一种无载体型超支化大分子聚合物及制备方法与流程
本发明属于高分子材料合成领域和功能性药物的制备方法,涉及一种无载体型超支化大分子聚合物及制备方法。
背景技术:
无载体纳米药物递送系统具有载药量高,制备策略简单,结构精确,药物释放稳定等优点,已经成为纳米药物发展的主要趋势。完全去除惰性的赋形剂不仅可以使载药量达100%,极大提高治疗效果,而且可以有效的避免了因为载体引发的毒性和免疫反应。
文献1“xiujuanxi,shiqihu,zhuxianzhouandyouqingshenetal.dendrimerswiththeprotocatechuicacidbuildingblockforanticancerdrugdelivery[j].journalofmaterialschemistryb,2016,4,5236-5245.”公开了一种以原儿茶酸单个药物为基元,构筑了具有树枝状形貌的聚合物药物,但是此方法合成路线较为繁琐,合成方法不够简单。
文献2“yajunhuang,xiaokangding,yuqi,bingranyuandfu-jianxu.reduction-responsivemultifunctionalhyperbranchedpolyaminoglycosideswithexcellentantibacterialactivity,biocompatibilityandgenetransfectioncapability[j].biomaterials,2016,106:134-143.”公开了一种以抗生素庆大霉素或妥布霉素或新霉素单个药物为基元,结合二硫键构筑具有树枝状形貌的聚合物药物,但是只采用了单一药物为基元,没有起到联合治疗疾病的作用。
文献3“xiaopinduan,jishengxiao,qiyinandyapinglietal.smartph-sensitiveandtemporal-controlledpolymericmicellesforeffectivecombinationtherapyofdoxorubicinanddisulfiram[j].acsnano,2013,7(7):5858-5869.”公开了一种采用阿霉素和二硫仑两种药物联合抗癌的策略,但是采用传统的载体进行物理包覆的方法,导致体系载药率不高,药物输送不稳定等缺点。
技术实现要素:
要解决的技术问题
为了避免现有技术的不足之处,本发明提出一种无载体型超支化大分子聚合物及制备方法,以疏水性甲氨蝶呤为基元的含有“二硫键”结构和两端为两个氨基的甲氨蝶呤单体与以疏水性药物苯丁酸氮芥为基元的含有“酯键”和末端为三个羧基修饰的苯丁酸氮芥单体通过双单体法合成的无载体型超支化大分子药物的制备方法。
技术方案
一种无载体型超支化大分子聚合物,其特征在于结构式为:
一种所述无载体型超支化大分子聚合物的制备方法,其特征在于步骤如下:
步骤1:冰浴条件下,按摩尔比1:2.5~3.5混合胱胺二盐酸盐和三乙胺,以甲醇为溶剂,搅拌20~40min;称取与胱胺二盐酸盐摩尔比为1︰0.3~0.35的二碳酸二叔丁酯溶解于甲醇后逐滴滴加到反应体系之中,滴加完毕后反应4~8h;随后加入与反应溶剂体积相等的去离子水,旋转蒸发仪蒸干溶剂,并用二氯甲烷作为溶剂溶解后加入饱和氯化钠溶液萃取,将下层有机相用无水硫酸钠干燥后通过旋转蒸发将滤液浓缩,过硅胶柱,取滤液,用旋转蒸发仪旋干溶剂,在真空干燥箱之中干燥36~60h,得到的淡黄色油状液体为单端叔丁氧羰基保护氨基的胱胺;
步骤2:冰浴条件下,按摩尔比1︰1.8~2.2︰1.8~2.2混合甲氨蝶呤、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和1-羟基苯并三唑,并加入n,n-二甲基甲酰胺作为溶剂,搅拌25~35min;之后将与甲氨蝶呤摩尔量比为1︰2.3~2.7的单端叔丁氧羰基保护氨基的胱胺溶于溶剂n,n-二甲基甲酰胺中,逐滴加入反应体系后反应10~14h;反应结束后,用旋转蒸发仪蒸干溶剂得到棕黄色油状液体,加入三氯甲烷作为溶剂溶解,加入饱和碳酸氢钠溶液萃取,将下层有机相用无水硫酸钠干燥后通过旋转蒸发将滤液旋干,再加入二氯甲烷作为溶剂溶解,旋转蒸发将液体浓缩,过硅胶柱,取滤液,用旋转蒸发仪蒸干溶剂后得到黄色固体粉末,即两端叔丁氧羰基保护的甲氨蝶呤;
步骤3:按摩尔比1︰0.4~0.6加入两端叔丁氧羰基保护的甲氨蝶呤和三氟乙酸,以二氯甲烷为溶剂,搅拌反应12~16h;反应结束后,用旋转蒸发仪蒸干溶剂,然后加入与溶剂二氯甲烷体积比为1︰2~2.5的1mol/l氢氧化钠溶液;最后用旋转蒸发仪旋干溶剂,在真空干燥箱中干燥46~50h,得到的黄色固体即为两端为两个氨基修饰的甲氨蝶呤单体;
步骤4:按摩尔比1︰1.1~1.2︰1.1~1.2加入苯丁酸氮芥、三羟甲基氨基甲烷、2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉,以乙醇为溶剂,回流加热搅拌于55~65℃下反应10~14h;反应结束后,用旋转蒸发仪蒸干溶剂,过硅胶柱,取滤液,用旋转蒸发仪蒸干溶剂后得到白色固体粉末,即末端为三个羟基修饰的苯丁酸氮芥;
步骤5:冰浴条件下,按摩尔比为1︰8~12混合末端为三个羟基修饰的苯丁酸氮芥和三乙胺,以四氢呋喃作为溶剂,搅拌30~40min;再将与三羟基苯丁酸氮芥摩尔量之比为1︰3.5~4.0的丁二酸酐溶于四氢呋喃中并逐滴加入到反应体系中,滴加完毕后搅拌反应10~14h;反应结束后,用旋转蒸发仪蒸干溶剂,加入二氯甲烷作为溶剂溶解,用饱和氯化铵溶液萃取;旋转蒸发将液体浓缩,过硅胶柱,取滤液,用旋转蒸发仪蒸干溶剂后得到白色固体粉末,即末端为三个羧基苯丁酸氮芥单体;
步骤6:冰浴条件下,按摩尔比为1:2.0~2.2︰2.0~2.2︰22~27混合末端为三个羧基苯丁酸氮芥单体、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐、1-羟基苯并三唑和三乙胺,以n,n-二甲基甲酰胺作为溶剂,搅拌25~35min;之后将与末端为三个羧基苯丁酸氮芥单体摩尔量比为1︰2.8~3.2的两端为两个氨基修饰的甲氨蝶呤单体溶于溶剂n,n-二甲基甲酰胺中,逐滴加入反应体系后反应70~75h;反应结束后,加入与反应液体积相同的去离子水,旋转蒸发将液体浓缩,然后将其转移到截留分子量为5000的透析袋在n,n-二甲基甲酰胺中透析2~3d;之后,旋转蒸发将液体进一步浓缩,加入浓缩后反应溶液体积25~35倍的冰四氢呋喃沉淀,得到棕黄色的固体即无载体型超支化大分子聚合物。
所述步骤1、2、3、5中所述萃取操作是指在分液漏斗中,将有机相与相应水溶液充分混合之后,收集有机相并重复三次。
所述步骤4中所述回流加热是指在恒温油浴条件下,单口瓶瓶口加上冷凝回流管并通冷凝水。
所述步骤6中所述滴加两端为两个氨基修饰的甲氨蝶呤单体的时间持续为15~20min。
所述步骤6中所述冰四氢呋喃是指:将四氢呋喃置于-18℃~-22℃冰箱冷冻室放置30min~1h后所得。
有益效果
本发明提出的一种无载体型超支化大分子聚合物及制备方法,以两种常见的疏水性药物甲氨蝶呤和苯丁酸氮芥为基元,对这两种疏水性药物进行结构修饰改性,使其结构中分别含有动态共价键“二硫键”和“酯键”,然后以“a2+b3”法聚合成为超支化大分子聚合物,最后自组装成为超支化大分子药物型的无载体纳米药物自递送系统,从而为无载体纳米药物递送系统的构筑提供了新的思路,并且解决了现有超支化聚合物在药物包载方面所存在的载药量低,细胞毒性大,对于肾脏等器官毒副作用大以及响应性不足等问题。
本发明的有益效果是:通过两端为两个氨基修饰的甲氨蝶呤单体与末端为三个羧基苯丁酸氮芥单体之间的酰胺化反应聚合得到超支化大分子药物,自组装形成无载体纳米药物自递送系统。该体系解决了现有超支化聚合物自组装体在药物包载方面所存在的载药量低、细胞毒性大等问题;同时为新型无载体纳米药物递送系统的构筑提供了思路和方法。
附图说明
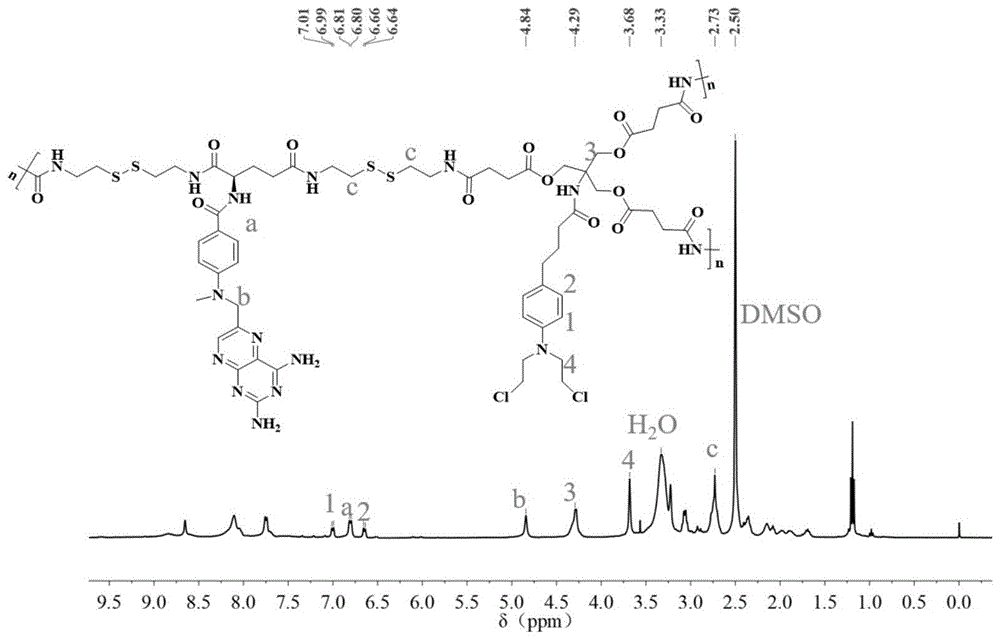
图1:无载体型超支化大分子药物的核磁共振氢谱图
具体实施方式
现结合实施例、附图对本发明作进一步描述:
本发明的无载体型超支化大分子聚合物,包括两个单体:
单体结构式之一:
单体结构式之二:
以“a2+b3”法聚合成为超支化大分子聚合物,结构式为:
制备方法:
步骤1:冰浴条件下,向干燥的单口烧瓶中按摩尔比1:2.5~3.5加入胱胺二盐酸盐和三乙胺,以甲醇为溶剂,搅拌20~40min;称取与胱胺二盐酸盐摩尔比为1:0.3~0.35的二碳酸二叔丁酯溶解于甲醇后逐滴滴加到反应体系之中,滴加完毕后反应4~8h;随后加入与反应溶剂体积相等的去离子水,旋转蒸发仪蒸干溶剂,并用二氯甲烷作为溶剂溶解后加入饱和氯化钠溶液萃取,将下层有机相用无水硫酸钠干燥后通过旋转蒸发将滤液浓缩,过硅胶柱,取滤液,用旋转蒸发仪旋干溶剂,在真空干燥箱之中干燥36~60h,得到的淡黄色油状液体为单端叔丁氧羰基保护氨基的胱胺;
步骤2:冰浴条件下,向干燥的单口烧瓶中按摩尔比1:1.8~2.2:1.8~2.2加入甲氨蝶呤、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和1-羟基苯并三唑,并加入n,n-二甲基甲酰胺作为溶剂,搅拌25~35min;之后将与甲氨蝶呤摩尔量比为1:2.3~2.7的单端叔丁氧羰基保护氨基的胱胺溶于溶剂n,n-二甲基甲酰胺中,逐滴加入反应体系后反应10~14h;反应结束后,用旋转蒸发仪蒸干溶剂得到棕黄色油状液体,加入三氯甲烷作为溶剂溶解,加入饱和碳酸氢钠溶液萃取,将下层有机相用无水硫酸钠干燥后通过旋转蒸发将滤液旋干,再加入二氯甲烷作为溶剂溶解,旋转蒸发将液体浓缩,过硅胶柱,取滤液,用旋转蒸发仪蒸干溶剂后得到黄色固体粉末,即两端叔丁氧羰基保护的甲氨蝶呤;
步骤3:向干燥的单口烧瓶中按摩尔比1:0.4~0.6加入两端叔丁氧羰基保护的甲氨蝶呤和三氟乙酸,以二氯甲烷为溶剂,搅拌反应12~16h;反应结束后,用旋转蒸发仪蒸干溶剂,然后加入与溶剂二氯甲烷体积比为1:2~2.5的1mol/l氢氧化钠溶液;最后用旋转蒸发仪旋干溶剂,在真空干燥箱中干燥46~50h,得到的黄色固体即为两端为两个氨基修饰的甲氨蝶呤单体;
步骤4:向干燥的单颈烧瓶中按摩尔比1:1.1~1.2:1.1~1.2加入苯丁酸氮芥、三羟甲基氨基甲烷、2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉,以乙醇为溶剂,回流加热搅拌于55~65℃下反应10~14h;反应结束后,用旋转蒸发仪蒸干溶剂,过硅胶柱,取滤液,用旋转蒸发仪蒸干溶剂后得到白色固体粉末,即末端为三个羟基修饰的苯丁酸氮芥;
步骤5:冰浴条件下,向干燥的单口烧瓶中按摩尔比为1:8~12加入末端为三个羟基修饰的苯丁酸氮芥和三乙胺,以四氢呋喃作为溶剂,搅拌30~40min;再将与cb-tris摩尔量之比为1:3.5~4.0的丁二酸酐溶于四氢呋喃中并逐滴加入到反应体系中,滴加完毕后搅拌反应10~14h。反应结束后,用旋转蒸发仪蒸干溶剂,加入二氯甲烷作为溶剂溶解,用饱和氯化铵溶液萃取;旋转蒸发将液体浓缩,过硅胶柱,取滤液,用旋转蒸发仪蒸干溶剂后得到白色固体粉末,即末端为三个羧基苯丁酸氮芥单体;
步骤6:冰浴条件下,向干燥的单口烧瓶中按摩尔比为1:2.0~2.2:2.0~2.2:22~27加入末端为三个羧基苯丁酸氮芥单体、1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐、1-羟基苯并三唑和三乙胺,以n,n-二甲基甲酰胺作为溶剂,搅拌25~35min;之后将与末端为三个羧基苯丁酸氮芥单体摩尔量比为1:2.8~3.2的两端为两个氨基修饰的甲氨蝶呤单体溶于溶剂n,n-二甲基甲酰胺中,逐滴加入反应体系后反应70~75h;反应结束后,加入与反应液体积相同的去离子水,旋转蒸发将液体浓缩,然后将其转移到截留分子量为5000的透析袋在n,n-二甲基甲酰胺中透析2~3d。之后,旋转蒸发将液体进一步浓缩,加入浓缩后反应溶液体积25~35倍的冰四氢呋喃沉淀,得到棕黄色的固体即无载体型超支化大分子药物。
实施例1:
称取250mg胱胺二盐酸盐置于100ml干燥的单口烧瓶之中,并加入5ml的甲醇和350mg三乙胺,在冰浴的条件下搅拌30min。称取88mg二碳酸二叔丁酯于烧杯中并用10ml甲醇溶解,用恒压滴液漏斗将其逐滴滴加到单口烧瓶之中,反应5h之后加入10ml的去离子水,终止反应。减压旋蒸除去反应溶剂,然后加入30ml饱和氯化钠溶液,用50ml的二氯甲烷萃取三次,有机相用无水硫酸钠干燥。所得固体直接上样过硅胶柱分离。用旋转蒸发仪蒸干溶剂,在真空干燥箱之中干燥2d,得到淡黄色油状液体单端叔丁氧羰基保护氨基的胱胺123mg。
称取90.9mg甲氨蝶呤、76.7mg1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和54.1mg1-羟基苯并三唑于50ml干燥的单口烧瓶之中,加入10ml的n,n-二甲基甲酰胺作为溶剂,在冰水浴的条件下搅拌30min,之后称取126mg单端叔丁氧羰基保护氨基的胱胺溶于5ml的n,n-二甲基甲酰胺,逐滴加入单口烧瓶之中。反应12h之后,用旋转蒸发仪蒸干溶剂,得到棕黄色油状液体。将棕黄色油状液体再溶于30ml的三氯甲烷之中,用饱和碳酸氢钠溶液萃取三次,再用无水硫酸钠干燥。最后用旋转蒸发仪蒸干溶剂,溶于二氯甲烷上样,柱分离。最终得到黄色固体粉末两端叔丁氧羰基保护的甲氨蝶呤97mg。
称取30mg两端叔丁氧羰基保护的甲氨蝶呤置于50ml干燥的单口烧瓶之中,加入15ml的二氯甲烷和1.2ml三氟乙酸,反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,加入24ml浓度为1mol/l的氢氧化钠溶液。最后用旋转蒸发仪蒸干溶剂,在真空干燥箱中干燥2d,得到黄色固体两端为两个氨基修饰的甲氨蝶呤单体20mg。
分别称取50mg苯丁酸氮芥、22mg三羟甲基氨基甲烷、45mg2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉和10ml的乙醇加入到50ml干燥的单口烧瓶之中,在60℃下回流反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,样品直接上样进行柱分离。柱分离,用旋转蒸发仪蒸干溶剂最终得到末端三个羟基修饰的苯丁酸氮芥单体白色固体粉末58mg。
称取20mg末端三个羟基修饰的苯丁酸氮芥单体和量取40μl三乙胺于50ml单口烧瓶之中,加入5ml的四氢呋喃作为溶剂,并将反应装置置于冰水浴中搅拌30min,再称取20mg丁二酸酐并将其溶于5ml四氢呋喃之中,逐滴加入到单口烧瓶之中,反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,再加入20ml的二氯甲烷再次溶解,用饱和氯化铵溶液萃取三次,用旋转蒸发仪将液体浓缩后进行柱分离,用旋转蒸发仪蒸干溶剂分得末端三个羧基修饰的苯丁酸氮芥单体白色固体粉末25mg。
称取20mg末端三个羧基修饰的苯丁酸氮芥单体,7.7mg的1-羟基苯并三唑,10.9mg的1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和量取100μl的三乙胺于100ml单口烧瓶之中,加入20mln,n-二甲基甲酰胺作为溶剂,在冰水浴的条件下搅拌30min,随后称取61mg两端为两个氨基修饰的甲氨蝶呤单体溶于5mln,n-二甲基甲酰胺之中,逐滴加入到单口烧瓶之中,过程持续约15min,滴加完毕后反应3d。反应结束后,加入20ml去离子水,用旋转蒸发仪将液体浓缩后用截留分子量为5000的透析袋在n,n-二甲基甲酰胺中透析2d。之后用旋转蒸发仪将液体进一步旋蒸使其浓缩至2-3ml,加入70ml冰的四氢呋喃沉淀,得到40mg棕黄色固体无载体型超支化大分子药物。
从图1的核磁谱图可以看出,本实例所制备的无载体型超支化大分子药物结果产物与所设计的预期产物结构一致。
实施例2:
称取400mg胱胺二盐酸盐置于100ml干燥的单口烧瓶之中,并加入8ml的甲醇和560mg三乙胺,在冰浴的条件下搅拌30min。称取140mg二碳酸二叔丁酯于烧杯中并用12ml甲醇溶解,用恒压滴液漏斗将其逐滴滴加到单口烧瓶之中,反应5h之后加入10ml的去离子水,终止反应。减压旋蒸除去反应溶剂,然后加入30ml饱和氯化钠溶液,用50ml的二氯甲烷萃取三次,有机相用无水硫酸钠干燥。所得固体直接上样过硅胶柱分离。用旋转蒸发仪蒸干溶剂,在真空干燥箱之中干燥2d,得到淡黄色油状液体单端叔丁氧羰基保护氨基的胱胺196mg。
称取100mg甲氨蝶呤、84.3mg1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和60mg1-羟基苯并三唑于50ml干燥的单口烧瓶之中,加入15ml的n,n-二甲基甲酰胺作为溶剂,在冰水浴的条件下搅拌30min,之后称取138.6mg单端叔丁氧羰基保护氨基的胱胺溶于10ml的n,n-二甲基甲酰胺,逐滴加入单口烧瓶之中。反应12h之后,用旋转蒸发仪蒸干溶剂,得到棕黄色油状液体。将棕黄色油状液体再溶于30ml的三氯甲烷之中,用饱和碳酸氢钠溶液萃取三次,再用无水硫酸钠干燥。最后用旋转蒸发仪蒸干溶剂柱分离,最终得到黄色固体粉末两端叔丁氧羰基保护的甲氨蝶呤117mg。
称取100mg两端叔丁氧羰基保护的甲氨蝶呤置于50ml干燥的单口烧瓶之中,加入40ml的二氯甲烷和4ml三氟乙酸,反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,加入80ml浓度为1mol/l的氢氧化钠溶液。最后用旋转蒸发仪蒸干溶剂,在真空干燥箱中干燥2d,得到黄色固体两端为两个氨基修饰的甲氨蝶呤单体66mg。
分别称取200mg苯丁酸氮芥、87mg三羟甲基氨基甲烷、178mg2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉和40ml的乙醇加入到100ml干燥的单口烧瓶之中,在60℃下回流反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,样品直接上样进行柱分离,用旋转蒸发仪蒸干溶剂最终得到末端三个羟基修饰的苯丁酸氮芥单体白色固体粉末183mg。
称取50mg末端三个羟基修饰的苯丁酸氮芥单体和量取100μl三乙胺于50ml单口烧瓶之中,加入10ml的四氢呋喃作为溶剂,并将反应装置置于冰水浴中搅拌30min,再称取50mg丁二酸酐并将其溶于8ml四氢呋喃之中,逐滴加入到单口烧瓶之中,反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,再加入20ml的二氯甲烷再次溶解,用饱和氯化铵溶液萃取三次,用旋转蒸发仪将液体浓缩后进行柱分离,用旋转蒸发仪蒸干溶剂分得末端三个羧基修饰的苯丁酸氮芥单体白色固体粉末65mg。
称取60mg末端三个羧基基修饰的苯丁酸氮芥单体,23.1mg的1-羟基苯并三唑,32.7mg的1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和量取300μl的三乙胺于100ml单口烧瓶之中,加入30mln,n-二甲基甲酰胺作为溶剂,在冰水浴的条件下搅拌30min,随后称取183mg两端为两个氨基修饰的甲氨蝶呤单体溶于8mln,n-二甲基甲酰胺之中,逐滴加入到单口烧瓶之中,过程持续约15min,滴加完毕后反应3d。反应结束后,加入45ml去离子水,用旋转蒸发仪将液体浓缩后用截留分子量为5000的透析袋在n,n-二甲基甲酰胺中透析2d。之后用旋转蒸发仪将液体进一步旋蒸使其浓缩至2-3ml,加入70ml冰的四氢呋喃沉淀,得到133mg棕黄色固体无载体型超支化大分子药物。
实施例3:
称取500mg胱胺二盐酸盐置于100ml干燥的单口烧瓶之中,并加入10ml的甲醇和700mg三乙胺,在冰浴的条件下搅拌30min。称取175mg二碳酸二叔丁酯于烧杯中并用15ml甲醇溶解,用恒压滴液漏斗将其逐滴滴加到单口烧瓶之中,反应5h之后加入10ml的去离子水,终止反应。减压旋蒸除去反应溶剂,然后加入30ml饱和氯化钠溶液,用50ml的二氯甲烷萃取三次,有机相用无水硫酸钠干燥。所得固体直接上样过硅胶柱分离,用旋转蒸发仪蒸干溶剂,在真空干燥箱之中干燥2d,得到淡黄色油状液体单端叔丁氧羰基保护氨基的胱胺257mg。
称取181.8mg甲氨蝶呤、153.3mg1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和108.1mg1-羟基苯并三唑于50ml干燥的单口烧瓶之中,加入20ml的n,n-二甲基甲酰胺作为溶剂,在冰水浴的条件下搅拌30min,之后称取252mg单端叔丁氧羰基保护氨基的胱胺溶于10ml的n,n-二甲基甲酰胺,逐滴加入单口烧瓶之中。反应12h之后,用旋转蒸发仪蒸干溶剂,得到棕黄色油状液体。将棕黄色油状液体再溶于30ml的三氯甲烷之中,用饱和碳酸氢钠溶液萃取三次,再用无水硫酸钠干燥。最后用旋转蒸发仪蒸干溶剂,柱分离。最终得到黄色固体粉末两端叔丁氧羰基保护的甲氨蝶呤200mg。
称取50mg两端叔丁氧羰基保护的甲氨蝶呤置于50ml干燥的单口烧瓶之中,加入20ml的二氯甲烷和2ml三氟乙酸,反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,加入40ml浓度为1mol/l的氢氧化钠溶液。最后用旋转蒸发仪蒸干溶剂,在真空干燥箱中干燥2d,得到黄色固体两端为两个氨基修饰的甲氨蝶呤单体30mg。
分别称取100mg苯丁酸氮芥、44mg三羟甲基氨基甲烷、90mg2-乙氧基-1-乙氧碳酰基-1,2-二氢喹啉和20ml的乙醇加入到50ml干燥的单口烧瓶之中,在60℃下回流反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,样品直接上样进行柱分离,用旋转蒸发仪蒸干溶剂最终得到末端三个羟基修饰的苯丁酸氮芥单体白色固体粉末100mg。
称取25mg末端三个羟基修饰的苯丁酸氮芥单体和量取50μl三乙胺于50ml单口烧瓶之中,加入5ml的四氢呋喃作为溶剂,并将反应装置置于冰水浴中搅拌30min,再称取24.8mg丁二酸酐并将其溶于5ml四氢呋喃之中,逐滴加入到单口烧瓶之中,反应12h。反应结束后,用旋转蒸发仪蒸干溶剂,再加入20ml的二氯甲烷再次溶解,用饱和氯化铵溶液萃取三次,用旋转蒸发仪将液体浓缩后进行柱分离,用旋转蒸发仪蒸干溶剂分得末端三个羧基修饰的苯丁酸氮芥单体白色固体粉末35mg。
称取40mg末端三个羧基修饰的苯丁酸氮芥单体,15.4mg的1-羟基苯并三唑,21.8mg的1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐和量取200μl的三乙胺于100ml单口烧瓶之中,加入25mln,n-二甲基甲酰胺作为溶剂,在冰水浴的条件下搅拌30min,随后称取122mga2单体溶于5mln,n-二甲基甲酰胺之中,逐滴加入到单口烧瓶之中,过程持续约15min,滴加完毕后反应3d。反应结束后,加入30ml去离子水,用旋转蒸发仪将液体浓缩后用截留分子量为5000的透析袋在n,n-二甲基甲酰胺中透析2d。之后用旋转蒸发仪将液体进一步旋蒸使其浓缩至2-3ml,加入70ml冰的四氢呋喃沉淀,得到91mg棕黄色固体无载体型超支化大分子药物。
- 还没有人留言评论。精彩留言会获得点赞!