含磷丙烯酸酯阻燃剂、阻燃EVA胶膜及其制备方法与流程
本发明涉及一种含磷丙烯酸酯阻燃剂及其制备方法,一种应用该阻燃剂的阻燃eva胶膜及其制备方法,属于高分子材料技术领域。
背景技术:
太阳能光伏组件,简称pv组件,是太阳能光伏发电系统中的核心部分。它是由预先排列好的一组建电池,超薄钢化透明玻璃,封装胶膜而构成。若干光伏组件用密封胶安装固定在框架上组成太阳能电池板,普遍规定太阳能电池板应用寿命是在30年,从而对组件用封装胶提出了很高的要求,能耐受高低温环境,防潮,抗腐蚀,耐紫外和耐湿热老化,以及良好的绝缘和阻燃性能。现有胶膜产品已具有很好的绝缘,抗老化和环境变化的性能,但是,不具有阻燃性能是太阳能光伏组件的一重大缺陷。
随着分布式发电的大幅度发展,越来越多的光伏组件应用在居民比较多的领域,如厂房,公共建筑,别墅,办公大楼等。伴随着晶硅太阳能电池的光电转换,会明显提高组件内部的温度,由于失配或热斑效应会带来过热而引起火灾的危险。
传统的胶膜改性,是在聚烯烃弹性体,如eva或poe中添加大量的小分子阻燃剂。如中国专利文献cn1401721公开了一种一种聚烯烃热熔胶粘剂及使用该热熔胶粘剂的复合结构胶片,其中添加了氢氧化物。中国专利文献cn102936483a公开了一种单组份有机硅太阳能光伏组件用密封胶及其制备方法,其中添加硅系阻燃剂,膨胀石墨,磷氮系小分子添加剂。在交联改性后的胶膜中添加这些添加剂,由于小分子添加剂和eva的相容性差,长期的紫外和湿热老化中,相界面间发生老化缺陷。随意地选择阻燃剂,一方面没有理想的阻燃效果,例如难以达到垂直燃烧ul-94v-0级;另一方面,势必影响胶膜其它组分之间的亲和性和协同性,进而造成粘结性降低,老化后性能衰减严重。
技术实现要素:
本发明要解决的技术问题是提供一种阻燃效果好,可以接枝反应到eva分子链上的含磷丙烯酸酯阻燃剂,以及该阻燃剂的制备方法,以及含有该阻燃剂的阻燃eva胶膜,以及该胶膜的制备方法。
本发明为解决上述技术问题提出的一种技术方案是:一种含磷丙烯酸酯阻燃剂,具有如式ⅰ所示的结构,
其中,r是氢原子、脂烃基或芳烃基。
上述r是c1~c8的烷基(可以是甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基等)、c3~c8的环烷基(可以是环丙基、环丁基、环戊基、环己基、环辛基等)、c2~c8的不饱和烃基(可以是乙烯基、丙烯基、烯丙基等)、c3~c8的环烯烃(可以是环丙烯、环丁烯、环戊烯等)或c6~c10的芳烃基(可以是苯基、甲苯基、二甲苯基、萘基等)。
本发明为解决上述技术问题提出的另一种技术方案是:一种含磷丙烯酸酯阻燃剂的制备方法,如式ⅱ所示的二乙基膦酸酯与如式ⅱ所示的醛类化合物在缚酸剂的作用下反应生成如式ⅳ所示的中间体化合物,
如式ⅳ所示的中间体化合物与如式ⅴ所示的甲基丙烯酰氯反应生成如式ⅰ所示的含磷丙烯酸酯,
上述醛类化合物优选多聚甲醛,三聚乙醛,丙醛,丁醛,或苯甲醛,萘甲醛。
上述缚酸剂为三乙胺、氟化钾或氟化铯,所述二乙基膦酸酯、醛类化合物、缚酸剂的摩尔比为1:1:1~5,生成中间体化合物的反应温度为0℃~30℃,反应时间1h~5h。
上述二乙基膦酸酯和甲基丙烯酰氯的摩尔比为1:1~1.1,生成含磷丙烯酸酯的反应温度为0℃~30℃,反应时间1h~12h。
生成含磷丙烯酸酯所得到的固体产物,分别采用6mol/l的盐酸水溶液、1mol/l的碳酸氢钠水溶液洗涤,分液后,有机层用硫酸镁干燥,再真空干燥;然后所得固体采用硅胶柱层析提纯即得到含磷丙烯酸酯成品,硅胶的粒径是70~230目,洗脱相是乙酸乙酯和石油醚按照质量比1:1混合的混合液。
本发明为解决上述技术问题提出的一种技术方案是:一种阻燃eva胶膜具有aba三层结构,表层包括以下质量百分比的组分:eva树脂96.5%~98.5%,纳米磷酸锆0.05%~0.1%,过氧化物0.5%~1%,交联助剂0.5%~1%,硅烷偶联剂0.2%~0.6%,紫外光吸收剂0.1%~0.5%,光稳定剂0.1%~0.5%;
芯层包括以下质量百分比的组分:eva树脂74%~82.5%,钛白粉7~8%,含磷丙烯酸酯阻燃剂9%~15%,过氧化物0.5%~1%,交联助剂0.5%~1%,硅烷偶联剂0.2%~0.6%,紫外光吸收剂0.1%~0.5%,光稳定剂0.1%~0.5%;
所述含磷丙烯酸酯阻燃剂具有如式ⅰ所示的结构,
其中,r是氢原子、脂烃基或芳烃基。
芯层厚度为400μm~500μm,表层厚度为50μm~100μm;所述过氧化物是2,5-二甲基-2,5二叔丁基过氧化己烷、过氧化-2-乙基己基碳酸叔戊酯和过氧化-2-乙基己基碳酸叔丁酯中的一种或多种;
所述交联助剂是异氰酸三烯丙酯、三(2-羟乙基)-异氰脲酸三丙烯酸酯、三羟甲基丙烷三丙烯酸酯、季戊四醇三丙烯酸酯和乙氧化三羟甲基丙烷三丙烯酸酯中的一种或多种;
所述硅烷偶联剂是乙烯基三甲氧基硅烷、乙烯基三乙酰氧基硅烷、乙烯基三(β-甲氧基乙氧基)硅烷中一种或多种;
所述光稳定剂是双(1-辛氧基-2,2,6,6-四甲基-4-哌啶基)癸二酸酯、双(2,2,6,6-四甲基-4-哌啶基)癸二酸酯和聚丁二酸(4羟基-2,2,6,6-四甲基-1哌啶乙醇脂)中的一种或多种;
所述紫外光吸收剂是2-[4,6-双(2,4-二甲基苯基)-1,3,5-三嗪-2-基]-5-(辛氧基)酚;
所述eva树脂中的va含量是20%~30%;
纳米磷酸锆的粒径为200nm~300nm。
本发明为解决上述技术问题提出的另一种技术方案是:一种上述的阻燃eva胶膜的制备方法,包括以下步骤:
步骤s1,将eva树脂和纳米磷酸锆混合挤出制成磷酸锆母粒,将eva树脂和钛白粉混合挤出制成钛白粉母粒;
步骤s2,采用挤出设备和多层挤出模头制备aba三层结构的胶膜;磷酸锆母粒用于与其他组分混合挤出制成表层,钛白粉母粒用于与其他组分混合挤出制成芯层。
上述步骤s1中采用立式双螺带混合机进行混合,驱动功率为3kw~120kw,混合时间为3min~10min;
所述步骤s1中采用同向啮合双螺杆挤出机进行挤出,同向啮合双螺杆挤出机的双螺杆转速为40r/min~60r/min,各段温度依次分别为140℃、150℃、170℃、180℃、180℃;
所述步骤s2中的表层制备采用同向啮合双螺杆挤出机,螺杆直径为95mm,螺杆转速为30r/min~60r/min,各段温度依次分别为75℃、80℃、84℃、85℃、85℃;
所述步骤s2中的芯层制备采用单螺杆挤出机,螺杆直径为130mm,螺杆转速为30r/min~60r/min,各段温度依次分别为75℃、80℃、84℃、85℃、85℃。
本发明具有积极的效果:
(1)本发明的含磷丙烯酸酯阻燃剂以二乙基膦酸酯、醛、丙烯酰氯为原料,三乙胺、氟化钾或氟化铯为缚酸剂,低温下反应即可得到含磷丙烯酸酯,合成方法简单,成本低。含磷丙烯酸酯阻燃剂可以与eva树脂、交联助剂在过氧化物引发下进行自由基接枝反应,不存在迁移析出和相容性差的缺陷。使得eva树脂在不改变胶膜原有粘结性和抗老化性能的前提下,胶膜本身具有一定的阻燃性,可被用于阻燃防火要求更高的光伏领域。
(2)本发明的eva胶膜含有表层含有纳米磷酸锆,通过在表层强剪切加工下,添加磷酸锆母粒可以减少团聚,阻燃剂配合纳米磷酸锆的阻燃抗熔滴效应,减缓热量和热质的传递,进一步提升了eva胶膜的阻燃性。
附图说明
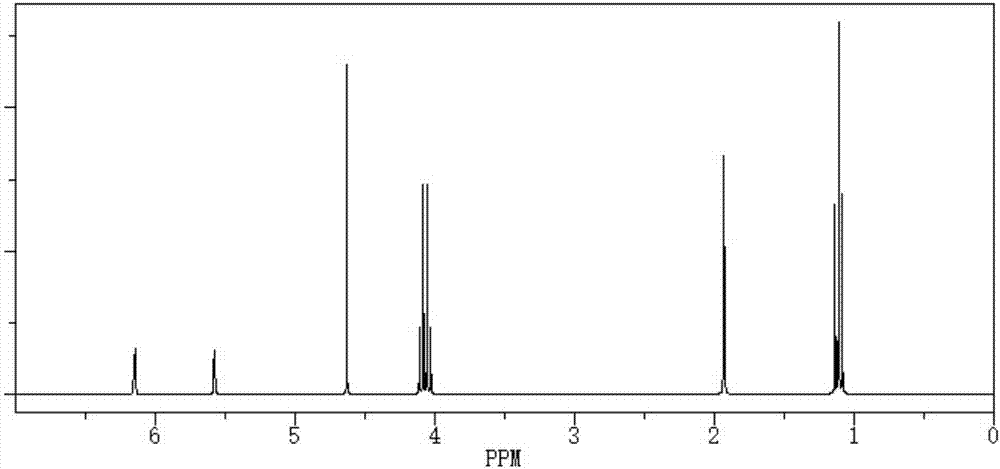
图1是本发明实施例1的含磷丙烯酸酯阻燃剂的核磁氢谱图。
图2是本发明实施例1的含磷丙烯酸酯阻燃剂的核磁碳谱图。
图3是本发明实施例1的含磷丙烯酸酯阻燃剂的红外光谱图。
图4是本发明应用例1的阻燃eva胶膜固化后的热失重曲线。
图5添加实施例1的阻燃eva胶膜的制备方法的流程示意图。
具体实施方式
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。例如:c1~c8的烷基是指碳链长度为1~8的烷基,如:甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、己基、庚基、辛基等。c3~c8的环烷基是指碳链长度为3~8的环烷基,如:环丙基、环丁基、环戊基、环己基、环辛基等。c2~c8的不饱和烃基是指碳链长度为2~8的含有双键或三键的烃基,如:乙烯基、丙烯基、烯丙基。c3~c8的环烯烃是指碳链长度为3~8的含有双键的环烃基,如:环丙烯、环丁烯、环戊烯。芳基是指碳链长度为6~10的芳香族烃,如:苯基、甲苯基、二甲苯基、萘基等。
实施例1
本实施例的含磷丙烯酸酯(diethoxyphosphoryl)methylmethacrylate-depmm的反应式如下:
制备方法是将二乙基膦酸酯(dep)滴加到多聚甲醛和三乙胺的(tea)的二氯甲烷混合溶液中。其中dep、多聚甲醛、三乙胺按摩尔比1:1:1的比例,反应温度为120℃,反应时间4h。反应完后,硫酸镁干燥后,旋转蒸发除去溶剂即可得到中间体化合物。
所得中间体化合物和800ppm的4-甲氧基苯酚溶解在氯仿溶剂中,4-甲氧基苯酚起调节ph值和增溶的作用不参加反应,丙烯酰氯的氯仿溶液在0℃滴加到中间体化合物和三乙胺氯仿溶液中。其中,二乙基膦酸酯和甲基丙烯酰氯的摩尔比为1:1.1,室温下反应时间12h。反应完后的固体混合物,用6mol/l的盐酸水溶液,1mol/l的碳酸氢钠水溶液洗涤,分液后,有机层用硫酸镁干燥,再真空干燥;然后所得固体采用硅胶柱层析提纯即得到含磷丙烯酸酯成品,硅胶的粒径是70~230目,洗脱相是乙酸乙酯和石油醚按照质量比1:1混合的混合液。产物进行氢谱检验和红外检验,产率71%。
产物的核磁氢谱图如图1所示,1hnmr:1.35(6h,t,j=7.1),1.97(3h,s),4.11(4h,m),4.41(2h,d,jhp=8.7),5.65(1h,s),6.18(1h,s)。其中化学位移在δ=1.35ppm是乙氧基上甲基ch3-特征峰,δ=4.11ppm是乙氧基上亚甲基ch2-的特征峰,δ=1.97ppm是膦酸酯上亚甲基ch2-特征峰,δ=5.65ppm为苯基次酸与羰基相连的-ch2-ch2-上质子的特征峰,δ=4.11ppm为膦酸酯上亚甲基ch2-特征峰苯环上质子的特征峰,δ=4.41ppm为乙烯基上亚甲基ch2-的特征峰,δ=6.18ppm为乙烯基上次甲基的特征峰。上述峰值说明,结构中既含有丙烯酸酯,又含有二乙基膦酸酯结构。
产物的核磁碳谱图如图2所示,13cnmr:16.0(2c,d,jcp=5.9),17.7,56.5(d,jcp=169.2),62.3(2c,jcp=6.5),126.3,134.9,165.7(d,j=8.5)。其中化学位移在δ=16ppm是二乙基的甲基上c的特征峰,δ=17.7ppm是乙烯基旁的甲基上c的特征峰,δ=56.5ppm是为乙基的亚甲基上c的特征峰,δ=62.3ppm是和膦酸酯相连的亚甲基上c的特征峰,以及δ=165.7ppm乙烯基上次甲基上c的特征峰,δ=126.3ppm乙烯基上亚甲基上c的特征峰;上述峰值说明,结构中既含有二乙基膦酸酯,又含有丙烯酸酯的结构。
产物的红外光谱图如图3所示,ir:2983,2864,1730,1640,1593,1268,1035,970,760。其中在波数2983cm-1处为ch3的c-h的伸缩振动吸收峰,在波数2864cm-1处为ch2的c-h的伸缩振动吸收峰,波数1730cm-1处为c=o的伸缩振动峰,波数1593cm-1处为ch2的伸缩振动吸收峰,波数1268cm-1处为p-o-c的伸缩振动吸收峰,波数1035cm-1处为ch2=ch-的伸缩振动吸收峰;波数970cm-1处为c-o-c弯曲振动吸收峰。上述峰值说明,结构中既含有二乙基膦酸酯,又含有丙烯酸酯的结构。
实施例2
本实施例的含磷丙烯酸酯1-(diethoxyphosphoryl)butylmethacrylate-depbmm的反应式如下:
制备方法是将二乙基膦酸酯(dep)滴加到丁醛和氟化铯(csf)的二氯甲烷混合溶液中。其中dep、丁醛、氟化铯按摩尔比1:1:2的比例,反应温度25℃,反应1h。反应完后,硫酸镁干燥后,旋转蒸发除去溶剂即可得到中间体化合物。
所得中间体化合物和800ppm的4-甲氧基苯酚溶解在氯仿溶剂中,丙烯酰氯的氯仿溶液在0℃滴加到中间体化合物和氟化铯氯仿溶液中。其中,二乙基膦酸酯和甲基丙烯酰氯的摩尔比为1:1.1,室温下反应时间12h。反应完后的固体混合物,用6mol/l的盐酸水溶液,1mol/l的碳酸氢钠水溶液洗涤,分液后,有机层用硫酸镁干燥,再真空干燥;然后所得固体采用硅胶柱层析提纯即得到含磷丙烯酸酯成品,硅胶的粒径是70~230目,洗脱相是乙酸乙酯和石油醚按照质量比1:1混合的混合液。产物进行氢谱检验和红外检验,产率82%。
实施例3
本实施例的含磷丙烯酸酯(diethoxyphosphoryl)(phenyl)methylmethacrylate-deppmm的反应式如下:
制备方法是将二乙基膦酸酯(dep)滴加到苯甲醛和氟化钾(kf)的二氯甲烷混合溶液中。dep、苯甲醛、氟化钾按摩尔比1:1:5的比例,反应温度25℃,反应1h。反应完后,硫酸镁干燥,旋转蒸发除去溶剂即可得到中间体化合物。
所得中间体化合物和800ppm的4-甲氧基苯酚溶解在氯仿溶剂中,丙烯酰氯的氯仿溶液在0℃滴加到中间体化合物和氟化钾氯仿溶液中。其中,二乙基膦酸酯和甲基丙烯酰氯的摩尔比为1:1.1,室温下反应时间12h。反应完后的固体混合物,用6mol/l的盐酸水溶液,1mol/l的碳酸氢钠水溶液洗涤,分液后,有机层用硫酸镁干燥,再真空干燥;然后所得固体采用硅胶柱层析提纯即得到含磷丙烯酸酯成品,硅胶的粒径是70~230目,洗脱相是乙酸乙酯和石油醚按照质量比1:1混合的混合液。产物进行氢谱检验和红外检验,产率83%。
实施例4
本实施例的含磷丙烯酸酯(diethoxyphosphoryl)(naphthalen-2-yl)methylmethacrylate-depnmm的反应式如下:
制备方法是将二乙基膦酸酯(dep)滴加到萘甲醛和氟化钾(kf)的二氯甲烷混合溶液。dep、萘甲醛、氟化钾按摩尔比1:1:5的比例,反应温度25℃,反应时间1h。反应完后,硫酸镁干燥,旋转蒸发除去溶剂即可得到中间体化合物。
所得中间体化合物和800ppm的4-甲氧基苯酚溶解在氯仿溶剂中,丙烯酰氯的氯仿溶液在0℃滴加到中间体化合物和氟化钾氯仿溶液中。其中,二乙基膦酸酯和甲基丙烯酰氯的摩尔比为1:1.1,室温下反应时间12h。反应完后的固体混合物,用6mol/l的盐酸水溶液,1mol/l的碳酸氢钠水溶液洗涤,分液后,有机层用硫酸镁干燥,再真空干燥;然后所得固体采用硅胶柱层析提纯即得到含磷丙烯酸酯成品,硅胶的粒径是70~230目,洗脱相是乙酸乙酯和石油醚按照质量比1:1混合的混合液。产物进行氢谱检验和红外检验,产率83%。
对实施例1至4所得的终产进行氢谱和红外光谱检测,所得化合物结构表征波谱数据如表1所示。
表1实施例1至4阻燃剂结构表征波谱数据
应用例1
本应用例的阻燃eva胶膜具有aba三层结构,表层包括以下质量百分比的组分:eva树脂96.5%,纳米磷酸锆0.1%,过氧化物1%,交联助剂1%,硅烷偶联剂0.6%,紫外光吸收剂0.4%,光稳定剂0.4%。
芯层包括以下质量百分比的组分:eva树脂74%,钛白粉8%,含磷丙烯酸酯阻燃剂15%,过氧化物1%,交联助剂1%,硅烷偶联剂0.6%,紫外光吸收剂0.2%,光稳定剂0.2%。
过氧化物是2,5-二甲基-2,5二叔丁基过氧化己烷。交联助剂是异氰酸三烯丙酯。光稳定剂是双(1-辛氧基-2,2,6,6-四甲基-4-哌啶基)癸二酸酯。紫外光吸收剂是2-[4,6-双(2,4-二甲基苯基)-1。硅烷偶联剂是乙烯基三甲氧基硅烷。
eva树脂中的va含量是30%。纳米磷酸锆的粒径为200nm~300nm。含磷丙烯酸酯阻燃剂采用实施例1的含磷丙烯酸酯1-(diethoxyphosphoryl)butylmethacrylate–depbmm。
如图5所示,本应用例的阻燃eva胶膜的制备方法,包括以下步骤:
步骤s1,将eva树脂和纳米磷酸锆混合挤出制成磷酸锆母粒,将eva树脂和钛白粉混合挤出制成钛白粉母粒;
步骤s2,采用挤出设备和多层挤出模头制备aba三层结构的胶膜;磷酸锆母粒用于与其他组分混合挤出制成表层,钛白粉母粒用于与其他组分混合挤出制成芯层。
步骤s1中采用立式双螺带混合机进行混合,驱动功率为120kw,混合时间为3min;
步骤s1中采用同向啮合双螺杆挤出机进行挤出,同向啮合双螺杆挤出机的双螺杆转速为60r/min,各段温度依次分别为140℃、150℃、170℃、180℃、180℃;
步骤s2中的表层制备采用同向啮合双螺杆挤出机,螺杆直径为95mm,螺杆转速为60r/min,各段温度依次分别为75℃、80℃、84℃、85℃、85℃;
步骤s2中的芯层制备采用单螺杆挤出机,螺杆直径为130mm,螺杆转速为60r/min,各段温度依次分别为75℃、80℃、84℃、85℃、85℃。
本应用例的阻燃eva胶膜固化后的热失重曲线,如图4所示。
应用例2
本应用例的阻燃eva胶膜具有aba三层结构,表层包括以下质量百分比的组分:eva树脂98.5%,纳米磷酸锆0.05%,过氧化物0.5%,交联助剂0.5%,硅烷偶联剂0.2%,紫外光吸收剂0.1%,光稳定剂0.15%。
芯层包括以下质量百分比的组分:eva树脂82.5%,钛白粉7%,含磷丙烯酸酯阻燃剂9%,过氧化物0.5%,交联助剂0.5%,硅烷偶联剂0.2%,紫外光吸收剂0.1%,光稳定剂0.2%。
过氧化物是过氧化-2-乙基己基碳酸叔戊酯。交联助剂是三(2-羟乙基)-异氰脲酸三丙烯酸酯。光稳定剂是双(2,2,6,6-四甲基-4-哌啶基)癸二酸酯。紫外光吸收剂是3,5-三嗪-2-基]-5-(辛氧基)酚。硅烷偶联剂是乙烯基三乙酰氧基硅烷。
eva树脂中的va含量是25%。纳米磷酸锆的粒径为200nm~300nm。含磷丙烯酸酯阻燃剂采用实施例1的含磷丙烯酸酯1-(diethoxyphosphoryl)butylmethacrylate–depbmm。
如图5所示,本应用例的阻燃eva胶膜的制备方法,包括以下步骤:
步骤s1,将eva树脂和纳米磷酸锆混合挤出制成磷酸锆母粒,将eva树脂和钛白粉混合挤出制成钛白粉母粒;
步骤s2,采用挤出设备和多层挤出模头制备aba三层结构的胶膜;磷酸锆母粒用于与其他组分混合挤出制成表层,钛白粉母粒用于与其他组分混合挤出制成芯层。
步骤s1中采用立式双螺带混合机进行混合,驱动功率为3kw,混合时间为10min;
步骤s1中采用同向啮合双螺杆挤出机进行挤出,同向啮合双螺杆挤出机的双螺杆转速为40r/min,各段温度依次分别为140℃、150℃、170℃、180℃、180℃;
步骤s2中的表层制备采用同向啮合双螺杆挤出机,螺杆直径为95mm,螺杆转速为30r/min,各段温度依次分别为75℃、80℃、84℃、85℃、85℃;
步骤s2中的芯层制备采用单螺杆挤出机,螺杆直径为130mm,螺杆转速为30r/min,各段温度依次分别为75℃、80℃、84℃、85℃、85℃。
应用例3
本应用例的阻燃eva胶膜具有aba三层结构,表层包括以下质量百分比的组分:eva树脂97.5%,纳米磷酸锆0.1%,过氧化物0.8%,交联助剂0.7%,硅烷偶联剂0.4%,紫外光吸收剂0.2%,光稳定剂0.3%。
芯层包括以下质量百分比的组分:eva树脂78%,钛白粉7.5%,含磷丙烯酸酯阻燃剂12%,过氧化物0.8%,交联助剂0.8%,硅烷偶联剂0.4%,紫外光吸收剂0.2%,光稳定剂0.2%。
过氧化物是过氧化-2-乙基己基碳酸叔丁酯。交联助剂是三羟甲基丙烷三丙烯酸酯。光稳定剂是聚丁二酸(4羟基-2,2,6,6-四甲基-1哌啶乙醇脂)。紫外光吸收剂是3,5-三嗪-2-基]-5-(辛氧基)酚。硅烷偶联剂是乙烯基三(β-甲氧基乙氧基)硅烷。
eva树脂中的va含量是20%。纳米磷酸锆的粒径为200nm~300nm。含磷丙烯酸酯阻燃剂采用实施例1的含磷丙烯酸酯(diethoxyphosphoryl)methylmethacrylate-depmm。
如图5所示,本应用例的阻燃eva胶膜的制备方法,包括以下步骤:
步骤s1,将eva树脂和纳米磷酸锆混合挤出制成磷酸锆母粒,将eva树脂和钛白粉混合挤出制成钛白粉母粒;
步骤s2,采用挤出设备和多层挤出模头制备aba三层结构的胶膜;磷酸锆母粒用于与其他组分混合挤出制成表层,钛白粉母粒用于与其他组分混合挤出制成芯层。
步骤s1中采用立式双螺带混合机进行混合,驱动功率为50kw,混合时间为5min;
步骤s1中采用同向啮合双螺杆挤出机进行挤出,同向啮合双螺杆挤出机的双螺杆转速为50r/min,各段温度依次分别为140℃、150℃、170℃、180℃、180℃;
步骤s2中的表层制备采用同向啮合双螺杆挤出机,螺杆直径为95mm,螺杆转速为45r/min,各段温度依次分别为75℃、80℃、84℃、85℃、85℃;
步骤s2中的芯层制备采用单螺杆挤出机,螺杆直径为130mm,螺杆转速为45r/min,各段温度依次分别为75℃、80℃、84℃、85℃、85℃。
应用例4
本应用例的阻燃eva胶膜其余部分与应用例1相同,不同之处在于:含磷丙烯酸酯阻燃剂采用实施例2的含磷丙烯酸酯1-(diethoxyphosphoryl)butylmethacrylate-depbmm。
应用例5
本应用例的阻燃eva胶膜其余部分与应用例2相同,不同之处在于:含磷丙烯酸酯阻燃剂采用实施例3的含磷丙烯酸酯(diethoxyphosphoryl)(phenyl)methylmethacrylate-deppmm。
应用例6
本应用例的阻燃eva胶膜其余部分与应用例3相同,不同之处在于:含磷丙烯酸酯阻燃剂采用实施例4的含磷丙烯酸酯(diethoxyphosphoryl)(naphthalen-2-yl)methylmethacrylate-depnmm。
二乙基膦酸酯、多聚甲醛、4-甲氧基苯酚、丁醛、苯甲醛、萘甲醛、三乙胺、氟化钾、氟化铯、硫酸镁,分析纯,购自国药集团化学试剂成都有限公司。碳酸氢钠,盐酸,石油醚,乙酸乙酯,氯仿,二氯甲烷,分析纯,采购自成都科龙试剂厂。柱层析硅胶,采购自国药集团化学试剂成都有限公司。本发明中所用试剂如无特殊说明浓度均为化学纯。
本发明提供的含磷丙烯酸酯结构和纯度,阻燃eva胶膜的厚度,热失重,剥离强度,体积电阻率、老化性能以及阻燃性能的测试标准如下:
薄膜厚度:采用iso:4593-1993标准,采用数显千分尺测定。
氧指数:采用gb/t2406-2009标准,采用jf-3氧指数测定仪测试。
拉伸性能:按gb/t1040-2006标准测试,采用instron1185型万能试验机,夹持长度为150mm,拉伸速度100mm/min。
热收缩率:采用gb/t13542.2-2009标准检测;从阻燃聚酰胺薄膜上取两块100×100mm的试样,并做纵向、横向标记,测定横向与纵向长度l0。将薄膜自然放置在135℃的烘箱中分别保持1小时,然后从烘箱中取出试样,试样冷却至室温。重新测定横向与纵向长度l1,则收缩率(%)=(l0-l1)/l0×100%。
垂直燃烧:按ansl-ul94-2009标准,采用czf-5型垂直燃烧仪测试。
采用上述的检测方法对应用例1至6制得的阻燃eva胶膜进行测试,其测试结果如表2所示。
表2阻燃eva胶膜性能数据表
注:md为纵向,td为横向。
显然,上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而这些属于本发明的精神所引伸出的显而易见的变化或变动仍处于本发明的保护范围之中。
- 还没有人留言评论。精彩留言会获得点赞!