低共熔溶剂萃取液相色谱法快速测定黄曲霉毒素B1、碱性橙II和碱性嫩黄O的方法与流程
本发明属于食品中非食用色素检测技术领域,尤其涉及一种低共熔溶剂萃取液相色谱法快速测定黄曲霉毒素B1、碱性橙II和碱性嫩黄O的方法。
背景技术:
碱性橙II和碱性嫩黄O均为芳香胺类碱性工业染料,主要用于麻、纸、皮革、草编织品、人造丝等的染色。碱性橙II和碱性嫩黄O对皮肤黏膜有轻度刺激,可引起结膜炎、皮炎和上呼吸道刺激症状,人体接触或吸食均会引起中毒,且很难降解代谢,这两种物质均具有致癌和致突变性质。根据GB 2760食品添加剂适用卫生标准规定,碱性橙II和碱性嫩黄O都严禁作为食品添加剂使用。由于在中性或偏碱性条件下,碱性橙II和碱性嫩黄O与蛋白质吸附较牢固,不易褪色,比其他水溶性染料如柠檬黄、日落黄等更易于在食品中染色,因此,一些不法商贩常将其用于食品的染色,使产品色泽鲜亮,欺骗消费者,危害消费者的身体健康。
GB 35/T 897-2009《食品中碱性橙、碱性嫩黄O和碱性桃红T含量的测定》中规定了碱性橙II和碱性嫩黄O的检测方法,该方法存在前处理过程繁琐复杂、有机溶剂使用量较多、耗时长等多方面的缺陷,而且该方法主要是针对固体样品,对于调味油这类基质和杂质均比较复杂的样品并不适用。
黄曲霉毒素(AFT)是公认的天然强致癌物质之一,其中以黄曲霉毒素B1(AFTB1)毒性最大,它们常存在于各种动物饲料和人类食品中。我国真菌毒素限量标准GB 2761《食品安全国家标准食品中真菌毒素限量》针对不同食品品种规定了黄曲霉毒素B1的限量指标。
GB/T 18979-2003《食品中黄曲霉毒素的测定免疫亲和层析净化高效液相色谱法和荧光光度法》和GB/T 5009.23-2006《食品中黄曲霉毒素B1、B2、G1、G2的测定》中均规定了黄曲霉毒素B1的检测方法,该方法存在前处理过程繁琐复杂、有机溶剂使用量较多、耗时长等多方面的缺陷。
技术实现要素:
针对以上技术问题,本发明公开了一种低共熔溶剂萃取液相色谱法快速测定黄曲霉毒素B1、碱性橙II和碱性嫩黄O的方法,该方法操作简单,检测快速,成本低廉。
对此,本发明采用的技术方案为:
一种低共熔溶剂萃取液相色谱法快速测定黄曲霉毒素B1、碱性橙II和碱性嫩黄O的方法,其包括以下步骤:
步骤S1:配制碱性橙II和碱性嫩黄O的混合标准液,并稀释成不同浓度的标准工作液,分别采用液相色谱仪进行检测得到不同浓度的碱性橙II和碱性嫩黄O标准工作液的液相色谱图,根据液相色谱图的峰面积和对应的浓度制作碱性橙II和碱性嫩黄O的校正曲线;配制黄曲霉毒素B1标准液,并分别稀释成不同浓度的黄曲霉毒素B1的标准工作液,分别采用液相色谱仪进行检测得到不同浓度黄曲霉毒素B1标准工作液的液相色谱图,根据液相色谱图的峰面积和对应的浓度制作黄曲霉毒素B1的校正曲线;
步骤S2:制备低共熔溶剂,所述低共熔溶剂包括氢键受体化合物和氢键供体化合物;
步骤S3:称取调味油样品,加入步骤S2中所述低共熔溶剂和烷烃试剂,进行震荡萃取,离心后去除上面的油层,剩余物采用液相色谱仪进行检测得到样品的色谱图;其中,所述低共熔溶剂和烷烃试剂的体积比大于0.0025;其中优选的,所述剩余物用烷烃洗涤后采用液相色谱仪进行检测,洗涤的目的是为了除油除杂更彻底;
步骤S4:将样品的色谱图与碱性橙II和碱性嫩黄O标准工作液的液相色谱图、以及黄曲霉毒素B1标准工作液的液相色谱图进行比较确定是否含有碱性橙II、碱性嫩黄O和黄曲霉毒素B1,如果确定含有碱性橙II和碱性嫩黄O,再根据样品的色谱图峰面积所对应的标准曲线上的浓度确定样品中碱性橙II和碱性嫩黄O的含量;如果确定含有黄曲霉毒素B1,再根据样品的色谱图峰面积所对应的标准曲线上的浓度确定样品中黄曲霉毒素B1的含量。
采用此技术方案,利用低共熔溶剂提取调味油样品中的碱性橙II和碱性嫩黄O,利用烷烃类试剂除去复杂的基质可达到样品净化的目的,再将低共熔溶剂上机检测,实现了快速检测,步骤简单,无需复杂的样品前处理过程,减少了溶剂的使用,同时又除去了调味油中复杂基质的干扰,保证了检测结果的准确性。
作为本发明的进一步改进,所述步骤S1还包括:配制黄曲霉毒素B1标准液,并分别稀释成不同浓度的黄曲霉毒素B1的标准工作液,分别采用液相色谱仪进行检测得到不同浓度黄曲霉毒素B1标准工作液的液相色谱图,根据液相色谱图的峰面积和对应的浓度制作黄曲霉毒素B1的校正曲线;
所述步骤S4还包括:将样品的色谱图与黄曲霉毒素B1标准工作液的液相色谱图进行比较确定是否含有黄曲霉毒素B1,如果确定含有黄曲霉毒素B1,再根据样品的色谱图峰面积所对应的标准曲线上的浓度确定样品中黄曲霉毒素B1的含量。
采用此技术方案,可以同时进行碱性橙II和碱性嫩黄O非食用色素和黄曲霉毒素B1的检测,处理简单,使用方案,检验快速,克服了现有检测方法的缺点与不足。
作为本发明的进一步改进,所述氢键供体化合物为尿素和醇类中的至少一种,所述氢键受体化合物与氢键供体化合物的摩尔比为1:(1~3)。
优选的,所述低共熔溶剂的用量按照每克调味油样品0.05~5.0mL。
作为本发明的进一步改进,所述氢键受体化合物为甜菜碱,所述甜菜碱与所述氢键供体化合物的摩尔比为1:1、1:2或1:3。
作为本发明的进一步改进,所述氢键供体化合物为丙三醇。
作为本发明的进一步改进,所述烷烃试剂包括正己烷、环己烷、辛烷、庚烷中的至少一种。
作为本发明的进一步改进,所述低共熔溶剂和烷烃试剂的体积比为0.5~5。
作为本发明的进一步改进,所述烷烃试剂为正己烷或正庚烷。
作为本发明的进一步改进,步骤S3中,所述萃取的方式为摇床震荡提取或超声波提取,所述提取温度在10-50℃。优选的,所述萃取的方式为摇床震荡提取。
作为本发明的进一步改进,步骤S3中,剩余物用烷烃洗涤除去残留的油,然后将低共熔溶剂直接上机或者定容到所需要的体积后上机检测。
与现有技术相比,本发明的有益效果为:
第一,本发明的技术方案,实用性强,对于辣椒油、番椒油、胭脂树橙油等粘度大、基质复杂的调味油样品,均可以进行碱性橙II、碱性嫩黄O以及黄曲霉毒素B1的准确检测。
第二,本发明的技术方案操作简单、成本低,本发明仅需要简单的样品提取过程,不需要复杂的前处理过程,提取所用的低共熔溶剂可以节省有机溶剂的使用,大大降低了实验成本。
第三,本发明的技术方案检测快速,由于本方法可以节省样品前处理时间,大大缩短了调味油中碱性橙II、碱性嫩黄O以及黄曲霉毒素B1检测所需要的时间。
第四,加标回收率高,检出限低,低共熔溶剂可以有效的从调味油中提取碱性橙II、碱性嫩黄O以及黄曲霉毒素B1,同时烷烃类试剂可以除去复杂的油基质,避免杂质干扰,因此能够保证足够高的回收率和较低的检出限,样品的回收率分别在82.3%-101%、81.6%-103%和75.6%-108%范围内,检出限分别可达0.02mg/kg、0.02mg/kg和1.0μg/kg。
附图说明
图1是本发明一种实施例的浓度为0.1μg/L的碱性橙II和碱性嫩黄O两种色素混合标准工作液色谱图。
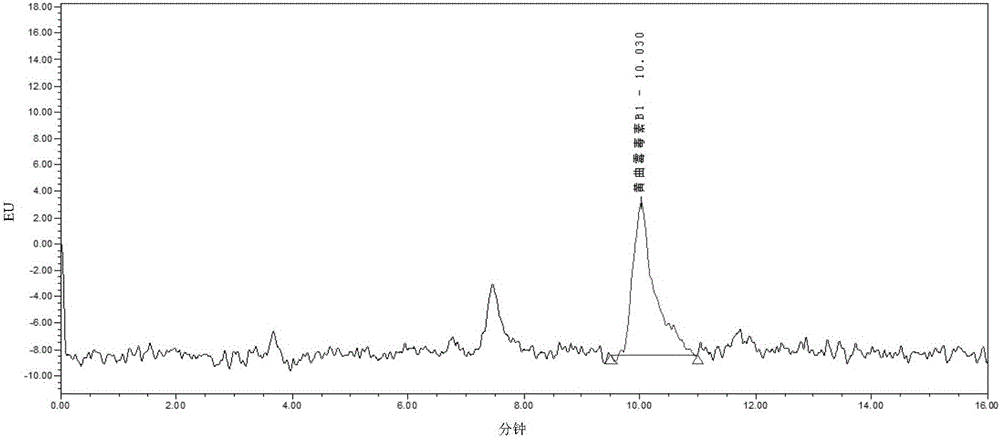
图2是本发明一种实施例浓度为0.5μg/L的黄曲霉毒素B1标准工作液色谱图。
图3是本发明一种实施例的碱性橙II标准工作液校正曲线。
图4是本发明一种实施例的碱性嫩黄O标准工作液校正曲线。
图5是本发明一种实施例的黄曲霉毒素B1标准工作液校正曲线。
具体实施方式
下面结合说明书附图和具体实施例对本发明作出进一步地详细阐述,所述实施例只用于解释本发明,并非用于限定本发明的范围。下述实施例中所使用的材料、试剂、仪器设备等,如无特殊说明,均为商业可得。
甲醇:色谱纯,购于默克公司;乙腈:色谱纯,购于默克公司;正己烷:色谱纯,购于默克公司;正庚烷:色谱纯,购于默克公司;乙酸铵:分析纯,购于上海安谱,冰乙酸:分析纯,上海凌峰;甜菜碱:分析纯,阿拉丁;尿素:分析纯,阿拉丁;丙三醇:分析纯,上海凌峰;丙二醇:分析纯,上海凌峰;水为超纯水。
碱性橙II标准品:纯度95.00%,购自Sigma-Aldrich。
碱性嫩黄O标准品:纯度86.00%,购自Sigma-Aldrich。
黄曲霉毒素B1标准品:纯度100.0%,购自百灵威。
高效液相色谱仪:配备四元梯度泵,脱气装置,微量进样器,紫外检测器和荧光检测器。
碱性橙II和碱性嫩黄O液相色谱条件参数如下:
色谱柱:Agilent 5TC-C18,150×4.6mm
柱温:25℃
检测波长:450nm
乙酸铵缓冲溶液:称取0.77g乙酸铵固体,加水溶解,转移至1000mL容量瓶中,再加入1.2mL冰醋酸,用超纯水定容至刻度,超声除去气泡,抽滤除去固体杂质。
进样量:25μL。
流速:1.0mL/min。
梯度洗脱条件:流动相A为乙腈,流动相B为乙酸铵缓冲溶液。
流动相的具体情况如表1所示。
表1
黄曲霉毒素B1液相色谱条件参数如下:
色谱柱:Agilent 5TC-C18,150×4.6mm
柱温:40℃
检测波长:激发波长360nm,发射波长420nm
进样量:20μL
流速:0.8mL/min
等度洗脱条件:甲醇:水=45:55
柱后衍生条件:0.05%碘溶液,流速0.2mL/min,反应温度70℃
按照以下步骤进行检测:
步骤S1:配制碱性橙II和碱性嫩黄O的混合标准液,并稀释成不同浓度的标准工作液,分别采用液相色谱仪进行检测得到不同浓度的碱性橙II和碱性嫩黄O标准工作液的液相色谱图,根据液相色谱图的峰面积和对应的浓度制作碱性橙II和碱性嫩黄O的校正曲线;并配制黄曲霉毒素B1标准液,并分别稀释成不同浓度的黄曲霉毒素B1的标准工作液,分别采用液相色谱仪进行检测得到不同浓度黄曲霉毒素B1标准工作液的液相色谱图,根据液相色谱图的峰面积和对应的浓度制作黄曲霉毒素B1的校正曲线;
步骤S2:制备低共熔溶剂。
低共熔溶剂DES-1的制备:称取20g甜菜碱和15.72g的丙三醇于100mL反应瓶中,升温到90℃并搅拌至澄清为止,冷却至室温备用。
低共熔溶剂DES-2的制备:称取20g甜菜碱和20.51g的尿素于100mL反应瓶中,升温到90℃并搅拌至澄清为止,冷却至室温备用。
低共熔溶剂DES-3的制备:称取20g甜菜碱和31.79g的乙二醇于100mL反应瓶中,升温到90℃并搅拌至澄清为止,冷却至室温备用。
步骤S3:称取调味油样品,加入步骤S2中所述低共熔溶剂和烷烃试剂,进行震荡萃取,离心后去除上面的油层,剩余物用烷烃洗涤后采用液相色谱仪进行检测得到样品的色谱图;其中,所述低共熔溶剂和烷烃试剂的体积比大于0.0025;
步骤S4:将样品的色谱图与碱性橙II和碱性嫩黄O标准工作液的液相色谱图、以及黄曲霉毒素B1标准工作液的液相色谱图进行比较确定是否含有碱性橙II和碱性嫩黄O,如果确定含有碱性橙II和碱性嫩黄O,再根据样品的色谱图峰面积所对应的标准曲线上的浓度确定样品中碱性橙II和碱性嫩黄O的含量;将样品的色谱图与黄曲霉毒素B1标准工作液的液相色谱图进行比较确定是否含有黄曲霉毒素B1,如果确定含有黄曲霉毒素B1,再根据样品的色谱图峰面积所对应的标准曲线上的浓度确定样品中黄曲霉毒素B1的含量。浓度为0.1μg/L的碱性橙II和碱性嫩黄O两种色素混合标准工作液色谱图如图1所示;浓度为0.5μg/L的黄曲霉毒素B1标准工作液色谱图如图2所示;碱性橙II标准工作液校正曲线如图3所示;碱性嫩黄O标准工作液校正曲线如图4所示;黄曲霉毒素B1标准工作液校正曲线如图5所示。
实施例1
样品:称取番椒油1g于15mL离心管中,加入0.5mL低共熔溶剂DES-1和8mL正己烷,摇床震荡提取10分钟,7500r/min离心2分钟,用塑料吸管吸取上面的油层弃去,再加入4mL正己烷,震荡洗涤1分钟,7500r/min离心2分钟,上面的油层用塑料吸管吸走弃去。残留的低共熔溶剂DES-1部分用甲醇定容至2.0mL,涡旋混匀,过0.22μm有机滤膜,上机测试。
加标样品1:称取番椒油1g于15mL离心管中,添加碱性橙II、碱性嫩黄O以及黄曲霉毒素B1的浓度水平均为2.0mg/kg,加入0.5mL低共熔溶剂DES-1和8mL正己烷,摇床震荡提取10分钟,7500r/min离心2分钟,用塑料吸管吸取上面的油层弃去,再加入4mL正己烷,震荡洗涤1分钟,7500r/min离心2分钟,上面的油层用塑料吸管吸走弃去。残留的低共熔溶剂DES-1部分用甲醇定容至2.0mL,涡旋混匀,过0.22μm有机滤膜,上机测试。
将样品的色谱图与碱性橙II和碱性嫩黄O标准工作液的色谱图、以及黄曲霉毒素B1标准工作液的液相色谱图进行比较确定是否含有碱性橙II和碱性嫩黄O、或同时含有黄曲霉毒素B1,再根据样品的色谱图峰面积所对应的标准曲线上的浓度确定样品中碱性橙II、碱性嫩黄O、黄曲霉毒素B1的含量,得到的结果如表2所示。
表2实施例1的检测结果表
实施例2
样品:称取辣椒油1g于15mL离心管中,加入0.3mL低共熔溶剂DES-2和5mL正庚烷,摇床震荡提取10分钟,7500r/min离心2分钟,用塑料吸管吸取上面的油层弃去,再加入5mL正庚烷,震荡洗涤1分钟,7500r/min离心2分钟,上面的油层用塑料吸管吸走弃去。残留的低共熔溶剂DES-2部分用甲醇定容至2.0mL,涡旋混匀,过0.22μm有机滤膜,上机测试。
加标样品2:称取辣椒油1g于15mL离心管中,添加碱性橙II、碱性嫩黄O以及黄曲霉毒素B1的浓度水平均为2.0mg/kg,加入0.3mL低共熔溶剂DES-2和5mL正庚烷,摇床震荡提取10分钟,7500r/min离心2分钟,用塑料吸管吸取上面的油层弃去,再加入5mL正庚烷,震荡洗涤1分钟,7500r/min离心2分钟,上面的油层用塑料吸管吸走弃去。残留的低共熔溶剂DES-2部分用甲醇定容至2.0mL,涡旋混匀,过0.22μm有机滤膜,上机测试。
将样品的色谱图与碱性橙II和碱性嫩黄O标准工作液的色谱图、以及黄曲霉毒素B1标准工作液的液相色谱图进行比较确定是否含有碱性橙II和碱性嫩黄O,以及是否同时含有黄曲霉毒素B1,再根据样品的色谱图峰面积所对应的标准曲线上的浓度确定样品中碱性橙II、碱性嫩黄O、黄曲霉毒素B1的含量,得到的结果如表3所示。
表3实施例2的检测结果表
实施例3
样品:称取姜黄油1g于15mL离心管中,加入0.5mL低共熔溶剂DES-3和5mL正庚烷,摇床震荡提取10分钟,7500r/min离心2分钟,用塑料吸管吸取上面的油层弃去,再加入5mL正庚烷,震荡洗涤1分钟,7500r/min离心2分钟,上面的油层用塑料吸管吸走弃去。残留的低共熔溶剂DES-3部分用甲醇定容至2.0mL,涡旋混匀,过0.22μm有机滤膜,上机测试。
加标样品3:称取姜黄油1g于15mL离心管中,添加碱性橙II、碱性嫩黄O以及黄曲霉毒素B1的浓度水平均为2.0mg/kg,加入0.5mL低共熔溶剂DES-3和5mL正庚烷,摇床震荡提取10分钟,7500r/min离心2分钟,用塑料吸管吸取上面的油层弃去,再加入5mL正庚烷,震荡洗涤1分钟,7500r/min离心2分钟,上面的油层用塑料吸管吸走弃去。残留的低共熔溶剂DES-3部分用甲醇定容至2.0mL,涡旋混匀,过0.22μm有机滤膜,上机测试。
将样品的色谱图与碱性橙II和碱性嫩黄O标准工作液的色谱图、以及黄曲霉毒素B1标准工作液的液相色谱图进行比较确定是否含有碱性橙II和碱性嫩黄O、或同时含有黄曲霉毒素B1,再根据样品的色谱图峰面积所对应的标准曲线上的浓度确定样品中碱性橙II、碱性嫩黄O、黄曲霉毒素B1的含量,得到的结果如表4所示。
表4实施例3的检测结果表
通过反复实验,含有碱性橙II、碱性嫩黄O、黄曲霉毒素B1的样品的回收率分别在82.3%-101%、81.6%-103%和75.6%-108%范围内,碱性橙II、碱性嫩黄O、黄曲霉毒素B1的检出限分别可达0.02mg/kg、0.02mg/kg和1.0μg/kg。
以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!