一种芦荟制品中主要化学成分的质量评价的方法与流程
本发明涉及药材及其制品质量标准领域,更具体地,涉及芦荟制品中主要化学成分的质量评价的方法。
背景技术:
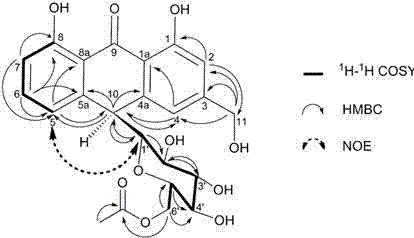
:蒽醌和蒽酮是库拉索芦荟中的主要化学成分,芦荟的基本生物活性均来源于蒽醌和蒽酮类成分,同时蒽醌和蒽酮类成分也是芦荟的毒性成分,由此该类成分是芦荟制品的功效以及毒性的标志性成分。长期以来芦荟中的蒽醌和蒽酮类成分一直是以芦荟苷为代表,并以芦荟苷a单一成分的量进行制品的质量控制,如《中国药典》2010版、食品原料用芦荟制品qb/t2489-2007、化妆品用芦荟汁、粉qb/t2488-2006以及国际芦荟科学协会使用的芦荟制品标准均是如此。而我们的研究表明芦荟中的蒽醌和蒽酮类成分众多,并且功能与毒性相差很大,如仅以芦荟苷a一种成分对芦荟制品进行质量控制目前已不能对芦荟制品的有效性、安全性进行科学全面的质量评价和对芦荟制品的生产过程进行科学控制与指导。此外,芦荟苷在溶液状态下很不稳定,最近我们的研究表明芦荟苷溶解于水后会立即发生分解与聚合,在ph7.11,室温条件下600ug/ml浓度的芦荟苷溶液1天后,芦荟苷降解了近50%,5天降解90%以上,降解产物经我们分析鉴定为芦荟大黄素与elgonica-dimer,经细胞毒理mtt实验,表明后二者毒性远大于芦荟苷,也就是说,芦荟提取物配成水溶液放置后芦荟苷的量在不断降低,芦荟大黄素与elgonica-dimer的量在不断增加,芦荟制品在生产过程中由于生产工艺不同,生产条件不同均会造成制品中所含蒽醌和蒽酮的成分种类与含量不同,因此仅仅测定芦荟苷的含量无法满足对制品有效性和安全性的质量控制。药理药效研究方面多为库拉索芦荟整体药材入药,但是不同的化合物、不同的官能团将赋予库拉索芦荟化学成分不同的物理化学性质、药理药效特征及可能的毒性。对于芦荟制品,科学可靠的质量控制方法应该同时测定其中芦荟苷和芦荟苷的分解与聚合产物以及相关蒽醌和蒽酮,这样才能确保芦荟制品的安全性、有效性。技术实现要素:本发明旨在克服上述现有技术无法满足对芦荟制品有效性和安全性的质量控制,提供一种芦荟制品中主要化学成分的质量评价的方法。为解决上述技术问题,本发明采用的技术方案是:一种芦荟制品中主要化学成分的质量评价的方法,通过检测芦荟样品中芦荟苷a、芦荟苷b、芦荟糖甙a、芦荟糖甙b、6’-乙酰化芦荟苷b、6’-乙酰化芦荟苷b和芦荟大黄素的含量总值来确定芦荟制品中蒽醌和蒽酮的总含量,从而评价芦荟制品的质量,所述芦荟为库拉索芦荟。同时测定库拉索芦荟制品中共有的、含量较高的7种蒽醌和蒽酮的总含量,相对于仅测量芦荟苷的含量,可以更全面地对芦荟制品中稳定存在的和分解与聚合后产生的蒽醌和蒽酮类成分含量进行检测,从而对芦荟制品的有效性、安全性进行更科学有效的质量评价。所述库拉索芦荟制品为芦荟叶皮粉或芦荟全叶烘干粉或芦荟全叶冻干粉或芦荟药材粉或芦荟精粉或芦荟凝胶冻干粉。进一步地,芦荟样品中蒽醌和蒽酮的总含量采用高效液相色谱测定。进一步地,根据对照品溶液峰面积绘制标准曲线,并根据标准曲线计算得到芦荟制品中蒽醌和蒽酮的总量。高效液相检测的色谱条件为:色谱柱为填料为5μmc18柱;梯度洗脱程序为:0~30min,40%~70%甲醇;30~32min,70%~80%甲醇;32~38min,40%甲醇;检测波长:芦荟大黄素225nm,其余6种蒽醌356nm;柱温:40℃;流速:1ml/min;进样量:10μl。流动相乙腈-水对芦荟苷b的分离度不及甲醇-水,并且酸水体系对峰形的改善效果不显著,故优选以甲醇-水为流动相。对于检测波长,芦荟大黄素属蒽醌类化合物,在225nm,256nm,286nm及429nm附近有最大吸收,其他6种化合物属蒽酮类化合物,在269nm,297nm和356nm附近有最大吸收。由于芦荟化学成分复杂,除蒽醌和蒽酮类化合物外还包含色酮等化合物,而色酮类化合物在225nm,249nm和303nm附近有最大吸收。因此,而对于芦荟大黄素,由于其在各类样品中含量均较低,为提高其检测的灵敏度,选择以225nm作为其检测波长,其他6种化合物为减少干扰,选择356nm作为检测波长。进一步地,所述色谱柱的规格为4.6mm×250mm或者是近似的尺寸。芦荟样品的制备为将芦荟样品粉加入适量甲醇,超声提取30min后经0.45μm微孔滤膜过滤得到。检测中用到的芦荟苷a、芦荟苷b、芦荟糖甙a、芦荟糖甙b、6’-乙酰化芦荟苷a、6’-乙酰化芦荟苷b和芦荟大黄素7种对照品中芦荟苷a和芦荟大黄素为购买得到,其他对照品均为库拉索芦荟通过自制得到。具体自制方法是通过高速逆流设备提取分离,再通过核磁共振、紫外、红外和高分辨质谱结构鉴定自制得到。其中6’-乙酰化芦荟苷a和6’-乙酰化芦荟苷b为提取发现的新化合物,其化学结构如下:其中6’-乙酰化芦荟苷a的r1=6’-acetyl-c-β-d-glc,r2=r3=h;6’-乙酰化芦荟苷b的r1=r3=h,r2=6’-acetyl-c-β-d-glc。其中,acetyl是乙酰基,c-β-d-glc为碳糖苷及其键型与糖的构型。与现有技术相比,本发明的有益效果是:相对于只检测芦荟苷a含量来代表芦荟制品中的蒽醌和蒽酮总含量,本发明还对芦荟苷分解聚合产生的含量较高、多数芦荟制品中所共有的芦荟苷b、芦荟糖甙a、芦荟糖甙b、6’-乙酰化芦荟苷b、6’-乙酰化芦荟苷b和芦荟大黄素蒽醌和蒽酮类成分进行检测得到芦荟制品中蒽醌和蒽酮的总含量,更为准确合理,方法学验证表示测定方法稳定性高、准确度好、回收率高,能够为芦荟产品质量更科学有效的控制评价提供新的标准。附图说明图1为7种对照品及库拉索芦荟精粉的hplc色谱图,其中a为对照品的hplc色谱图,b为356nm检测波长下的库拉索芦荟精粉的hplc色谱图,c为225nm波长下的库拉索芦荟精粉的hplc色谱图。图2为6’-乙酰化芦荟苷a的结构及重要的1h–1hcosy,hmbc和noe相关信号。图3为6’-乙酰化芦荟苷a的高分辨质谱图。图4为6’-乙酰化芦荟苷a的红外光谱图。图5为6’-乙酰化芦荟苷a的核磁共振氢谱图(cd3od)。图6为6’-乙酰化芦荟苷a的核磁共振碳谱图(cd3od)。图7为6’-乙酰化芦荟苷a的1h–1hcosy谱图(cd3od)。图8为6’-乙酰化芦荟苷a的hsqc谱图(cd3od)。图9为6’-乙酰化芦荟苷a的hmbc谱图(cd3od)。图10为6’-乙酰化芦荟苷a的noesy谱图(cd3od)。图11为6’-乙酰化芦荟苷b的结构及重要的1h–1hcosy,hmbc和noe相关信号。图12为6’-乙酰化芦荟苷b的高分辨质谱图。图13为6’-乙酰化芦荟苷b的红外光谱图。图14为6’-乙酰化芦荟苷b的核磁共振氢谱图(cd3od)。图15为6’-乙酰化芦荟苷b的核磁共振碳谱图(cd3od)。图16为6’-乙酰化芦荟苷b的1h–1hcosy谱图(cd3od)。图17为6’-乙酰化芦荟苷b的hsqc谱图(cd3od)。图18为6’-乙酰化芦荟苷b的hmbc谱图(cd3od)。图19为6’-乙酰化芦荟苷b的noesy谱图(cd3od)。具体实施方式下面结合具体实施方式对本发明作进一步的说明,但实施例并不对本发明做任何形式的限定。除非另有说明,本发明实施例采用的原料试剂为常规购买的原料试剂。仪器与试药:lc-20at型高效液相色谱仪(hplc)(日本岛津公司),配备lc-20ad二元泵、sil-20a自动进样器及uv-vis双波长检测器;sb25-12d型超声波清洗器(宁波新芝生物科技股份有限公司);ab135-s型十万分之一天平(德国瑞士梅特勒公司);色谱甲醇(韩国skchemicals公司);超纯水(purelabultra超纯水系统,英国elga公司);所有其他试剂均为分析纯(天津大茂化学试剂厂);芦荟苷a标准品(成都曼思特生物科技有限公司);芦荟大黄素对照品(上海同田生物技术有限公司);其他对照品为自制,hplc检测为单一峰,面积归一化法计算纯度大于98.0%;不同批次的库拉索芦荟样品均为市售产品。实施例1精密称取库拉索芦荟叶皮粉1g,置于25.0ml锥形瓶中加入规定量甲醇,超声提取30min,冷却至室温后以甲醇补足原重,经0.45μm微孔滤膜过滤,取滤液待测;分别精密称取7种对照品适量,置容量瓶中,以甲醇定容,配成浓度分别为1.608、1.624、1.025、0.425、1.125、0.484、0.156mg/ml的储备液,取适量储备液,加入甲醇逐步稀释得到一系列浓度的工作液:1.芦荟苷b:20.100、40.200、100.500、201.000、301.500、502.500μg/ml;2.芦荟苷a:20.300、40.600、101.500、203.000、304.500、507.500μg/ml;3.芦荟糖甙b:5.125、10.250、25.625、51.250、76.875、128.125μg/ml;4.6’-乙酰化芦荟苷b:1.062、2.125、5.312、10.625、15.938、26.562μg/ml;5.芦荟糖甙a:5.625、11.250、28.125、56.250、84.375、140.625μg/ml;6.6’-乙酰化芦荟苷a:0.968、1.936、4.840、9.680、14.520、24.200μg/ml;7.芦荟大黄素:0.195、0.390、0.975、1.950、2.925、4.875μg/ml。将对照品和芦荟样品分别注入高效液相色谱检测,色谱条件为:色谱柱为4.6mm×250mm、5μm的aglienttc-c18柱;梯度洗脱程序为:0~30min,40%~70%甲醇;30~32min,70%~80%甲醇;32~38min,40%甲醇;检测波长:芦荟大黄素225nm,其余6种蒽醌356nm;柱温:40℃;流速:1ml/min;进样量:10μl。根据对照品溶液峰面积绘制标准曲线,并根据标准曲线计算得到7种蒽醌和蒽酮的总量,分别检测了两个批次的芦荟叶皮粉。式中:xi为样品中蒽醌i的含量,单位为毫克每克(mg/g);ai为色谱图中蒽醌i的峰面积;bi为蒽醌i回归方程中的截距值;ki为蒽醌i回归方程中的斜率;v为样品提取时所用的甲醇的体积,单位为毫升(ml);m为称取样品的质量,单位为克(g)。对新化合物对照品6’-乙酰化芦荟苷a和6’-乙酰化芦荟苷b的结构鉴定如下:6’-乙酰化芦荟苷a的理化性质、光谱数据及归属:本品为黄色无定形粉末,能溶于水,易溶于甲醇,不溶于氯仿。比旋度[α]20d=+5.2°(c0.63,meoh);uv(meoh)λmax:269、297、356nm;ir(kbr)νmax:3423(br)、2920、2872、1728、1622、1236、1101、1032cm–1;positiveesimsm/z461.08[m+h]+;hresimsm/z483.1285[m+na]+(c23h24o10na,计算值483.1262);1h-nmr(cd3od,400mhz)和13c-nmr(cd3od,100mhz)数据见表1,光谱图如图2,高分辨质谱图如图3,红外光谱图如图4,核磁共振氢谱图如图5,核磁共振碳谱图如图6,1h–1hcosy谱图如图7,hsqc谱图如图8,hmbc谱图如图9,noesy谱图如图10。检测发现其生物活性与芦荟苷相近。6’-乙酰化芦荟苷b的理化性质、光谱数据及归属:本品为黄色无定形粉末,能溶于水,易溶于甲醇,不溶于氯仿。比旋度[α]20d=–53.8°(c0.66,meoh);uv(meoh)λmax:269,297,356nm;ir(kbr)νmax:3403(br),2876,1721,1612,1286,1233,1069cm–1;positiveesimsm/z461.05[m+h]+;hresimsm/z483.1279[m+na]+(c23h24o10na,计算值483.1262);1h-nmr(cd3od,400mhz)和13c-nmr(cd3od,100mhz)数据见表1,光谱图如图11,高分辨质谱图如图12,红外光谱图如图13,核磁共振氢谱图如图14,核磁共振碳谱图如图15,1h–1hcosy谱图如图16,hsqc谱图如图17,hmbc谱图如图18,noesy谱图如图19。检测发现其生物活性与芦荟苷相近。表16’-乙酰化芦荟苷a和6’-乙酰化芦荟苷b的1h-nmr和13c-nmr数据(400mhz,δinppm,jinhz).实施例2实施例2与实施例1的区别在于库拉索芦荟样品为芦荟全叶烘干粉,检测了3个批次的芦荟全叶烘干粉。实施例3实施例3与实施例1的区别在于将0.25g芦荟全叶冻干粉置于50.0ml锥形瓶制备待测样品,检测了3个批次的芦荟全叶冻干粉。实施例4实施例4与实施例1的区别在于将0.25g芦荟药材粉置于50.0ml锥形瓶制备待测样品,检测了3个批次的芦荟药材粉。实施例5实施例5与实施例1的区别在于0.15g芦荟精粉置于50.0ml锥形瓶制备待测样品,检测了2个批次的芦荟精粉。实施例5实施例6与实施例1的区别在于芦荟制品为芦荟凝胶冻干粉,检测了3个批次的芦荟凝胶冻干粉。实施例1-5的检测结果如下表2所示:表2表不同的库拉索芦荟样品中7种蒽醌和蒽酮类化合物的含量从上表数据可以看出芦荟苷a、芦荟苷b、芦荟糖甙a、芦荟糖甙b、6’-乙酰化芦荟苷b、6’-乙酰化芦荟苷b和芦荟大黄素为不同芦荟制品中共有成分,其含量相加可以跟准确的检测芦荟制品中的蒽醌和蒽酮总含量。对本发明提供的7种相关蒽醌和蒽酮的多成分定量测定方法进行了方法学验证:1.标准曲线和线性范围按上述高效液相色谱条件,分别对不同浓度的工作液进行分析,重复测定3次,取峰面积平均值,分别以7种化合物的浓度(μg·ml-1)为横坐标(x),峰面积平均值为纵坐标(y),进行线性回归计算。7种对照品的回归方程、线性范围及相关系数具体结果见表3。2.日内精密度分别取高、中、低浓度的供试品,按上述高效液相色谱条件,在同一天连续进样6次,记录色谱图,以峰面积计算rsd。7种化合物的日内测定的rsd均小于3%,符合要求,具体结果见表4。3.日间精密度分别取高、中、低浓度的供试品,按上述高效液相色谱条件,连续三天进样,记录色谱图,以峰面积计算rsd。7种化合物的日间测定的rsd均小于3%,符合要求。具体结果见表4。4.稳定性精密称取同一批库拉索样品,制备供试品溶液,分别于0、2、4、8、12、24h进样进行检测,计算供试品中7种目标化合物的含量,并计算rsd值。稳定性实验结果表明,供试品溶液在4oc时,在24h内稳定性良好,见表5。5.回收率精密称取已知含量的库拉索芦荟样品,分别精密加入高、中、低浓度的混合对照品溶液,制得供试品溶液,进样分析,按下列公式计算7个蒽醌的加样回收率,以评估方法的准确度。加样回收率结果见表6。结果表明,7种化合物的平均加样回收率在96.0%~104.3%范围内,rsd值均小于3%,表明方法的准确度良好。回收率(%)=(测得量-初始量)/加入量×100%表37种化合物的标准曲线的回归方程线性范围及相关系数表4方法的日内及日间精密度(%rsd)表5稳定性试验结果(n=6)化合物rtrsd(%)含量(mg/g)rsd(%)10.940.023.74791.6421.000.004.79921.9131.120.031.46071.5341.170.050.19061.3651.230.041.23091.4861.270.080.09882.2471.590.100.03361.48表6加样回收率试验(n=3)化合物原含量(μg)加入量(μg)测得量(μg)回收率(%)rsd(%)12670.51810.54484.6100.21.02681.13621.06270.799.10.62654.55431.58194.4102.01.023652.31828.55472.099.51.93666.93657.07396.5102.01.83630.55485.58998.797.91.23833.4376.71226.2104.30.4836.7753.41611.2102.80.3828.41130.01996.3103.42.04126.453.1179.7100.42.1126.9106.3234.9101.71.8125.6159.4287.4101.51.65693.6413.21121.6103.61.3696.4826.51521.099.82.4689.41239.71979.0104.00.8677.237.8114.298.02.577.575.6150.096.01.376.7113.4186.696.91.6719.09.828.698.32.619.119.538.599.22.318.929.348.099.31.2当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1