奥沙利铂杂质C及其制备方法和应用与流程
本发明涉及化学合成技术领域,特别涉及一种奥沙利铂杂质c的制备方法。
背景技术:
奥沙利铂是继顺铂、卡铂之后的第三代铂类抗癌药物,主要通过产生烷化结合物作用于dna,形成链内和链间交联,从而抑制dna的合成及复制。临床主要用于治疗经氟尿嘧啶治疗失败后的结(直)肠癌转移患者以及胃癌、卵巢癌、非小细胞肺癌等癌症。1996年在法国批准上市,2002年获得美国fda的认可。奥沙利铂收载于欧洲药典9.0版,国内收载于中国药典2015年版。
奥沙利铂杂质c化学名为(oc-6-33)-[(1r,2r)-环己烷-1,2-二胺-n,n][乙二酸(2-)-o1,o2]二羟基铂。欧洲药典采用对照品法对其含量进行控制,而中国药典采用自身对照法对其进行定量。目前国内尚无奥沙利铂杂质c对照品,只能购买欧洲药典的杂质c对照品,价格昂贵。而且由于西林瓶及瓶塞的吸附,标示装量25mg的杂质c对照品实际只能称取15mg左右,造成极大的浪费。奥沙利铂杂质c结构式如下:
《药物分析杂志》2011.31(7)第1358页,山东省药品检验所的牛冲、徐志洲、李涛、李军等(其中部分也是本申请的发明人),采用h2o2作为氧化剂,通过氧化制备杂质c,但其产率低,只有83%,纯度为99.5%,且加入的双氧水易引入其他物质导致纯度与欧洲药典对照品相比较低,并且收率也较低。
技术实现要素:
为了解决以上现有技术中以奥沙利铂为原料,采用h2o2作为氧化剂制备奥沙利铂杂质c中存在的收率低、纯度低的问题,本申请公开了一种收率高、纯度高的奥沙利铂杂质c的制备方法。
本发明是通过以下步骤得到的:
一种奥沙利铂杂质c,是通过以下步骤得到的:
(1)氧化:取奥沙利铂,超声溶解,在30℃的条件下持续向奥沙利铂溶液中充入氧气,密封,静置1-1.5小时,重复充氧静置操作4-10次,保持氧气过量,促使反应的进一步进行,最终确保奥沙利铂与氧气的摩尔比小于1:1;
(2)粗品制备:将步骤(1)中得到的溶液减压浓缩至原体积的二分之一,水浴蒸干,即得杂质c粗品;
(3)重结晶:取杂质c粗品,加入约180倍水,水浴加热至80℃,超声使溶解,过滤,滤液减压浓缩至二分之一,4℃冰箱放置2-3小时,析出结晶,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至体积的二分之一,4℃冰箱放置2-3小时,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至体积的二分之一,4℃冰箱放置2-3小时,过滤,结晶用水洗涤数次,合并各次所得结晶,得奥沙利铂杂质c精制品,干燥即得奥沙利铂杂质c。
以奥沙利铂为起始原料,原料含量大于99%,其中草酸、奥沙利铂二水合物及其他杂质的总和均低于0.1%,引入的杂质少。
采用o2作为氧化剂,不引入其他杂质,无须后续的分离提纯工作,且与双氧水相比,o2氧化作用温和,不存在过度氧化。双氧水工艺,由于双氧水为浓度30%的水溶液,会引入其他杂质,后续分离提纯损失大,导致收率低,且双氧水氧化能力较强,会导致其他氧化分解产物的产生,影响终产品的纯度。
奥沙利铂水溶性差,如配制的溶液过稀,可导致奥沙利铂水解为草酸和二水合奥沙利铂,故需控制奥沙利铂与水的比例,适当超声助溶。
奥沙利铂在水溶液中易水解生成二水合奥沙利铂(杂质b),温度对水解过程影响较大,本发明控制温度在25~35℃(30℃尤为合适),可有效防止其水解。
所述的奥沙利铂杂质c,优选步骤(1)中奥沙利铂与氧气的摩尔比小于1:1。
所述的奥沙利铂杂质c,优选步骤(1)和(3)中超声功率500w,频率40khz。
所述的奥沙利铂杂质c,优选步骤(2)和(3)中减压浓缩工艺为温度:80℃,真空度:100mbar,转速:60转/分钟。防止样品受热分解,采用减压浓缩,控制温度不超过80℃,同时控制真空度在100mbar以加快浓缩速度。
所述的奥沙利铂杂质c,优选步骤(3)中奥沙利铂杂质c精制品105℃干燥4小时。减压干燥方式,不能完全去除结晶水,为确保水分完全去除,选用高温干燥方式。为防止样品受热降解,干燥温度控制在100~110℃之间,最终确定105℃,干燥4小时。
所述的奥沙利铂杂质c作为对照品,对奥沙利铂及注射用奥沙利铂中杂质c进行测定中的应用,为奥沙利铂原料及制剂质量标准的提升及产品的安全性提供技术支撑,具有良好的经济社会效益。
所述的应用,优选包括以下步骤:
(1)溶液的制备:
对照品溶液:称取奥沙利铂杂质c对照品10mg,置100ml量瓶中,加水溶解并稀释至刻度,摇匀,量取5ml,置100ml量瓶中,加水稀释至刻度,摇匀;
供试品溶液:取供试品,采用水制备供试品溶液,每1ml供试品溶液中含5mg奥沙利铂;
系统适用性溶液:取奥沙利铂对照品10mg,置10ml量瓶中,加水适量,超声使溶解,加3%双氧水2ml,用水稀释至刻度,摇匀,立即进样;
(2)高效液相色谱法,色谱条件:十八烷基硅烷键合色谱柱;流动相为磷酸溶液:乙腈=1000:5;检测波长为210nm,柱温为30℃,流速为1.0ml/min;
(3)测定方法
取系统适用性溶液、供试品溶液及对照品溶液10μl注入液相色谱仪,记录色谱图,以外标法计算供试品中杂质c的含量。
所述的应用,优选检测时配制溶液中杂质c的含量不超过0.1%。
所述的应用,优选步骤(2)中磷酸溶液是通过以下操作配制得到的:
取0.6ml稀磷酸溶液,用水稀释至1000ml,用氢氧化钠溶液或磷酸调ph至3.0±0.05。
奥沙利铂溶于水中后,加入h2o2溶液,是在同相水介质中发生的反应,此反应从原理上来说,反应是很充分的。向奥沙利铂水溶液中通入氧气氧化,是在不同相水相和气相之间发生的反应,从原理上看,不会比同相反应更充分。但在本申请技术方案中,采用持续通入氧气氧化的操作,保持氧气浓度始终处于过量状态,从而促使氧化反应不断的进行;同时由于氧气不带入任何杂质,且氧化作用温和,虽然反应时间长,但使得奥沙利铂杂质c的收率更高,产物纯度也更高,所以说,采用氧气氧化,取得了意料不到的技术效果。
本发明的有益效果:
(1)以药用奥沙利铂为起始原料,引入的杂质少,在双氧水氧化工艺上进行了优化,采用o2作为氧化剂,不引入其他杂质,无须后续的分离提纯工作;不存在过度氧化,产率及纯度均有明显提高;
(2)通过对起始物料的选择、水溶液浓度、氧气用量、氧化条件等的设定,使反应过程中奥沙利铂的其他杂质如杂质a、杂质b等不会氧化,同时也不会导致奥沙利铂水解产生杂质a和杂质b,大大提高了杂质c的纯度;
(3)易于制备,反应步骤少,制备成本低、产率高、制备方法简单,氧气充足的条件下,奥沙利铂几乎全部转化为杂质c,可极大的降低奥沙利铂杂质c对照品的成本,制备的杂质c的纯度优于双氧水氧化工艺,与欧洲药典奥沙利铂杂质c对照纯度相当,为奥沙利铂的杂质鉴定和检测、质量标准的提高及产品的质量控制提供技术支持。
附图说明
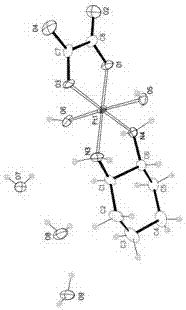
图1:奥沙利铂杂质cx-单晶衍射pka图,
图2:奥沙利铂杂质cx-单晶衍射telp30图,
图3:奥沙利铂杂质c欧洲药典对照品液相色谱图,
图4:本发明制备得到的奥沙利铂杂质c液相色谱图。
具体实施方式
下面结合具体实施例对本发明进行进一步说明:
实施例1
(1)氧化取奥沙利铂原料5.0g,置3000ml量瓶中,加水1000ml,超声(功率:500w,频率:40khz)10分钟使溶解,在30℃的条件下瓶中充入过量氧气,密封,静置1小时,如此充氧静置4次。
(2)粗品制备将上述溶液用buchir210旋转蒸发仪(温度:80℃,真空度:100mbar,转速:60转/分钟)减压浓缩至500ml,水浴上蒸干,即得杂质c粗品5.52g。
(3)重结晶取杂质c粗品,加水1000ml,水浴加热至80℃,超声(功率:500w,频率:40khz)30分钟使溶解,过滤,滤液用buchir210旋转蒸发仪(温度:80℃,真空度:100mbar,转速:60转/分钟)减压浓缩至400ml,4℃冰箱放置2小时,析出结晶,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至200ml,4℃冰箱放置2小时,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至50ml,4℃冰箱放置2小时,过滤,结晶用水洗涤数次。合并每次所得结晶,得奥沙利铂杂质c精制品。
(4)取奥沙利铂杂质c精制品,105℃减压干燥4小时,得4.79g淡黄色粉末,最终产率为88.21%。
(5)含量测定对本发明方法制得的奥沙利铂杂质c进行含量测定,经测定,杂质c的含量为99.79%,rsd为0.1%(n=9)。
实施例2
(1)氧化取奥沙利铂原料5.0g,置3000ml量瓶中,加水1000ml,超声(功率:500w,频率:40khz)10分钟使溶解,在30℃的条件下瓶中充入过量氧气,密封,静置1小时,如此充氧静置10次。
(2)粗品制备将上述溶液用buchir210旋转蒸发仪(温度:80℃,真空度:100mbar,转速:60转/分钟)减压浓缩至500ml,水浴上蒸干,即得杂质c粗品5.5g。
(3)重结晶取杂质c粗品,加水1000ml,水浴加热至80℃,超声(功率:500w,频率:40khz)30分钟使溶解,过滤,滤液用buchir210旋转蒸发仪(温度:80℃,真空度:100mbar,转速:60转/分钟)减压浓缩至400ml,4℃冰箱放置2小时,析出结晶,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至200ml,4℃冰箱放置2小时,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至50ml,4℃冰箱放置2小时,过滤,结晶用水洗涤数次。合并每次所得结晶,得奥沙利铂杂质c精制品。
(4)取奥沙利铂杂质c精制品,105℃减压干燥4小时,得4.81g淡黄色粉末,最终产率为88.58%。
(5)含量测定对本发明方法制得的奥沙利铂杂质c进行含量测定,经测定,杂质c的含量为99.82%,rsd为0.1%(n=9)。
对比例1:
(1)氧化取奥沙利铂原料5.0g,置3000ml量瓶中,加水1000ml,超声(功率:500w,频率:40khz)10分钟使溶解,在30℃的条件下瓶中充入过量氧气,密封,静置1小时,如此充氧静置2次。
(2)粗品制备将上述溶液用buchir210旋转蒸发仪(温度:80℃,真空度:100mbar,转速:60转/分钟)减压浓缩至500ml,水浴上蒸干,即得杂质c粗品5.4g。
(3)重结晶取杂质c粗品,加水1000ml,水浴加热至80℃,超声(功率:500w,频率:40khz)30分钟使溶解,过滤,滤液用buchir210旋转蒸发仪(温度:80℃,真空度:100mbar,转速:60转/分钟)减压浓缩至400ml,4℃冰箱放置2小时,析出结晶,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至200ml,4℃冰箱放置2小时,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至50ml,4℃冰箱放置2小时,过滤,结晶用水洗涤数次。合并每次所得结晶,得奥沙利铂杂质c精制品。
(4)取奥沙利铂杂质c精制品,105℃减压干燥4小时,得4.03g淡黄色粉末,最终产率为74.22%。
(5)含量测定对本发明方法制得的奥沙利铂杂质c进行含量测定,经测定,杂质c的含量为97.04%,rsd为0.2%(n=9)。
对比例2:
(1)氧化取奥沙利铂原料5.0g,置3000ml量瓶中,加水1000ml,超声(功率:500w,频率:40khz)10分钟使溶解,在25℃的条件下瓶中充入过量氧气,密封,静置1小时,如此充氧静置4次。
(2)粗品制备将上述溶液用buchir210旋转蒸发仪(温度:80℃,真空度:100mbar,转速:60转/分钟)减压浓缩至500ml,水浴上蒸干,即得杂质c粗品5.4g。
(3)重结晶取杂质c粗品,加水1000ml,水浴加热至80℃,超声(功率:500w,频率:40khz)30分钟使溶解,过滤,滤液用buchir210旋转蒸发仪(温度:80℃,真空度:100mbar,转速:60转/分钟)减压浓缩至400ml,4℃冰箱放置2小时,析出结晶,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至200ml,4℃冰箱放置2小时,过滤,结晶用水洗涤数次;合并滤液及洗液,减压浓缩至50ml,4℃冰箱放置2小时,过滤,结晶用水洗涤数次。合并每次所得结晶,得奥沙利铂杂质c精制品。
(4)取奥沙利铂杂质c精制品,105℃减压干燥4小时,得4.23g淡黄色粉末,最终产率为77.90%。
(5)含量测定对本发明方法制得的奥沙利铂杂质c进行含量测定,经测定,杂质c的含量为98.23%,rsd为0.1%(n=9)。
最终确定的反应条件为,反复充氧气至少4次,水溶液的温度保持30℃。
应用例:
(1)实验组:使用本发明制备的杂质c作为对照品以外标法测定奥沙利铂及注射用奥沙利铂中杂质c。
对照品:自制杂质c(含量99.8%),乙腈为色谱纯,水为纯化水,起始试剂为分析纯。
对照品溶液:精密称取杂质c对照品10mg,置100ml量瓶中,加水溶解并稀释至刻度,摇匀。精密量取5ml,置100ml量瓶中,加水稀释至刻度,摇匀。
供试品溶液:取供试品,采用水制备供试品溶液,每1ml供试品溶液中含5mg奥沙利铂。
系统适用性溶液:取奥沙利铂对照品10mg,置10ml量瓶中,加水适量,超声使溶解,加3%双氧水2ml,用水稀释至刻度,摇匀,立即进样。出峰顺序依次为过氧化氢、杂质c和奥沙利铂。
色谱条件:岛津lc-20a高效液相色谱仪,色谱柱为waterssymmetryc18柱(4.6×150mm,5μm)。流动相为磷酸溶液(0.6ml稀磷酸,稀释至1000ml,用磷酸或氢氧化钠溶液调ph至3.0)-乙腈(1000:5);检测波长为210nm。
测定法:取系统适用性溶液10μl,注入液相色谱仪,记录色谱图,杂质c与奥沙利铂峰分离度应大于10。在分别取供试品溶液和对照品溶液各10μl,注入液相色谱仪,记录色谱图,以外标法计算杂质c的含量,杂质c不得过0.1%。
(2)欧洲药典对照组:使用欧洲药典杂质c对照品以外标法测定奥沙利铂及注射用奥沙利铂中杂质c。
对照品:奥沙利铂杂质c,欧洲药典,分类号:y0000273,批号:6.0。
溶液制备、色谱条件及测定方法同(1)实验组。
(3)中国药典对照组:使用中国药典方法采用自身对照法测定奥沙利铂及注射用奥沙利铂中杂质c。
供试品溶液:取供试品,采用水制备供试品溶液,每1ml供试品溶液中含2mg奥沙利铂。
对照溶液:精密量取供试品溶液1ml,置100ml量瓶中,加水溶解并稀释至刻度,摇匀。精密量取5ml,置50ml量瓶中,加水稀释至刻度,摇匀。
系统适用性、色谱条件同(1)实验组
测定法:测定法:取系统适用性溶液10μl,注入液相色谱仪,记录色谱图,杂质c与奥沙利铂峰分离度应大于10。在分别取供试品溶液和对照溶液各10μl,注入液相色谱仪,记录色谱图,供试品溶液色谱图中,如有与系统适用性溶液第二个色谱峰保留时间一致杂质峰,其峰面积不得过对照溶液主峰面积的4.6倍(0.1%)。
(4)测定结果,见表1。
表1实验组、欧洲药典对照组、中国药典对照组供试品测定结果。
由表1可知,采用自制对照品的实验组与欧洲药典对照品对照组结果基本一致,中国药典由于采用加校正因子的自身对照法,结果有一定的偏差。可见,本发明制备的杂质c对照品可用于原料及制剂的质量控制,方法更科学合理,杂质含量更接近真实值。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受实施例的限制,其它任何未背离本发明的精神实质与原理下所做的改变、修饰、组合、替代、简化均应为等效替换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!