一种制备阿法替尼的中间体及其对映异构体的检测方法与流程
本发明属于药物技术领域,具体涉及一种制备阿法替尼的关键中间体及其对映异构体的含量检测方法。
背景技术:
手性药物是指由具有药理活性的手性化合物组成的药物。由于药物分子所作用的受体或靶位是由氨基酸、核苷、膜等组成的手性蛋白质和核酸大分子等。它们对与其结合的药物分子的空间立体构型(手性)有一定的要求。因此,手性药物的两个对映体往往在生物体内的药理活性、代谢过程、代谢速率及毒性等存在显著的差异。以1960年代的一个著名的事件来说明:外消旋的沙利度胺(thalidomide)曾是有力的镇静剂和止吐药,尤其适合在早期妊娠反应中使用。但很快就发现它是极强的致畸剂。进一步研究表明,致畸性由该药的(S)-异构体引起,而(R)-异构体被认为即使在高剂量时在动物中也不引起畸变。
正是由于手性药物的不同立体异构体在药效、药代及毒理等方面都可能存在差异,美国FDA在其关于开发立体异构体新药的政策中要求在对手性药物进行药理毒理研究时,应分别获得该药物的各立体异构体,进行必要的比较研究,以确定拟进一步开发的药物。我国食品药品监督管理局也于2006年12月发布的《手性药物质量控制研究技术指导原则》,在其中指出手性药物研究的基本思路,其中包括“在原料药制备工艺研究时,应根据手性中心的引入方式,采取有效的过程控制手段,严格控制手性原料与每步反应产物的光学纯度;”及“质量研究时,应结合工艺与各手性中心的稳定性确定需研究控制的立体异构体杂质”等。
阿法替尼(式1)为手性药物,是由德国的勃林格殷格翰公司研发的一种多靶点小分子药物,其属于表皮生长因子受体(EGFR)和人表皮受体(HER2)酪氨酸激酶的不可逆抑制剂,也是首个用于表皮生长因子受体抑制剂治疗失败后的肺癌治疗药物。临床上可用于晚期非小细胞肺癌及晚期乳腺癌、肠癌的治疗。2013年7月在美国以Gilotrif为商品名获得批准,
2013年9月在欧盟获得批准上市。作为手性药物,阿法替尼对映异构体的含量直接影响产品的质量。目前已有一些专利及文献提供了阿法替尼对映异构体的含量检测方法,如文献1(高效液相色谱法测定马来酸阿法替尼对映异构体的含量,马丽萍等,《中南药学》,2016年3月第14卷第3期,303~305页)及专利CN104391058等。
N4-(3-氯-4-氟苯基)-7-[[(3S)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺(式2)是合成阿法替尼的关键中间体,该中间体的结构与阿法替尼成品的结构相似度较大,且引入了手性结构,是有可能在产品中引入其手性杂质的源头所在。国家局发布的《手性药物质量控制研究技术指导原则》明确规定了“在原料药制备工艺研究时,应根据手性中心的引入方式,采取有效的过程控制手段,严格控制手性原料与每步反应产物的光学纯度”,因此阿法替尼作为手性药物,有必要对其关键中间体(式2)的对映异构体(式3)的含量进行控制,以保证该药品的质量安全、有效、可控。现有技术仅有对阿法替尼成品的对映异构体含量的检测方法,由于阿法替尼(式1)与(式2)所示的关键中间体取代基差异较大,两化合物在色谱柱中的吸附及解吸附能力不同,从而造成阿法替尼成品的对映异构体的检测方法不能用来检测其关键中间体对映异构体的含量,因此出于“严格控制手性原料与每步反应产物的光学纯度”的需要,需要开发一种适合检测阿法替尼关键中间体(式2)的对映异构体(式3)含量方法,从而保证阿法替尼成品的光学纯度。
技术实现要素:
有鉴于此,本发明旨在提供一种阿法替尼关键中间体(式2)及其对映异构体(式3)含量的检测方法,从而有效控制手性药物阿法替尼成品的光学纯度。
为了实现上述目的,本发明提供如下技术方案:
一种采用双高效液相色谱、可同时测定阿法替尼关键中间体及其对映异构体含量的方法,采用的色谱条件为:用纤维素-三(3,5-二甲基苯基氨基甲酸酯)为填充剂,流动相是正己烷、异丙醇、乙腈的混合溶液,体积比是700~800:150~250:50,进行等度检测。
作为进一步的优选方案,流动相正己烷、异丙醇、乙腈的混合溶液的体积比是750:200:50。
作为进一步的优选方案,本发明使用紫外检测器进行检测,紫外检测波长为255~265nm,优选260nm。本发明提到的检测波长是根据方法耐用性实验确定的,即本方法的检测波长在5nm范围内调整,测试结果基本没有影响。
作为进一步的优选方案,洗脱流速为0.5~2.0mL/min,优选0.9~1.1mL/min,更优选0.9mL/min。本发明洗脱流速设定为一般技术人员公知的常识,常见的范围一般为0.5ml/min至2ml/min。
作为进一步的优选方案,检测样品的进样量为1μL~20μL,优选10μL。
作为进一步的优选方案,色谱柱柱温为25℃~35℃,优选25℃。本发明提到的柱温是根据方法耐用性实验确定的,即本方法在柱温在25~35℃范围内调整,测试结果没有显著影响。
作为进一步的优选方案,选择的是型号为Lux Chiral Cellulose-1,规格250*4.6mm,5μm的色谱柱。
作为进一步的优选方案,本发明采用纤维素-三(3,5-二甲基苯基氨基甲酸酯)为填充剂的色谱柱,流动相是正己烷、异丙醇、乙腈的混合溶液,体积比是700~800:250~150:50进行等度检测,使用紫外检测器进行检测,紫外检测波长为255~265nm,洗脱流速为0.5~2.0mL/min,检测样品的进样量为1μL~20μL,柱温为25℃~35℃。
作为进一步的优选方案,本发明采用型号为Lux Chiral Cellulose-1,规格为250*4.6mm,5μm的色谱柱,流动相是正己烷、异丙醇、乙腈的混合溶液,其体积比是750:200:50,进行等度检测,使用紫外检测器进行检测,紫外检测波长为260nm,洗脱流速为0.9mL/min,检测样品的进样量为10μL,柱温为25℃。
在本发明中所述的“阿法替尼关键中间体”是化学名称为“N4-(3-氯-4-氟苯基)-7-[[(3S)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺”的化合物(式2),其对映异构体是化学名称为“N4-(3-氯-4-氟苯基)-7-[[(3R)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺”的化合物(式3)。
本发明的有益效果是提供了阿法替尼关键中间体(式2)及其对映异构体(式3)含量检测方法,填补了该药在制备过程中质量控制的技术空白。使用本发明方法,阿法替尼关键中间体(式2)及其对映异构体(式3)的分离度可达2.0以上,保证了中间体及其异构体杂质含量检测的精确度及准确度,进而控制了阿法替尼手性杂质引入的源头,保证了阿法替尼成品的光学纯度。
附图说明
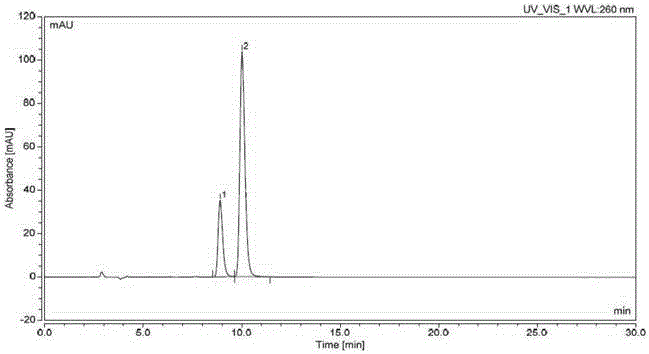
图1:实施例1系统适用性试验色谱图;
图2:实施例2系统适用性试验色谱图;
图3:实施例3系统适用性试验色谱图;
注:谱图中峰1为式3化合物,峰2为式2化合物。
具体实施方案
下述是结合具体实施例和实验例,进一步阐述本发明。但这些实施例限于说明本发明而不是用于限制本发明的范围。
实施例1
色谱条件:用纤维素-三(3,5-二甲基苯基氨基甲酸酯)为填充剂(参考色谱柱:Lux ChiralCellulose-1(250*4.6mm,5μm)),柱温为25℃,以正己烷-异丙醇-乙腈(750:200:50)为流动相,流速为每分钟0.9ml,检测波长为260nm,分析时间为30分钟。
检测方法:取N4-(3-氯-4-氟苯基)-7-[[(3S)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺化合物(式2)10mg,精密称定,置20ml量瓶中,加异丙醇溶解并稀释至刻度,摇匀,作为供试品溶液。取N4-(3-氯-4-氟苯基)-7-[[(3R)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺化合物(式3)适量,加异丙醇溶解并定量稀释制成每1ml中含2.5μg的溶液,作为对照品溶液。取式2化合物10mg与式3化合物2mg置同一200ml量瓶中,加异丙醇溶解并稀释至刻度,摇匀,作为系统适用性溶液。精密量取供试品溶液10μl注入液相色谱仪,记录色谱图,按外标法以峰面积计算。
方法验证
A.系统适用性实验:
取系统适用性溶液,照上述色谱条件,连续进针5针,系统适用性溶液中式3化合物峰面积的RSD为0.13%,式2化合物峰面积的RSD为0.16%,分离度大于3.27,系统适用性试验符合要求。具体数据见表1,系统适用性色谱图见附图1。
表1.系统适用性考察结果
B.耐用性试验
耐用性研究是通过对色谱条件作较小的改动,以确定这些改动是否会对图谱有较大的影响。按照下表进行耐用性研究,在所改变的条件下,分别进行系统适用性考察,其中标准条件用的是重复性试验的实验结果,结果见表2。
表2耐用性研究结果
C.定量限、检测限
溶液的配制:取式3化合物约10mg,精密称定,置100mL量瓶中,配制成每1mL约含式3化合物0.05μg/mL的溶液,进样分析,结果以信噪比为10:1计,其定量限浓度为0.46μg/mL,取定量限溶液,进样10μL,结果以信噪比3:1计,式3化合物的检测限浓度为0.19μg/mL。
D.线性
称取式3化合物,配制成4.63μg/mL、0.93μg/mL、0.46μg/mL、0.23μg/mL、0.09μg/mL的溶液,进行线性实验;该方法的线性回归方程为y=0.8248x-0.0172;相关系数R2=0.9999,结果表明式3化合物在0.09~5μg/mL的范围内峰面积与浓度线性关系良好。
E.回收率
称取式3化合物对照品配制成浓度为100μg/mL的储备溶液,量取储备溶液用异丙醇分别稀释成2.5μg/mL、1.0μg/mL、0.25μg/mL的异丙醇溶液。取10份式2化合物样品每份约10mg,精密称定,其中一份用异丙醇溶解制备成未加标的样品溶液,其余9份分别用3份2.5μg/mL、1.0μg/mL、0.25μg/mL的式3化合物的异丙醇溶液溶解并稀释至刻度,按照供试品溶液制备方法,进样检测,记录色谱图。以外标法计算测得量,进而计算回收率。结果表明,低、中、高水平的回收率RSD值为4.02%,平均回收率为100.52%,表明本方法的准确度良好。
实施例2
色谱条件:用纤维素-三(3,5-二甲基苯基氨基甲酸酯)为填充剂(参考色谱柱:Lux ChiralCellulose-1(250*4.6mm,5μm)),柱温为25℃,以正己烷-异丙醇-乙腈(700:250:50)为流动相,流速为每分钟0.9ml,检测波长为260nm,分析时间为30分钟。
检测方法:取N4-(3-氯-4-氟苯基)-7-[[(3S)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺化合物(式2)10mg,精密称定,置20ml量瓶中,加异丙醇溶解并稀释至刻度,摇匀,作为供试品溶液。取N4-(3-氯-4-氟苯基)-7-[[(3R)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺化合物(式3)适量,加异丙醇溶解并定量稀释制成每1ml中含2.5μg的溶液,作为对照品溶液。取式2化合物10mg与式3化合物2mg置同一200ml量瓶中,加异丙醇溶解并稀释至刻度,摇匀,作为系统适用性溶液。精密量取供试品溶液10μl注入液相色谱仪,记录色谱图,按外标法以峰面积计算。
方法验证
A.系统适用性实验:
取系统适用性溶液,照上述色谱条件,连续进针5针,系统适用性溶液中式3化合物峰面积的RSD为1.95%,式2化合物峰面积的RSD为1.93%,分离度大于2.50,系统适用性试验符合要求。具体数据见表3,系统适用性1色谱图见附图2。
B.定量限、检测限
溶液的配制:取式3化合物约10mg,精密称定,置100mL量瓶中,配制成每1mL约含式3化合物0.05μg/mL的溶液,进样分析,结果以信噪比为10:1计,杂质A的定量限为0.46μg/mL,取定量限溶液,进样10μL,结果以信噪比3:1计,式3化合物的检测限为0.19μg/mL。
表3.系统适用性考察结果
C.线性
称取式3化合物,配制成4.63μg/mL、0.93μg/mL、0.46μg/mL、0.23μg/mL、0.09μg/mL的溶液,进行线性实验;该方法的线性回归方程为y=1.4236x-0.23584;相关系数R2=0.9997,结果表明式3化合物在0.09~5μg/mL的范围内峰面积与浓度线性关系良好。
D.回收率
称取式3化合物对照品配制成浓度为100μg/mL的储备溶液,量取储备溶液用异丙醇分别稀释成2.5μg/mL、1.0μg/mL、0.25μg/mL的异丙醇溶液。取10份式2化合物样品每份约10mg,精密称定,其中一份用异丙醇溶解制备成未加标的样品溶液,其余9份分别用3份2.5μg/mL、1.0μg/mL、0.25μg/mL的式3化合物的异丙醇溶液溶解并稀释至刻度,按照供试品溶液制备方法,进样检测,记录色谱图。以外标法计算测得量,进而计算回收率。结果表明,低、中、高水平的回收率RSD值为3.89%,平均回收率为101.42%,表明本方法的准确度良好。
实施例3
色谱条件:用纤维素-三(3,5-二甲基苯基氨基甲酸酯)为填充剂(参考色谱柱:Lux Chiral Cellulose-1(250*4.6mm,5μm)),柱温为25℃,以正己烷-异丙醇-乙腈(800:150:50)为流动相,流速为每分钟0.9ml,检测波长为260nm,分析时间为30分钟。
检测方法:取N4-(3-氯-4-氟苯基)-7-[[(3S)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺化合物(式2)10mg,精密称定,置20ml量瓶中,加异丙醇溶解并稀释至刻度,摇匀,作为供试品溶液。取N4-(3-氯-4-氟苯基)-7-[[(3R)-四氢-3-呋喃基]氧基]-4,6-喹唑啉二胺化合物(式3)适量,加异丙醇溶解并定量稀释制成每1ml中含2.5μg的溶液,作为对照品溶液。取式2化合物10mg与式3化合物2mg置同一200ml量瓶中,加异丙醇溶解并稀释至刻度,摇匀,作为系统适用性溶液。精密量取供试品溶液10μl注入液相色谱仪,记录色谱图,按外标法以峰面积计算。
方法验证
A.系统适用性实验:
取系统适用性溶液,照上述色谱条件,连续进针5针,系统适用性溶液中式3化合物峰面积的RSD为0.60%,式2化合物峰面积的RSD为0.45%,分离度大于3.60,系统适用性试验符合要求。具体数据见表4,系统适用性1色谱图见附图3。
表4.系统适用性考察结果
B.定量限、检测限
溶液的配制:取式3化合物约10mg,精密称定,置100mL量瓶中,配制成每1mL约含式3化合物0.05μg/mL的溶液,进样分析,结果以信噪比为10:1计,杂质A的定量限为0.46μg/mL,取定量限溶液,进样10μL,结果以信噪比3:1计,式3化合物的检测限为0.19μg/mL。
C.线性
称取式3化合物,配制成4.63μg/mL、0.93μg/mL、0.46μg/mL、0.23μg/mL、0.09μg/mL的溶液,进行线性实验;该方法的线性回归方程为y=0.6252x-2.9741;相关系数R2=0.9998,结果表明式3化合物在0.09~5μg/mL的范围内峰面积与浓度线性关系良好。
D.回收率
称取式3化合物对照品配制成浓度为100μg/mL的储备溶液,量取储备溶液用异丙醇分别稀释成2.5μg/mL、1.0μg/mL、0.25μg/mL的异丙醇溶液。取10份式2化合物样品每份约10mg,精密称定,其中一份用异丙醇溶解制备成未加标的样品溶液,其余9份分别用3份2.5μg/mL、1.0μg/mL、0.25μg/mL的式3化合物的异丙醇溶液溶解并稀释至刻度,按照供试品溶液制备方法,进样检测,记录色谱图。以外标法计算测得量,进而计算回收率。结果表明,低、中、高水平的回收率RSD值为4.61%,平均回收率为102.72%,表明本方法的准确度良好。
应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!