通用无标记LIC载体pMF-LIC及其构建方法与应用与流程
本发明涉及基因工程
技术领域:
,具体涉及一种通用无标记lic载体pmf-lic及其构建方法与应用。
背景技术:
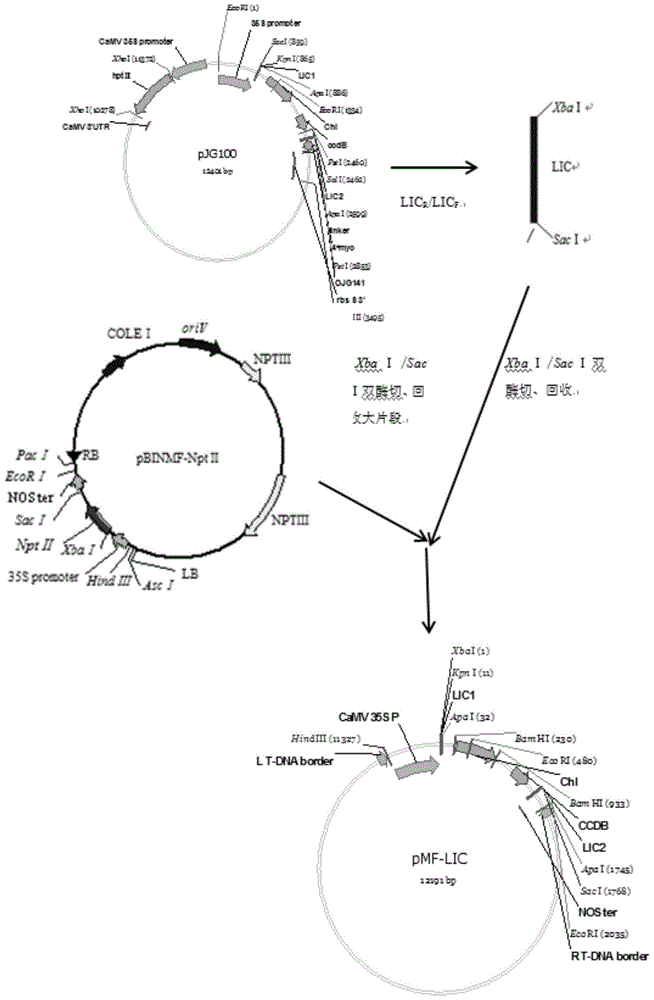
:谈到转基因技术,大家就会想到转基因生物安全性的问题。当今,大众对转基因生物的安全性问题非常关注,关注主要集中于对转基因过程中使用的一些筛选标记基因,这些标记基因多数为一些编码抗生素抗性和除草剂抗性的基因。因此,获得一些无标记基因的转基因植物,可能会以提高大众对转基因产品的接受度。到目前为止,已经获得了一些无标记基因的转化植物,但一些方法都是在转化时仍使用标记基因,然后得到阳性转化体后通过各种手段再将筛选标记基因去除,费时费力。另外一些学者在基因转化过程中就使用了无选择标记基因的转化系统,一步到位即可得到无标记基因的转化体。但不管哪种获得无标记转化体的载体,在外源基因克隆的过程中都依赖限制性内切酶,然后再依赖连接酶将外源基因和载体连接。这样将外源基因克隆到载体的方法受到基因序列中原有的酶切位点的限制,在应用中受到一定的局限。同时,如果实验室想将不同基因的克隆到载体,就要考虑每个基因和载体的酶切位点,就需要利用不同的酶切位点,需要购买不同的内切酶,通过多次的酶切和连接来克隆不同的基因,费时费钱。技术实现要素:针对现有技术中的缺陷,有必要构建一个不仅在转化过程中,只导入外源目的基因,不携带选择标记基因的生物安全的无标记载体,而且该无标记载体应该在克隆外源基因过程中,不受酶切位点限制,可以一次操作后克隆任何外源基因的通用无标记载体。本发明目的在于提供一种通用无标记lic载体pmf-lic,其是采用质粒pmf-nptii和lic序列构建而成。通用无标记lic载体pmf-lic的构建方法包括步骤:以质粒pjg100为模板,以seqidno.1(licr:5’-gactctagaggtaccttcgacgacaagaccgg-3’)和seqidno.2(licf:5’-tgggagctcgaggagaagagccgggcccctac-3’)为引物对进行pcr扩增,得到目的片段lic序列;将目的片段lic序列用限制性内切酶xbai和saci双酶切,再与经同样双酶切的pmf-nptii质粒连接,获得通用无标记lic载体pmf-lic。本发明另一种目的在于提供一种携带抗晚疫病基因r1的重组载体,其是采用通用无标记lic载体pmf-lic和抗晚疫病基因r1构建而成。携带抗晚疫病基因r1的重组载体的构建方法包括步骤:以携带抗晚疫病基因r1的pcld04541-r1为模板,以seqidno.3(r1r:5’-cgacgacaagaccgtaccatgaatttcaacaatgaattgtctgatc-3’)和seqidno.4(r1f:5’-gaggagaagagccgtctatcttatttctgcaagaatattttttac-3’)为引物对进行pcr扩增,得到抗晚疫病基因r1;将抗晚疫病基因r1用限制性内切酶apai酶切,再与经同样酶切的通用无标记lic载体pmf-lic连接,获得携带抗晚疫病基因r1的重组载体。本发明的另一种目的在于提供一种携带抗晚疫病基因r1的重组载体的微生物转化体,其是以农杆菌为受体而转化得到;优选地,农杆菌为农杆菌agl0。携带抗晚疫病基因r1的重组载体的微生物转化体的构建方法包括步骤:采用三亲融合的方法,将携带抗晚疫病基因r1的重组载体转入农杆菌中,得到携带抗晚疫病基因r1的重组载体的微生物转化体。本发明还保护通用无标记lic载体pmf-lic、携带抗晚疫病基因r1的重组载体或携带抗晚疫病基因r1的重组载体的微生物转化体在提高植物抗晚疫病效果中的应用。优选地,植物为马铃薯,优选为马铃薯品种desiree。本发明的另一种目的在于提供一种培育提高抗晚疫病效果的植物的方法,包括使用通用无标记lic载体pmf-lic的步骤;或者,包括使用携带抗晚疫病基因r1的重组载体的步骤;或者,包括使用携带抗晚疫病基因r1的重组载体的微生物转化体的步骤。本发明的另一种目的在于提供一种转基因马铃薯品种的构建方法,包括步骤:将携带抗晚疫病基因r1的重组载体的微生物转化体采用农杆菌介导的方法侵染马铃薯品种desiree,得到转基因马铃薯品种。pcr片段和载体在t4dna聚合酶的3’-5’外切酶活性和一种特定的dntp存在的条件下反应,在其末端生成10~15个碱基的粘性末端。因此,处理过的pcr片段和载体可依靠生成的粘性末端重组,在这个过程中没有用到连接酶。使用这种克隆方法免去了对插入片段序列的考虑,在载体上可以克隆任何需要的基因。而且整个连接过程都不需要使用限制性内切酶和连接酶,pcr片段和载体是在体外条件下连接,二者处理后生成的粘性末端退火互补形成双链环状分子,经转化进入大肠杆菌细胞后,细菌的修复系统修复这些突出和缺口而得到正确重组子。本发明提供的通用无标记lic载体pmf-lic,具有如下的有益效果:(1)安全:用该载体进行基因转化,得到的转基因材料,除了引入目的基因外,不含任何载体的骨架序列;(2)简便:外源待克隆的pcr片段不需要酶切和连接反应,简化了各种酶切位点在引物设计时的思考麻烦;(3)批处理:多个基因在实验过程中可以一同操作,同时同样处理所有的样品,省时省力;(4)高效:该方法不依赖连接酶,很大程度上简化了dna重组过程,且完全靠粘性末端配对退火,载体自连背景低;另外,也不用考虑载体末端的磷酸化等现状;(5)省钱:该方法只需t4dna聚合酶和datp及dttp,以及带有额外10-15bp附加碱基的引物,而且克隆过程中省去了连接酶,也没有使用价格不菲的特殊重组酶。本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。附图说明图1为本发明实施例中pmf-lic构建示意图;图2为本发明实施例中lic序列pcr扩增结果图;图3为本发明实施例中pmf-lic质粒xbai/saci双酶切及saci单酶切鉴定结果图;图4为本发明实施例中载体pmf-lic酶切图;图5为本发明实施例中pmf-lic-r1质粒单、双酶切图;图6为本发明实施例中转r1基因(r1)与对照(ck)无菌苗接种发病症状结果图。具体实施方式下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。以下实施例仅用于更加清楚地说明本发明的技术方案,因此只是作为示例,而不能以此来限制本发明的保护范围。下述实施例中的实验方法,如无特殊说明,均为常规方法。下述实施例中所用的试验材料,如无特殊说明,均为自常规试剂商店购买得到的。以下实施例中的定量试验,均设置三次重复实验,数据为三次重复实验的平均值或平均值±标准差。(一)构建通用无标记lic载体pmf-lic通用无标记lic载体pmf-lic的构建示意图如图1所示。1、目的片段lic序列的扩增、回收、纯化以质粒pjg100为模板,利用引物对licr/licf(序列如表1所示)进行pcr扩增,扩增目的片段lic序列,大小为1.8kb,lic序列pcr扩增结果如图2所示,其中,1:pjg100扩增;m:1kbdnamaker。表1引物表引物序列licr5’-gactctagaggtaccttcgacgacaagaccgg-3’licf5’-tgggagctcgaggagaagagccgggcccctac-3’r1r5’-cgacgacaagaccgtaccatgaatttcaacaatgaattgtctgatc-3’r1f5’-gaggagaagagccgtctatcttatttctgcaagaatattttttac-3’mfr1r5’-atgaatttcaacaatgaattgtctgatctg-3’mfr1f5’-gaaggttggccatttgatcactcaaaccaacc-3’2、质粒pmf-nptii和lic序列双酶切pmf-nptii质粒提取参照天跟质粒提取试剂盒,用限制性内切酶xbai和saci双酶切质粒pmf-nptii和目的片段lic序列的回收产物。3、质粒与lic序列连接并转化使用t4dnaligase酶,将回收的lic序列和pmf-nptii酶切载体于16℃过夜连接。然后用热激法转化db3.1感受态细胞,并涂布平板。4、通用无标记载体pmf-lic阳性克隆筛选以平板上的单菌落为模板,以引物对licr/licf(序列如表1所示)进行菌落pcr筛选出阳性克隆。经菌落pcr筛选出的阳性克隆子经培养后提取质粒,进行质粒pcr检测和质粒的xbai/saci双酶切及saci单酶切检测。pmf-lic质粒xbai/saci双酶切及saci单酶切鉴定结果如图3所示,其中,1:水;2-9:依次为阳性克隆子双酶切、单酶切;m:1kbplusdnamaker。经菌落pcr、质粒pcr及质粒单、双酶切鉴定获得重组载体pmf-lic。(二)构建携带抗晚疫病基因r1的重组载体以携带抗晚疫病基因r1的pcld04541-r1为模板,以引物对r1r/r1f(序列如表1所示)进行pcr扩增抗晚疫病基因r1。用限制性内切酶apai将pmf-lic进行酶切(载体pmf-lic酶切图如图4所示,其中,1:pmf-lic质粒;2:apai酶切质粒),并用t4dna聚合酶预处理,这样一次预处理后的载体就可以高通量克隆任何外源基因,不受基因和载体酶切位点的限制。外源基因也要进行t4dna聚合酶预处理,处理体系如表2所示。表2t4dna聚合酶预处理体系试剂体积(μl)载体pmf-lic/序列1.00/x(~50ng)5×buffer1.00100mmdttp0.25t4dna聚合酶0.10ddh2oupto5.00体系分别加配完后进行lic连接,运行程序如下:结束后,取5μllic连接产物用热激法转化大肠杆菌top10感受态细胞中。37℃倒置过夜培养,获得单克隆菌斑。挑取lb平板上的单菌落进行菌落pcr检测(检测引物为mfr1r/mfr1f,序列如表1所示)、质粒酶切(pmf-lic-r1质粒单、双酶切如图5,其中,1:pmf-r1质粒;2:单酶切;3:双酶切;m:1kbdnamaker)验证获得含有外源基因的阳性克隆。(三)转基因植物获得及检测采用三亲融合的方法,将携带抗晚疫病基因r1的重组载体转入农杆菌agl0中。然后采用农杆菌介导的方法侵染马铃薯品种desiree,并通过pcr检测获得阳性转化体。(四)转基因马铃薯抗性评价扩繁pcr鉴定的阳性转基因苗和野生型对照,待扩繁苗长到3~4周时取部分苗进行早期无菌苗接种鉴定。先制备晚疫病病原菌游动孢子,将生长2周的各生理小种的孢子囊用无菌水冲洗到灭菌培养皿中,4℃冰箱放置4~6h使其释放游动孢子,然后镜检游动孢子浓度并稀释调整到终浓度为5×104个/ml。转r1基因株系分别接种亲和性生理小种99018(1.4)和非亲和性生理小种89148-9(0),野生型接种生理小种89148-9(0)。接种6天后统计发病情况,结果如图6所示。图6为转r1基因(r1)与对照(ck)无菌苗接种发病症状结果图,其中,a为对照ck接种亲和性生理小种89148-9,b为转基因株系r1接种非亲和生理小种89148-9,c为转基因株系接种亲和生理小种99018,亲和性小种接种后都表现为高度感病(sp),非亲和小种则表现为抗病(hr)。上述试验每组处理重复3次。由图6可以说明,抗晚疫病基因r1已经成功由pmf-lic载体转入马铃薯品种desiree中,而且抗晚疫病效果明显。需要注意的是,除非另有说明,本申请使用的技术术语或者科学术语应当为本发明所属领域技术人员所理解的通常意义。除非另外具体说明,否则在这些实施例中阐述的部件和步骤的相对步骤、数字表达式和数值并不限制本发明的范围。在这里示出和描述的所有示例中,除非另有规定,任何具体值应被解释为仅仅是示例性的,而不是作为限制,因此,示例性实施例的其他示例可以具有不同的值。最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围,其均应涵盖在本发明的保护范围当中。sequencelisting<110>内蒙古科技大学<120>通用无标记lic载体pmf-lic及其构建方法与应用<130>1<160>6<170>patentinversion3.3<210>1<211>32<212>dna<213>人工序列<400>1gactctagaggtaccttcgacgacaagaccgg32<210>2<211>32<212>dna<213>人工序列<400>2tgggagctcgaggagaagagccgggcccctac32<210>3<211>46<212>dna<213>人工序列<400>3cgacgacaagaccgtaccatgaatttcaacaatgaattgtctgatc46<210>4<211>45<212>dna<213>人工序列<400>4gaggagaagagccgtctatcttatttctgcaagaatattttttac45<210>5<211>30<212>dna<213>人工序列<400>5atgaatttcaacaatgaattgtctgatctg30<210>6<211>32<212>dna<213>人工序列<400>6gaaggttggccatttgatcactcaaaccaacc32当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1