由二氧化碳、芳香肼与多聚甲醛电合成1,3,4‑噁二唑‑2(3H)‑酮衍生物的方法与流程
本发明属于1,3,4-噁二唑-2(3H)-酮衍生物合成技术领域,具体涉及以二氧化碳、芳香肼、多聚甲醛为原料,电化学一步合成1,3,4-噁二唑-2(3H)-酮衍生物的方法。
背景技术:
1,3,4-噁二唑-2(3H)-酮衍生物是一类重要的有机化合物,具有药物活性,在药物合成中有重要应用。例如,5-甲氧基-3-(3-苯氧苯基)-1,3,4-噁二唑-2(3H)-酮(俗称7600化合物)可作为HSL(Hormone Sensitive Lipase)的抑制剂;3-苯-1,3,4-噁二唑-2(3H)-酮的部分衍生物可用作FAAH(Fatty Acid Amide Hydrolase)的抑制剂。
已经报道的1,3,4-噁二唑酮衍生物合成工艺,一般涉及两步或三步:以含有吡啶的N-甲基吡咯烷酮为溶剂,用肼类及酰氯合成酰肼类中间产物;用二氯甲烷作溶剂,酰肼类中间产物进一步与光气或CO相互反应得到目标产物。例如,Kiss等人采用两步法制得1,3,4-噁二唑-2(3H)-酮衍生物[L.E.Kiss,H.S.Ferreira,A.Beliaev,L.et al.,Med.Chem.Commun.,2011,2,889]:
1)将肼类化合物酰化制得酰肼类中间产物
2)酰肼类化合物与光气(碳酰氯)反应生成目标产物
Point等人采用苯胺类化合物通过三步法合成1,3,4-噁二唑-2(3H)-酮衍生物[V.Point,K.V.P.Pavan Kumar,S.Marc,et al.,European Journal of Medicinal Chemistry,2012,58,452]:
a)苯胺类还原为肼类化合物
b)肼类化合物酰化
c)与光气(碳酰氯)作用得到目标产物
兰州大学陈保华课题组则利用苯肼类化合物和CO在钯催化下得到了1,3,4-噁二唑-2(3H)-酮衍生物,其合成过程分为两步[王昱,陈保华.兰州大学硕士学位论文,2015];
1)酰肼的合成
(a)芳香肼的酰化
(b)羧酸化合物的酰化
2)酰肼与CO反应合成1,3,4-噁二唑-2(3H)-酮衍生物
以上所述的方法,涉及到有毒的光气或CO作为C1合成原料,也涉及到了包括二氯甲烷在内的挥发性大、毒性高的有机溶剂使用,对环境不友好;另外,合成步骤多,产物分离提纯麻烦,这些不足很大程度上制约了这些合成方法的实际应用。
利用二氧化碳替代光气或CO作为C1合成材料,合成1,3,4-噁二唑-2(3H)-酮衍生物,不仅能降低生产成本,而且对环境有利。这是一条具有工业应用价值,有挑战性的合成路线。至今为止,这样的合成路线未见文献报道。
电化学方法利用最清洁的反应试剂“电子”,原位产生所需的催化剂或者反应活性中间体等,在温和的反应条件下可以实现普通化学方法难以完成的反应,电化学方法被誉为环境友好的绿色合成方法,具有广阔的发展前景。
技术实现要素:
本发明的目的在于克服现有技术不足,提供一种由二氧化碳、芳香肼与多聚甲醛电合成1,3,4-噁二唑-2(3H)-酮衍生物的方法。
本发明采用以下技术方案。
一种由二氧化碳、芳香肼与多聚甲醛电合成1,3,4-噁二唑-2(3H)-酮衍生物的方法,包括以下步骤:
(1)在装有惰性阳极、阴极的无隔膜单室电解池中,依次加入电解溶剂、催化剂、导电盐、芳香肼、多聚甲醛和碱,得混合液;
(2)通入二氧化碳,密封;
(3)常温下进行电解反应;
(4)反应完成后,对电解液进行减压蒸馏、有机溶剂萃取,得到1,3,4-噁二唑-2(3H)-酮衍生物。
优选的,步骤(1)所述惰性阳极为铂或石墨;所述阴极为镍、银、铜、铂、锡、钛、铁、锌、铝和铬中的一种或两种以上的合金。
优选的,步骤(1)所述电解溶剂为甲醇、乙醇、异丙醇、N,N-二甲基甲酰胺、乙腈或二甲基亚砜。
优选的,步骤(1)所述催化剂为卤素的钠盐、钾盐、无机铵盐和有机铵盐中的一种以上;所述卤素为氯、溴或碘;所述催化剂与芳香肼的摩尔比为(0.1~0.3):1。
优选的,步骤(1)所述导电盐为四丁基四氟硼酸铵;所述导电盐在混合液中的浓度为0.05~0.5mol/L。
优选的,步骤(1)所述芳香肼为苯肼、苯环芳香肼和杂环芳香肼中的一种;所述苯环芳香肼的取代基为–F、–Cl、–Br、–CN、–CF3、–CH3、–OCH3、硝基和烷基的单取代或多取代基团;所述杂环芳香肼为含氮、氧或硫的杂环芳香肼;所述芳香肼和多聚甲醛的摩尔比为1:(1~2)。
优选的,步骤(1)所述碱为碳酸钠、碳酸钾、碳酸铯、叔丁醇钾或1,8-二氮杂二环十一碳-7-烯;所述碱与芳香肼的摩尔比为(0.1~1):1。
优选的,步骤(2)中通入二氧化碳直至电解池内压力为0.1~3MPa。
优选的,步骤(3)所述电解的方式为恒电流电解模式,电流为0.02~0.1A;当每摩尔芳香肼需通入的电量(Q)为96500~482500库仑时,停止电解。电解时间t与芳香肼的摩尔量和电解的电流强度I有关,由Q=I*t可估算电解时间。
优选的,步骤(4)所述有机溶剂为乙醚或乙酸乙酯。
本发明的原理如下所示:
式中,R为H、F、Cl、Br、CN、CF3、OCH3、硝基或烷基(烷基中C的数量为1-3)在内的取代基团。
与现有技术相比,本发明具有如下的优点及有益效果:
(1)本发明以廉价易得的二氧化碳替代有毒的光气或一氧化碳作为C1材料,既可以降低成本,又可以避免对环境造成危害;
(2)本发明通过电极的电化学反应原位生成催化剂、有机碱活性中间体,能有效地催化或促进二氧化碳、芳香肼与多聚甲醛一步转化为目标产物1,3,4-噁二唑-2(3H)-酮衍生物,1,3,4-噁二唑-2(3H)-酮衍生物的产率为52%~87%;
(3)本发明工艺简单有效、产品容易分离提纯;
(4)本发明为1,3,4-噁二唑-2(3H)-酮衍生物的合成以及二氧化碳的转化利用,提供了一条有应用价值的途径,具有经济、环保双重效益。
附图说明
图1为实施例1制备的3-苯-1,3,4-噁二唑-2(3H)-酮的核磁共振氢谱图。
图2为实施例1制备的3-苯-1,3,4-噁二唑-2(3H)-酮的核磁共振碳谱图。
图3为实施例10制备的3-(4-氯苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振氢谱图。
图4为实施例10制备的3-(4-氯苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振碳谱图。
图5为实施例11制备的3-(4-三氟甲基苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振氢谱图。
图6为实施例11制备的3-(4-三氟甲基苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振碳谱图。
图7为实施例12制备的3-(3-氯-4-氟苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振氢谱图。
图8为实施例12制备的3-(3-氯-4-氟苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振碳谱图。
图9为实施例13制备的3-(2,4,6-三氯苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振氢谱图。
图10为实施例13制备的3-(2,4,6-三氯苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振碳谱图。
图11为实施例14制备的3-(4-甲基苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振氢谱图。
图12为实施例14制备的3-(4-甲基苯)-1,3,4-噁二唑-2(3H)-酮的核磁共振碳谱图。
具体实施方式
以下结合实施例对本发明作进一步说明,但本发明要求保护的范围并不限于此。
实施例1
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
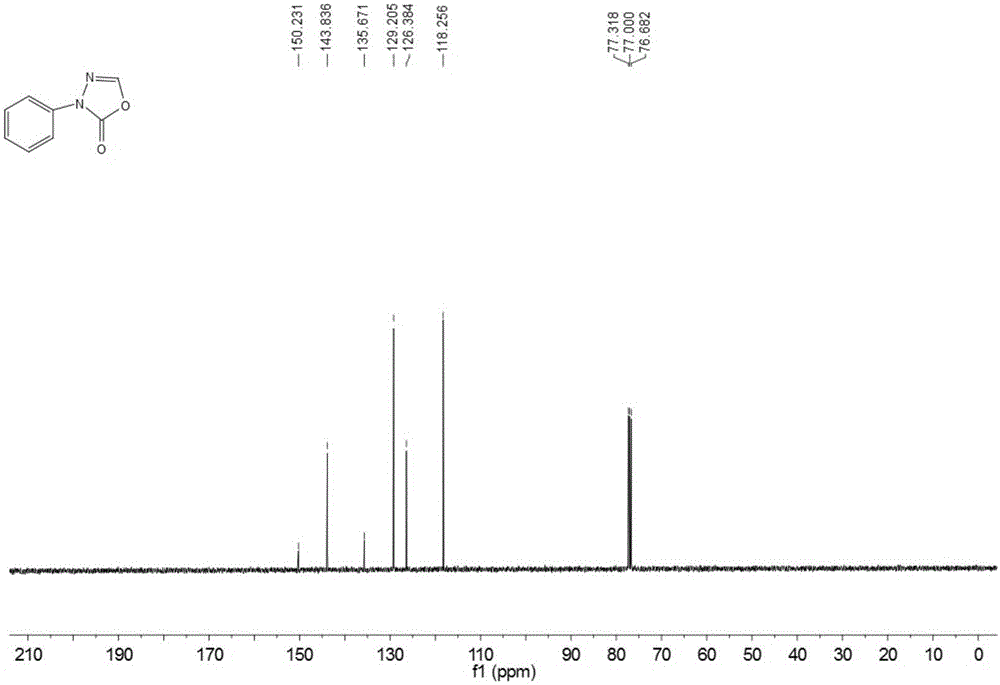
所得产品的结构由1H NMR、13C NMR和GC-MS确定,如图1、图2所示。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为87%。1HNMR(400MHz,CDCl3):δ7.85(d,J=7.9Hz,2H),7.71(s,1H),7.45(t,J=7.7Hz,2H),7.28(t,J=7.5Hz,1H).13C NMR(100MHz,CDCl3):δ150.2,143.8,135.7,129.2,126.4,118.3。
实施列2
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、1.5mmol多聚甲醛(聚合度为3)、0.15mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至1.5MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为85%。
实施例3
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.15mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目 标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为79%。
实施例4
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的乙腈,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、3.0mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为80%。
实施例5
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的乙醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为60%。
实施例6
以石墨电极为阳极,金属铜为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的乙醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为72%。
实施例7
以石墨电极为阳极,金属铝为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol四丁基碘化铵、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为82%。
实施例8
以石墨电极为阳极,不锈钢为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol碳酸铯、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为79%。
实施例9
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol DBU(1,8-二氮杂二环十一碳-7-烯)、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-苯-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定。3-苯-1,3,4-噁二唑-2(3H)-酮的分离产率为70%。
实施例10
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol 对氯苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-(4-氯苯)-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定,如图3、图4所示。3-(4-氯苯)-1,3,4-噁二唑-2(3H)-酮的分离产率为70%。1H NMR(400MHz,CDCl3):δ7.84–7.80(m,2H),7.72(s,1H),7.44–7.40(m,2H).13C NMR(100MHz,CDCl3):δ150.0,144.0,134.3,131.9,129.4,119.4。
实施例11
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol4-三氟甲基苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-(4-三氟甲基苯)-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定,如图5、图6所示。3-(4-三氟甲基苯)-1,3,4-噁二唑-2(3H)-酮的分离产率为81%。1H NMR(400MHz,CDCl3):δ8.02(d,J=8.6Hz,2H),7.76(s,1H),7.72(d,J=8.7Hz,2H).13C NMR(100MHz,CDCl3):δ149.9,144.3,138.4,128.8,128.44,128.1,127.8,126.6,126.6,126.5,126.5,125.1,122.4,117.9。
实施例12
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol3-氯-4-氟苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-(3-氯-4-氟苯)-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定,如图7、图8所示。3-(3-氯-4-氟苯)-1,3,4-噁二唑-2(3H)-酮的分离产率为81%。1H NMR(400MHz,CDCl3):δ7.95(dd,J=6.3,2.7Hz,1H),7.79–7.75(m,2H),7.22(t,J=8.7Hz,1H).13C NMR(100MHz,CDCl3):δ157.4,154.9,149.9,144.1,132.2,132.2,121.9,121.8,120.5,117.8,117.8,117.2,116.9。
实施例13
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol2,4,6-三氯苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-(2,4,6-三氯苯)-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定,如图9、图10所示。3-(2,4,6-三氯苯)-1,3,4-噁二唑-2(3H)-酮的分离产率为79%。1H NMR(400MHz,CDCl3):δ7.85(s,1H),7.49(s,2H).13C NMR(100MHz,CDCl3):δ150.6,145.3,137.3,135.9,128.8,128.5。
实施例14
以石墨电极为阳极,金属镍为阴极,在100mL带有聚四氟乙烯内衬的不锈钢单室高压电解池中,依次加入20mL的甲醇,0.45mmol碘化钠、10mmol四丁基四氟硼酸铵、1.5mmol对甲基苯肼、1.5mmol多聚甲醛(聚合度为3)、1.5mmol叔丁醇钾、磁力搅拌子,再通入二氧化碳至3MPa,密封;恒电流模式下,通0.05A的直流电,室温下电解5h;电解结束后,慢慢放掉未反应的二氧化碳气体,电解液在减压下蒸馏,然后用乙醚萃取三次,乙醚挥发后得到目标产物3-(4-甲基苯)-1,3,4-噁二唑-2(3H)-酮。
所得产品的结构由1H NMR、13C NMR和GC-MS确定,如图11、图12所示。3-(4-甲基苯)-1,3,4-噁二唑-2(3H)-酮的分离产率为52%。1H NMR(400MHz,CDCl3):δ7.69(t,J=5.4Hz,3H),7.22(d,J=8.1Hz,2H),2.35(s,3H).13C NMR(100MHz,CDCl3):δ150.3,143.7,136.2,133.2,129.6,118.3,20.8。
- 还没有人留言评论。精彩留言会获得点赞!