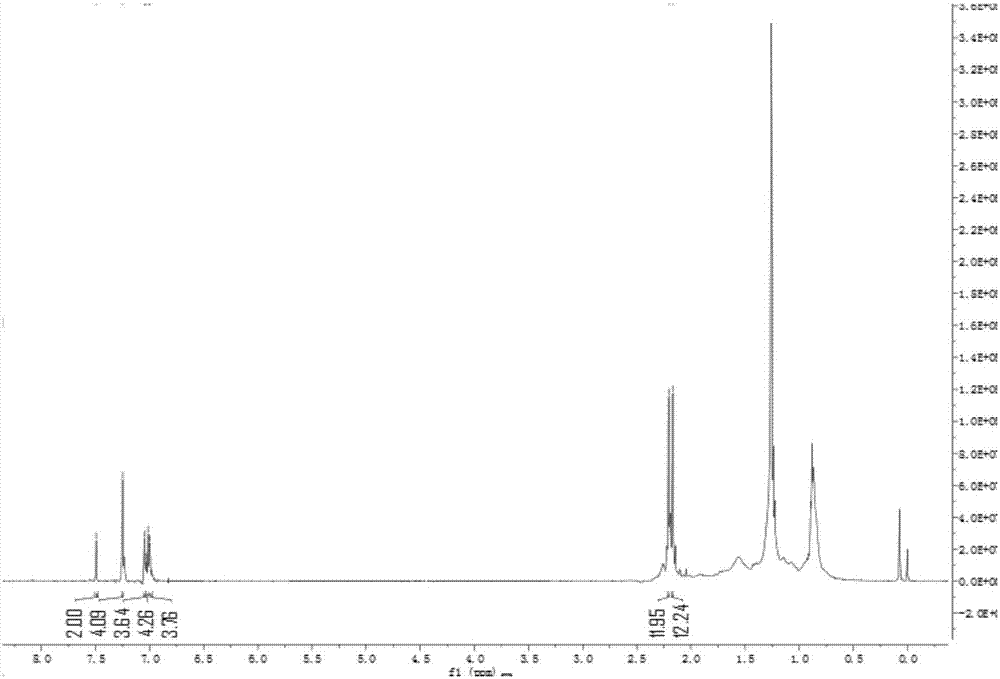
图1为本发明实施例1所得产物idtt核磁共振氢谱图(nmrvarianmercuryplus400型测试条件:500mhz);
图2为本发明实施例2所得产物idtt-dm核磁共振氢谱图(nmrvarianmercuryplus400型测试条件:500mhz);
图3为本发明实施例3所得产物idtt-m核磁共振氢谱图(nmrvarianmercuryplus400型测试条件:500mhz)。
具体实施方式
以下结合具体实施例,进一步阐明本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。
实施例1:
在氮气环境下,将1-(4-溴苯基)己烷(578mg,2.4mmol)溶解在10ml除水的四氢呋喃中,在-78℃下加入2.4m正丁基锂的己烷溶液(1ml,2.4mmol),反应1h后,将2,5-二溴对苯二甲酸酯(200mg,0.4mmol)加入反应中,室温下反应16h后将反应用水淬灭,乙酸乙酯萃取,无水硫酸钠干燥旋干,在氮气环境下将粗产物用二氯甲烷溶解,0℃下加入三氟化硼乙醚溶液(1ml,0.8mmol),室温下反应1h后加入甲醇5ml,之后反应16h。反应结束后,用水淬灭,二氯甲烷萃取,用柱色谱法分离,得产物idtt(223.8mg)。
本发明的合成方法所涉及反应方程式图如下:
如图1所示,该结构有九类化学环境不同的氢,分别位于7.50(2h),7.28(2h),7.25(2h),7.18(8h),7.07(8h),2.55(8h),1.60-1.53(8h),1.35-1.29(24h),0.86(12h)ppm,与文献报道的谱图一致,可见该化合物是正确的,得到的化合物结构为:
实施例2:
在氮气环境下,将4-溴-邻二甲苯(444mg,2.4mmol)溶解在10ml除水的四氢呋喃中,在-78℃下加入2.4m正丁基锂的己烷溶液(1ml,2.4mmol),反应1h后,将2,5-二溴对苯二甲酸酯(200mg,0.4mmol)加入反应中,室温下反应16h后将反应用水淬灭,乙酸乙酯萃取,无水硫酸钠干燥旋干,在氮气环境下将粗产物用二氯甲烷溶解,0℃下加入三氟化硼乙醚溶液(1ml,0.8mmol),室温下反应1h后加入甲醇5ml,之后反应16h。反应结束后,用水淬灭,二氯甲烷萃取,用柱色谱法分离,得产物idtt-dm(238.5mg)。
如图2所示,该结构有八类化学环境不同的氢,由于部分氢存在重叠问题,所以氢谱上面只显示了五种类别的氢,分别位于7.49(2h),7.25(4h),6.97-7.05(12h),2.21(12h),2.17(12h)ppm,与文献报道的谱图一致,可见该化合物是正确的,得到的化合物结构为:
实施例3:
在氮气环境下,将4-溴甲苯(410mg,2.4mmol)溶解在10ml除水的四氢呋喃中,在-80℃下加入2.4m正丁基锂的己烷溶液(1ml,2.4mmol),反应1h后,将2,5-二溴对苯二甲酸酯(200mg,0.4mmol)加入反应中,室温下反应16h后将反应用水淬灭,乙酸乙酯萃取,无水硫酸钠干燥旋干,在氮气环境下将粗产物用二氯甲烷溶解,0℃下加入三氟化硼乙醚溶液(1ml,0.8mmol),室温下反应1h后加入甲醇5ml,之后反应16h。反应结束后,用水淬灭,二氯甲烷萃取,用柱色谱法分离,得产物idtt-d(221.7mg)。
如图3所示,该结构有六类化学环境不同的氢,由于部分氢存在重叠问题,所以氢谱上面只显示了五种类别的氢,分别位于7.53(2h),7.29(4h),7.22(8h),7.12(8h),2.20(12h)ppm,与文献报道的谱图一致,可见该化合物是正确的,得到的化合物结构为:
实施例4:
在氮气环境下,将1-(4-溴苯基)己烷(578mg,2.4mmol)溶解在10ml除水的四氢呋喃中,在-80℃下加入2.4m正丁基锂的己烷溶液(1ml,2.4mmol),反应1h后,将2,5-二溴对苯二甲酸酯(200mg,0.4mmol)加入反应中,室温下反应16h后将反应用水淬灭,乙酸乙酯萃取,无水硫酸钠干燥旋干,在氮气环境下将粗产物用二氯甲烷溶解,0℃下加入三氟化硼乙醚溶液(1ml,0.8mmol),室温下反应1h后加入甲醇5ml,之后过夜反应16-20h。反应结束后,用水淬灭,二氯甲烷萃取,用柱色谱法分离,得产物idtt(200.6mg)。
实施例5:
在氮气环境下,将1-(4-溴苯基)己烷(578mg,2.4mmol)溶解在10ml除水的四氢呋喃中,在-78℃下加入2.4m正丁基锂的己烷溶液(1ml,2.4mmol),反应1.5h后,将2,5-二溴对苯二甲酸酯(200mg,0.4mmol)加入反应中,室温下反应16h后将反应用水淬灭,乙酸乙酯萃取,无水硫酸钠干燥旋干,在氮气环境下将粗产物用二氯甲烷溶解,0℃下加入三氟化硼乙醚溶液(1ml,0.8mmol),室温下反应1h后加入甲醇5ml,之后过夜反应16-20h。反应结束后,用水淬灭,二氯甲烷萃取,用柱色谱法分离,得产物idtt(190.6mg)。
实施例6:
在氮气环境下,将1-(4-溴苯基)己烷(578mg,2.4mmol)溶解在10ml除水的四氢呋喃中,在-78℃下加入2.4m正丁基锂的己烷溶液(1ml,2.4mmol),反应1h后,将2,5-二溴对苯二甲酸酯(200mg,0.4mmol)加入反应中,室温下反应16h后将反应用水淬灭,乙酸乙酯萃取,无水硫酸钠干燥旋干,在氮气环境下将粗产物用二氯甲烷溶解,5℃下加入三氟化硼乙醚溶液(1ml,0.8mmol),室温下反应1h后加入甲醇5ml,之后过夜反应16-20h。反应结束后,用水淬灭,二氯甲烷萃取,用柱色谱法分离,得产物idtt(201.3mg)。
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
- 还没有人留言评论。精彩留言会获得点赞!