一种白及药材HPLC指纹图谱的建立方法与流程
本发明涉及一种白及药材hplc指纹图谱的建立方法,属于药品技术的领域。
背景技术:
白及药材为兰科植物白及bletillastriata(thunb.)reichb.f.的干燥块茎。味苦、甘、涩,性微寒。归肺、肝、胃经。具有收敛止血,消肿生肌的功效。用于咯血,吐血,外伤出血,疮疡肿毒,皮肤皲裂等症。
中药成分复杂,且大部分中药的有效成分不完全明确,其中已知的有效成分往往不能代表其中药的全部疗效,凭借某一种化学成分定性和定量的传统中药质量评价方法的有效性和专属性渐渐受到质疑。要想了解评价中药质量整体性能,就要运用新的方法,对中药建立hplc指纹图谱,指纹图谱不是强调个体特异性,而是强调同一药材群体的相似性、整体性(共有特征性)。与传统质量控制模式的区别在于,指纹图谱是综合地看问题,也就是强调中药质量的“完整面貌”即整体性,反映药材综合质量信息。指纹图谱分析强调的是准确的辨认,而不是精密的计算,强调的是相似,而不是相同。在不可能将中药复杂成分都搞清楚的情况下,指纹图谱的作用是反映了中药内在质量的均一性和稳定性。
中药指纹图谱是一种综合的,可量化的鉴定手段,它是建立在中药化学成分系统研究的基础上,主要用于评价中药材以及中药制剂半成品质量的真实性、优良性和稳定性。中药及其制剂均为多组分复杂体系,因此评价其质量应采用与之相适应的,能提供丰富鉴别信息的检测方法,建立中药指纹图谱将能较为全面地反映中药及其制剂中所含化学成分的种类与数量,进而对药品质量进行整体描述和评价。在此基础上,如果进一步开展谱效学研究,可使中药质量与其药效真正结合起来,有助于阐明中药作用机理。
技术实现要素:
本发明目的在于,提供一种白及药材hplc指纹图谱的建立方法。该方法能较为全面地反映白及药材中所含化学成分的种类与数量,进而对其质量进行整体描述和评价,可将白及药材质量与其药效真正结合起来,有助于阐明其作用机理为白及药材的技术提升与深度开发提供了依据。且用该方法检测到的白及药材指纹图谱化学成分相对较多、各特征峰高度比例适中,基线较平稳,分离度、峰形和柱效好。
为解决上述技术问题,本发明采用如下技术方案实现:一种白及药材hplc指纹图谱的建立方法,包括以下步骤:
(1)对照品溶液的制备:精密称取1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯对照品6-6.5mg,置10ml棕色容量瓶中,用乙腈和0.05%-0.15%磷酸水溶液以2:2-2:4的比例混合溶解并稀释至刻度,摇匀,即得对照品溶液;
(2)供试品溶液的制备:精密称取白及药材粉末0.5-1.5g,置100ml具塞锥形瓶中,加入15-25ml分析甲醇,称定重量,超声提取25-35min,取出,放冷,用分析甲醇补足减失的重量,摇匀,过滤,以0.45μm微孔滤膜过滤,即得供试品溶液;
(3)色谱条件:色谱柱为diamonsilc18(250mm×4.6mm,5μm),流动相:乙腈(a)-水(b),检测波长为260-280nm,流速为0.5-1.5ml·min-1,柱温28-32℃,进样量10μl,洗脱时间为40-50min;
(4)指纹图谱的制作:按上述方法制备供试品溶液,在上述色谱条件下进样,记录谱图得到白及药材的hplc指纹图谱;
(5)指纹谱图的确认:按照上述提供的方法,对多批次白及药材建立了hplc指纹图谱,通过分析比较确定了15个共有峰,这些共有峰构成了白及药材的指纹特征,作为白及药材的指纹图谱。
前述的白及药材hplc指纹图谱的建立方法,所述对照品溶液这样制备:精密称取1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯(militarine)对照品6.80mg,置10ml棕色容量瓶中,用乙腈和0.1%磷酸水溶液以2:3的比例混合溶解并稀释至刻度,摇匀,即得0.680mg·ml-1的对照品溶液。
前述的白及药材hplc指纹图谱的建立方法,所述供试品溶液这样制备:精密称取白及药材粉末1g,置100ml具塞锥形瓶中,加入20ml分析甲醇,称定重量,超声提取30min,取出,放冷,用分析甲醇补足减失的重量,摇匀,过滤,以0.45μm微孔滤膜过滤,即得供试品溶液。
前述的白及药材hplc指纹图谱的建立方法,所述(3)色谱条件为:色谱柱为diamonsilc18(250mm×4.6mm,5μm),流动相:乙腈(a)-水(b),检测波长为270nm,流速为1ml·min-1,柱温30℃,进样量10μl,洗脱时间为45min;
前述的白及药材hplc指纹图谱的建立方法,所述洗脱程序为:0~5min,90-95%b;5~25min,80~90%b;25~40min,80~65%b,40~45min,65~30%b。
前述的白及药材hplc指纹图谱的建立方法,所述15个共有峰,以4号峰为参照峰,其余峰相对保留时间分别为:
0.422-0.423,0.641-0.642,0.687-0.689,0.920-0.921,1.395-1.397,1.459-1.461,1.579-1.580,1.626-1.628,1.661-1.662,1.765-1.768,1.869-1.871,1.913-1.915,1.972-1.974,2.012-2.015,2.038-2.042。
发明人进行了大量的实验,以下是部分实验研究
实验例
1仪器、试剂、药材、评价软件
1.1仪器
ultimate-3000高效液相色谱仪(赛默飞世尔科技公司),千分之一电子天平,十万分之一电子天平,高速多功能粉碎机(上海冰都电器有限公司),ht-220a柱温箱(天津市恒奥科技发展有限公司),drhh-2数显恒温水浴锅(上海双捷实验设备有限公司),数控超声波清洗器(昆山市超声仪器有限公司),循环水多用真空泵(8hz-0111型,上海予英仪器有限公司)。
1.2试剂
1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯(militarine)对照品(成都洛玛生物科技有限公司,批号:chb160626);乙腈(tediacompany,usa,批号:16065011),磷酸(重庆川江化学试剂厂),娃哈哈纯净水(杭州娃哈哈集团有限公司授权大理娃哈哈食品有限公司制造);其余试剂均为分析纯。
1.3药材
白及药材共十批,经贵阳中医学院实验师刘刚鉴定为兰科植物白及bletillastriata(thunb.)reichb.f.的干燥块茎(见表1.1)。
表1.110批白及药材的产地
1.4评价软件
“中药色谱指纹图谱相似度评价系统”2004年a版(国家药典委员会)
2方法与结果
2.1色谱条件的确定
2.1.1色谱柱
色谱柱为diamonsilc18(250mm×4.6mm,5μm)
2.1.2流动相的选择
本实验经查阅相关文献,选择了以下几种流动相系统进行实验比较,流动相系统见表2.1,hplc图谱见图1。
表2.1不同种流动相洗脱系统
由图1可得以下结论:
(1)甲醇-水:0-33min前基本无峰出现,所以峰都集中在后面,且分离不好。
(2)甲醇-0.1%乙酸水:基线不平稳,所以峰都集中在后面,分离不好,且大多峰有拖尾现象。在0-5min内有色谱峰出现,后仅出现几个小峰,包括倒峰。
(3)乙腈-水:20min之前基线不平稳,与乙腈-0.1%磷酸水溶液比较,明显后者优于前者。
(4)乙腈-0.1%磷酸水溶液水:色谱峰多,且峰形对称,大部分峰达到基线分离。
结论:流动相最终确定乙腈-0.1%磷酸水溶液进行梯度洗脱。
2.1.3流动相梯度洗脱程序的选择
本实验通过调整乙腈-0.1%磷酸水溶液溶液的比例,选择3种流动相梯度洗脱进行比较:①(t:0~60b%:5~100)、②(t:0~3~53b%:15~24~49)、③(t:0~5~25~45b%:5~20~35~60),t为洗脱时间,b为乙腈。hplc图谱见图2。
结论:由图2中的3种不同洗脱梯度得出的色谱图看,明显梯度③的效果较①②好,最终确定的洗脱梯度为③(t:0~5~25~45b%:5~20~35~60),测定达到较良好的色谱图。供试品中主要色谱峰在洗脱时间内已经分离,当乙腈比例大于60%后已无色谱峰出现,因此,乙腈最高比例为60%,洗脱时间确定为45min。
2.1.4检测波长的选择
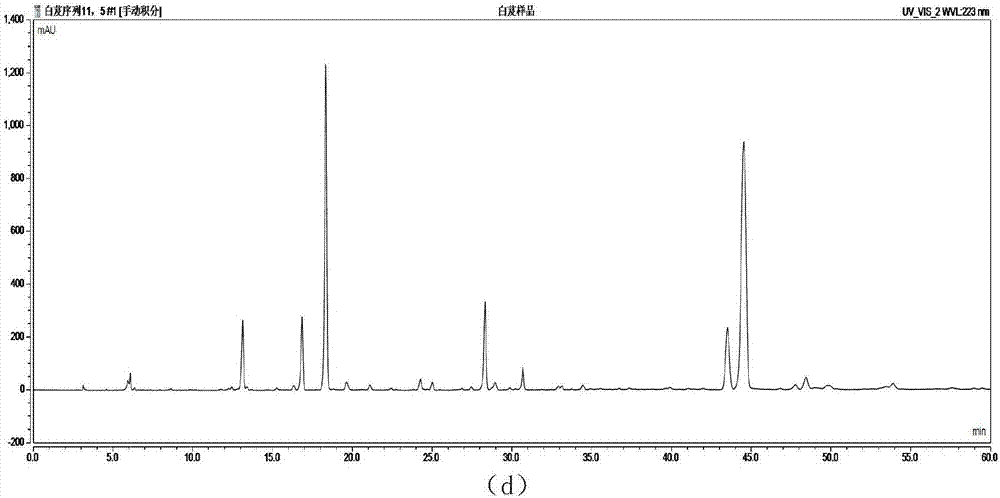
在实验的基础上,我们在建立中药指纹图谱时选择检测波长的原则和思路:(1)对于各种有效成分研究得比较透彻,药效成分已知的中药,应选择药效成分的最大吸收波长作为检测波长;(2)对于有效成分还不明确和未知的中药,应选择色谱峰出峰数目最多的检测波长。本实验利用紫外可见分光光度计全波长扫描功能对样品试液的较大吸收波长进行选择,选择了223nm、270nm2个波长进行测定。见图3。
由图2可以看出:
(1)在223nm检测波长下,出峰的数目减少。
(2)在270nm检测波长下,吸收峰较多,各峰吸收较好,峰形好,峰面积大。
结论:比较两波长来看,两者都有溶剂峰,但在270nm波长下,出峰更多。峰形好,峰面积大。所以选择波长270nm作为白及药材hplc指纹图谱的检测波长。
2.1.5柱温的考察
对于液相色谱来说,柱温高可加快分离过程,但分辨率可能下降。柱温低,分辨率提高,但分离过程时间加长。现对25℃、30℃和35℃进行考察,见图4。
由图.3可以得知:30℃时色谱峰出峰较多,且峰形对称,达到分离要求;25℃时色谱峰数目与30℃时一样,但25℃时在27-32min之间分离不好;35℃时,在27-32min之间出峰数减少。所以最终选择柱温为30℃。
3提取方法的优选
3.1提取方法的比较
本实验先后比较了用回流法、超声法提取的供试品溶液的色谱峰。超声法为精密称定白及药材粉末(过三号筛50目)1g,精密加入分析甲醇20ml,超声30min,回流法为精密称定白及药材粉末(过三号筛50目)1g,精密加入分析甲醇20ml,回流30min(保持微沸状态,70℃),放冷,称重,补足失重,滤过,取续滤液适量,以0.45μm微孔滤膜滤过,即得。所得色谱图见图5。
结论:从色谱图中可以看出,回流法和超声法出峰数目一样,分离效果也差不多,但在10-15min之间的两个峰的面积超声法明显大于回流法,而且出于考虑超声提取法操作较回流提取法更简便、节省时间,所以优选超声提取法。
3.2提取时间的考察
为了节约时间与能源,经查阅相关文献资料,本实验先后选择了不同的提取时间(20min、30min、45min、)进行提样,比较其色谱峰数。见图6。
结论:根据图5可以看出:三个图谱相比较,出峰数目一样,但20min和45min的图谱中色谱峰均在27-31min分离效果较30min的图谱中相应位置的峰的分离效果差,所以选择提取时间为30min。
3.3色谱条件的确定
通过上述实验结果,确定最终色谱条件为:色谱柱为diamonsilc18(250mm×4.6mm,5μm),流动相洗脱程序如表2.2所示,检测波长为270nm,流速为1ml·min-1,柱温30℃,进样量10μl,洗脱时间为45min。提取方法为:精密称取白及药材粉末(过三号筛50目)1g,加入分析甲醇20ml,称定重量,超声提取30min,放冷,用分析甲醇补足减失的重量,以0.45μm微孔滤膜过滤,即得。此条件下,测得白及药材样品的hplc谱图(见图8)。用1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯(militarine)对照品对其进行对照通过保留时间定性可以看出20.127min的色谱峰是1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯(militarine)(见图7)。
4溶液的制备
4.1对照品溶液的制备
精密称取1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯(militarine)对照品6.80mg,置10ml棕色容量瓶中,用乙腈和0.1%磷酸水溶液以2:3的比例混合溶解并稀释至刻度,摇匀。即得0.680mg·ml-1的对照品溶液。
4.2供试品溶液的制备
精密称取白及药材粉末(过三号筛50目)1g,置100ml具塞锥形瓶中,加入20ml分析甲醇,称定重量,超声提取30min,取出,放冷,用分析甲醇补足减失的重量,摇匀,过滤,以0.45μm微孔滤膜过滤,即得供试品溶液。
5方法学考查
5.1精密度试验
精密称取来自贵州省贵阳市花果园药材市场銧铭药行的白及药材粉末1g,依照4.2项下制备供试品溶液,按3.3项下确定的色谱条件进行测定,连续进样6次并记录色谱图。直观观察指纹图谱的全貌无明显差别。指纹图谱见图9。采用国家食品药品监督管理局推荐的“中药色谱指纹图谱相似度评价软件”进行评价,得各色谱图的相关系数(取平均数)。相关系数见表5.1。militarine含量较高,色谱峰峰形好,将其设为参照峰,设其相对保留时间和峰面积为1,计算各共有峰对它的相对峰面积和相对保留时间的rsd值。结果见表5.1.1和5.1.2。
表5.1精密度试验相似度结果
由表5.1可以看出,精密度实验的相关系数(取平均数)均大于0.95,相似度计算结果显示该仪器的精密度良好。符合指纹图谱的检测要求。
表5.1.1精密度试验白及药材共有峰的相对面积及rsd值
表5.1.2精密度试验白及药材共有峰的相对保留时间及rsd值
由表5.1.1和5.1.2知,精密度试验白及药材特征图谱共有峰的相对峰面积和相对保留时间的rsd值分别在(0.32~4.94,0.00~1.12)之间,说明该仪器的精密度良好。
5.2稳定性试验
精密称取来自贵州省贵阳市花果园药材市场銧铭药行的白及药材粉末1g,依照4.2项下制备供试品溶液,分别在0、3、5、9、12、15、24h按照3.3项下确定的色谱条件进行测定,直观观察指纹图谱的全貌无明显差别。指纹图谱见图10。
采用国家食品药品监督管理局推荐的“中药色谱指纹图谱相似度评价软件”进行评价,得各色谱图的相关系数(取平均数)。相关系数见表5.2。将militarine设为参照峰,设其相对保留时间和峰面积为1,计算各共有峰对它的相对峰面积和相对保留时间的rsd值。结果见表5.2.1和表5.2.2。
表5.2稳定性试验相似度结果
由表5.2可以看出,各指纹图谱的相关系数(取平均数)均大于0.95,结果表明24小时内样品溶液的成分是稳定的。符合指纹图谱的检测要求。
表5.2.1稳定性试验白及药材共有峰的相对峰面积及rsd值
表5.2.2稳定性试验白及药材共有峰的相对保留时间及rsd值
由表5.2.1和5.2.2知,稳定性试验白及药材特征图谱共有峰的相对峰面积和相对保留时间的rsd值分别在(0.12~4.55,0.00~0.44)之间,说明24小时内样品溶液的成分是稳定的。
5.3重复性试验
精密称取来自贵州省贵阳市花果园药材市场銧铭药行的白及药材粉末1g,6份,依照4.2项下制备供试品溶液,按3.3项下确定的色谱条件进行测定,直观观察指纹图谱的全貌无明显差别。指纹图谱见图11.采用国家食品药品监督管理局推荐的“中药色谱指纹图谱相似度评价软件”进行评价,得各色谱图的相关系数(取平均数)。相关系数见表5.3,将militarine设为参照峰,设其相对保留时间和峰面积为1,计算各共有峰对它的相对峰面积和相对保留时间的rsd值。结果见表5.3.1和5.3.2
表5.3重复性试验相似度结果
由表5.3可以看出,重复性实验的相关系数(取平均数)均大于0.95,相似度计算结果显示该方法的重复性很好。符合指纹图谱的检测要求。
表5.3.1重复性试验白及药材共有峰的相对峰面积及rsd值
表5.3.2重复性试验白及药材共有峰的相对保留时间及rsd值
由表5.3.1和5.3.2知,重复性试验白及药材特征图谱共有峰的相对峰面积和相对保留时间的rsd值分别在(1~4.61,0.06~0.19)之间,说明该方法的重复性好。
6十批白及药材的hplc指纹图谱的相似度
将十批白及药材的色谱数据导入指纹图谱相似度评价系统,用平均数法对相似性进行分析,手动匹配后得到数据及图谱(见表6、见图12)。将militarine设为参照峰,设其相对保留时间和峰面积为1,计算各共有峰对它的相对峰面积和相对保留时间的rsd值。结果见表6.1和6.2。
表610批白及药材hplc指纹图谱相似性分析结果
由表6可见,各指纹图谱的相关系数均大于0.9,即十批药材与对照指纹图谱的相似性良好,说明由平均数法得到的共有模式图具有代表性,可反映白及药材的指纹特征。
表6.1十批白及药材共有峰的相对峰面积及rsd值
表6.2十批白及药材共有峰的相对保留时间及rsd值
由表6.1和6.2知,十批白及药材共有峰相对保留时间的rsd值小(0.00~0.13),但相对峰面积的rsd值较大,说明十批白及药材特征图谱中的主要成分基本一致,但各成分含量区别较大。
6.1十批白及药材的hplc指纹图谱共有峰
根据10批白及药材指纹图谱的检测结果,进行分析比对,总结出指纹图谱中的共有峰有15个(见图13),其中1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯(militarine)(4号峰)对照品的色谱峰明显,且达到了基线分离。部分特征峰的峰形为:1、2、3、5、7、8、9、11、12、15号峰也达到了基线分离;6,10,号峰均由未完全分离的两个峰组成;13,14号峰为未完全分离的色谱峰;其中,8号峰最高,4号峰仅次于8号峰。
7结果
本实验选取10批不同产地的白及药材进行hplc指纹图谱研究,首先采用“中药色谱指纹图谱相似度评价软件”2004年a版(国家药典委员会)对10批不同产区的药材进行相似度评价,建立hplc指纹图谱共有模式,结果10批白及药材的相似性无明显差异,10批药材特征图谱中共有峰的相对保留时间的rsd值较小,而相对峰面积的rsd值较大,说明本发明方法能较为全面地反映白及药材中所含化学成分的种类与数量,进而对其质量进行整体描述和评价,可将白及药材质量与其药效真正结合起来,有助于阐明其作用机理为白及药材的技术提升与深度开发提供了依据。且用该方法检测到的白及药材指纹图谱化学成分相对较多、各特征峰高度比例适中,基线较平稳,分离度、峰形和柱效好。
与现有技术相比,本发明方法可快速、准确地鉴别产品的真伪优劣,具有方法简便、稳定、精密度高、重现性好等优点;用该方法还能方法能较为全面地反映白及药材中所含化学成分的种类与数量,进而对其质量进行整体描述和评价,可将白及药材质量与其药效真正结合起来,有助于阐明其作用机理为白及药材的技术提升与深度开发提供了依据。且用该方法检测到的白及药材指纹图谱化学成分相对较多、各特征峰高度比例适中,基线较平稳,分离度、峰形和柱效好。
附图说明
图1是4种流动相系统对白及分离情况色谱图:(a)甲醇-水;(b)甲醇-0.1%磷酸水溶液;(c)乙腈-水;(d)乙腈-0.1%磷酸水溶液;
图2是不同洗脱程序对百级分离情况色谱图:(a)洗脱程序①;(b)洗脱程序②;(c)洗脱程序③;
图3是供试液不同检测波长下色谱图:(a)波长为223nm;(b)波长为270nm;
图4是不同柱温色谱图:(a)(柱温为25℃),(b)(柱温为30℃),(c)(柱温为35℃);
图5是不同提取方法色谱图比较:(a)超声提取;(b)回流提取;
图6是不同提取时间色谱图比较:(a)20min;(b)30min;(c)45min;
图7是对照品1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯(militarine)的hplc色谱图;
图8是白及供试品的hplc色谱图;
图9是精密度试验hplc指纹图谱的叠加图;
图10是稳定性试验hplc指纹图谱的叠加图;
图11是重复性试验hplc指纹图谱的叠加;
图12是10批药材的指纹色谱图叠加图;
图13是10批白及药材(炮制后)指纹图谱的共有模式图(取平均数)。
下面结合实施例对本发明作进一步的说明。
具体实施方式
实施例1:
一种白及药材hplc指纹图谱的建立方法,包括以下步骤:
(1)对照品溶液这样制备:精密称取1,4-二[4-(葡萄糖氧)苄基]-2-异丁基苹果酸酯(militarine)对照品6.80mg,置10ml棕色容量瓶中,用乙腈和0.1%磷酸水溶液以2:3的比例混合溶解并稀释至刻度,摇匀,即得0.680mg·ml-1的对照品溶液;
(2)供试品溶液这样制备:精密称取白及药材粉末1g,置100ml具塞锥形瓶中,加入20ml分析甲醇,称定重量,超声提取30min,取出,放冷,用分析甲醇补足减失的重量,摇匀,过滤,以0.45μm微孔滤膜过滤,即得供试品溶液;
(3)色谱条件为:色谱柱为diamonsilc18(250mm×4.6mm,5μm),流动相:乙腈(a)-水(b),检测波长为270nm,流速为1ml·min-1,柱温30℃,进样量10μl,洗脱时间为45min;所述洗脱程序为:0~5min,90-95%b;5~25min,80~90%b;25~40min,80~65%b,40~45min,65~30%b;
(4)按上述方法制备供试品溶液,在上述色谱条件下进样,记录谱图得到白及药材的hplc指纹图谱;
(5)指纹谱图的确认:按照上述提供的方法,对多批次白及药材建立了hplc指纹图谱,通过分析比较确定了15个共有峰,这些共有峰构成了白及药材的指纹特征,作为白及药材的指纹图谱;
所述15个共有峰,以4号峰为参照峰,其余峰相对保留时间分别为:
0.422-0.423,0.641-0.642,0.687-0.689,0.920-0.921,1.395-1.397,1.459-1.461,1.579-1.580,1.626-1.628,1.661-1.662,1.765-1.768,1.869-1.871,1.913-1.915,1.972-1.974,2.012-2.015,2.038-2.042。
- 还没有人留言评论。精彩留言会获得点赞!