一种基于固相法分离N-糖肽和O-糖肽的方法与流程
本发明涉及生物分子分析试剂技术领域,具体涉及一种基于固相法分离n-糖肽和o-糖肽的方法。
背景技术:
:
蛋白质糖基化的分析通常包括多糖的组成和结构、糖基化位点和糖肽序列分析。基质辅助激光解吸电离-飞行时间质谱(maldi-tof)和液相色谱-电喷雾电离质谱(lc-esi)是蛋白质糖基化研究的主要分析方法。蛋白质糖基化分为n-糖基化和o-糖基化两种。n-糖基化发生在n-x-s/t(x=除脯氨酸,丝氨酸和苏氨酸以外的任何氨基酸)序列中的天冬酰胺(n)上。n-多糖可以用糖苷水解酶(例如pngasea和pngasef)从蛋白上切割下来进行进一步的研究。o-糖基化发生在没有特定序列规则的丝氨酸和苏氨酸上。
自然界尚未发现一种通用的糖苷水解酶可以将o-多糖从蛋白上切割下来。因此,o-多糖一般是用碱性化学方法处理,将之从蛋白上解离出来。在这种情况下,会产生过多的副反应,严重影响对o-多糖的组成和结构的分析。但是,最近发现了一些内切蛋白酶,可以识别o-多糖位点,并在o-多糖位点切割糖肽,有助于o-糖基化分析、o-多糖谱解析、o-聚糖位点测定等。例如,operator(o-糖肽切割酶)是一种内切蛋白酶,该酶源自黏液阿克曼(akkermansiamuciniphila),并在大肠杆菌中表达。该酶含有his标签,分子量为42kda。可催化天然黏蛋白型o-糖基化蛋白中与o-聚糖直接相邻的肽键的水解。operator高度特异性地在丝氨酸或苏氨酸的o-多糖n端消化蛋白质,但是不会对带有n-多糖的天冬酰胺消化。同时,在对该酶的活性位点进行变异后,能够保留识别o-多糖位点的亲和力,但是失去了对o-糖肽进行切割的能力。(yang,s.;onigman,p.;wu,w.w.;sjogren,j.;nyhlen,h.;shen,r.-f.;cipollo,j.anal.chem.2018,90,8261-8269)。本发明利用了变异后的operator的这种能力,实现了o-糖肽的特异性富集。
技术实现要素:
本发明的目的在于提供一种基于固相法分离n-糖肽和o-糖肽的方法,以解决现有技术中将o-糖肽从蛋白上解离出来时会产生副反应的缺陷。
一种基于固相法分离n-糖肽和o-糖肽的方法,所述方法包括如下步骤:
将糖蛋白偶联到固相树脂上;
通过衍生化试剂对固相在树脂上的糖蛋白唾液酸进行衍生化并清洗,加入胰蛋白酶降解处理后,得到多肽混合物;
将获得的多肽溶解后,对n-糖肽和o-糖肽进行分离。
进一步的,将糖蛋白偶联到固相树脂上的方法包括如下步骤:
将糖蛋白溶解在0.3~2.0ml的缓冲液a中,加入醛基树脂并在室温下混合2-6小时;
添加10-30μl1mnacnbh3后继续混合2-6小时;
用300-600μl缓冲液b冲洗两次并进行烷基化处理。
进一步的,所述烷基化处理的方法包括如下步骤:
用含有5~15mmdtt,2-10m尿素,0.5-2m碳酸氢铵的溶液进行还原处理;
加入10~20mm的碘乙酰胺并在20~50℃和避光环境中振荡0.5~2.0个小时。
进一步的,通过衍生化试剂对固定在树脂上的糖蛋白唾液酸进行衍生化并清洗,其具体步骤为:
用200~300mm1-羟基苯并三唑水合物和200~300mmedc.hcl(n-(3-二甲基氨基丙基)-n-乙基碳二亚胺盐酸盐)的乙醇溶液,在37℃恒温与固定在树脂上的糖蛋白唾液酸反应1小时;
分别用100%乙醇,1mnacl和hplc纯净水将样品洗涤三次。
进一步的,在对固定在树脂上的糖蛋白唾液酸进行衍生化并清洗后,加入胰蛋白酶降解处理后去除溶剂,得到多肽的方法包括如下步骤:
用1m氯化钠,10%乙腈溶液冲洗后,加入胰蛋白酶在碱性环境下通过100mm碳酸氢铵和1m尿素溶液的降解固相化的糖蛋白;
收集产生的多肽后,采用亲水色谱柱对糖肽(n-糖肽和o-糖肽的混合物)进行富集并用乙腈的水溶液(40%乙腈,60%水)清洗;
用离心干燥仪除去溶剂得到多肽。
进一步的,将获得的多肽溶解后,对n-糖肽和o-糖肽进行分离的方法包括如下步骤:
将多肽溶解在300-500μl缓冲液c中与树脂混合后振荡1小时,然后在600~1200rpm离心1-5min,收集上层清液;
用300-800μl的缓冲液b中对树脂进行了二次重洗;
将洗脱液汇集,并用反相c18纯化,用离心干燥仪除去溶剂并重新溶解在50μl的0.2%甲酸溶液中,获得n-糖肽;
分别用1mnacl500μl、10%乙腈溶液500μl和纯水500μl对树脂进行洗涤;
将400-500μl含有100单位的o-糖肽切割酶的20mmnh4hco3溶液和树脂混合后在37℃振荡8-15小时后,离心取上层清液;
用200-400μl10%乙腈溶液清洗2-4次,并洗脱液收集在一起;
用离心干燥仪除去洗脱液中的溶剂,重新溶解在0.2%的甲酸溶液,获得o-糖肽。
进一步的,所述缓冲液a包括10mm柠檬酸钠,50mm碳酸钠,其ph=10;
所述缓冲液b包括50mm的pbs,其ph=7.4;
所述缓冲液c包括50mm的nh4hco3溶液,其ph=8.0。
进一步的,所述固相树脂为亲水性固体材料,包括硅胶、琼脂糖和表面包被亲水性基团的磁珠中的一种或多种组合。
本发明的优点在于:该种基于固相纯化的方法可以采用过量衍生试剂的方法实现100%的唾液酸衍生化,降低常见的副反应,从而高效分离n-糖肽和o-糖肽,减少不含糖基修饰的多肽背景污染,有助于精确识别n-糖基化和o-糖基化位点,在下列各种场景(但不仅限于这些场景)有广泛应用:
(1)在蛋白药物的研发与生产过程中,用于分析多糖来提高蛋白药物的疗效以及质量控制;
(2)对各种涉及糖基化异常的复杂疾病(包括癌症,老年痴呆症等)进行研究并识别各种疾病的特征生物标记物;
(3)对各种病毒(包括冠状病毒,寨卡病毒和登革热病毒等)进行研究并开发疫苗。
附图说明
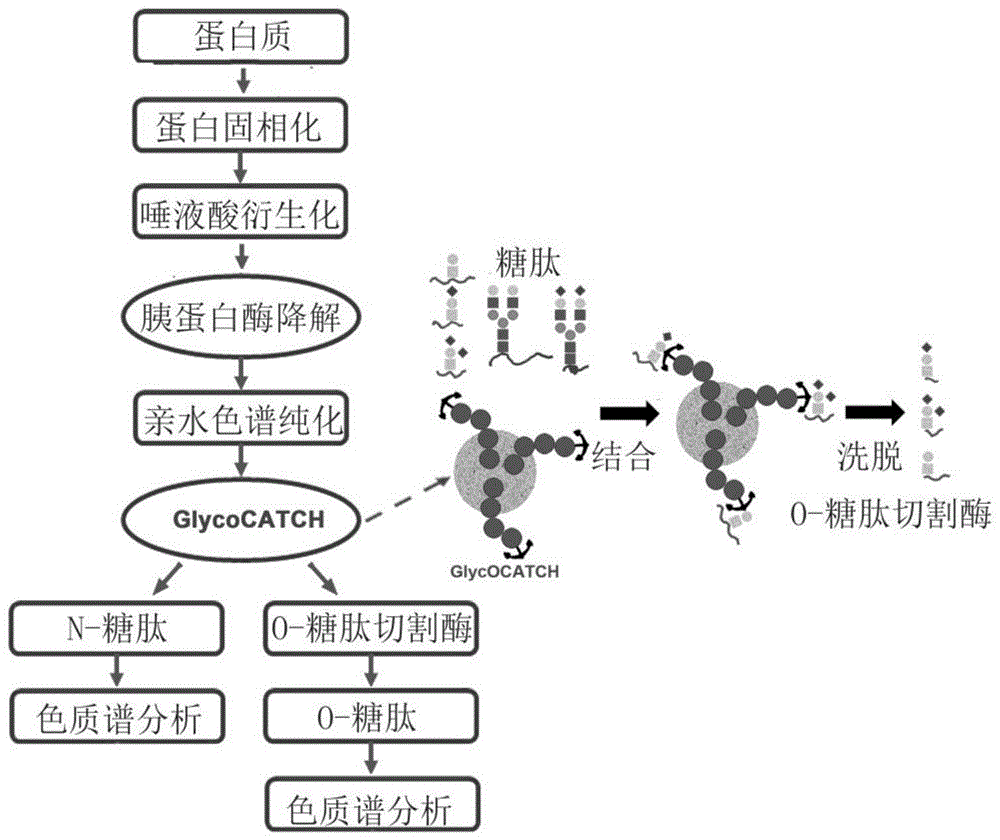
图1为本发明中方法的工作流程示意图。
图2为本发明中血型糖蛋白a(gpa)的糖基化分析示意图。
图3为本发明中a型流感病毒h1n1神经氨酸酶野生型和n295s突变型o-糖基化的比较示意图。
具体实施方式
为使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面结合具体实施方式,进一步阐述本发明。
如图1至图所示,一种基于固相法分离n-糖肽和o-糖肽的方法,所述方法包括如下步骤:
步骤一:将糖蛋白偶联到固相树脂上:
将糖蛋白溶解在0.3~2.0ml的缓冲液a(10mm柠檬酸钠,50mm碳酸钠,ph=10.0)中,加入醛基树脂并在室温下混合2-6小时;
添加10-30μl1mnacnbh3后继续混合2-6小时;
用300-600μl缓冲液b(50mmpbs,ph=7.4)冲洗两次并进行烷基化处理;
其中,固相树脂为亲水性固体材料,包括硅胶、琼脂糖和表面包被亲水性基团的磁珠中的一种或多种组合;
烷基化处理的方法为,用含有5~15mmdtt,2-10m尿素,0.5-2m碳酸氢铵的溶液进行还原处理;
加入10~20mm的碘乙酰胺并在20~50℃和避光环境中振荡0.5~2.0个小时;
步骤二:通过衍生化试剂对固定在树脂上的糖蛋白唾液酸进行衍生化并清洗,加入胰蛋白酶降解处理后去除溶剂,得到多肽;
用衍生化试剂(200~300mm1-羟基苯并三唑水合物和200~300mmedc.hcl(n-(3-二甲基氨基丙基)-n-乙基碳二亚胺盐酸盐)的乙醇溶液),在37℃恒温与固定在树脂上的糖蛋白唾液酸进行衍生化反应1小时;
分别用100%乙醇,1mnacl和hplc纯净水将样品洗涤三次;
用1m氯化钠,10%乙腈溶液冲洗后,加入蛋白酶在碱性环境下通过100mm碳酸氢铵和1m尿素溶液的降解固相化的糖蛋白;
收集产生的多肽后,采用亲水色谱柱对糖肽(n-糖肽和o-糖肽的混合物)进行富集并用乙腈的水溶液(40%乙腈,60%水)清洗;
用离心干燥仪除去溶剂。
步骤三:将获得的多肽溶解后,对n-糖肽和o-糖肽进行分离:
将多肽溶解在300-500μl缓冲液c与树脂混合后振荡1小时,然后在600~1200rpm离心1-5min,收集上层清液;
用300-800μl的缓冲液b对树脂进行了二次重洗;
将洗脱液汇集,并用c18纯化,用离心干燥仪除去溶剂并重新溶解在50μl的0.2%甲酸溶液中,获得n-糖肽;
分别用1mnacl500μl、10%乙腈溶液500μl和纯水500μl对树脂进行洗涤。
将400-500μl20-30mm碳酸氢铵含有100单位的o-糖肽切割酶的溶液和树脂混合后在37℃振荡8-15小时后,离心取上层清液;
用200-400μl10%乙腈溶液清洗2-4次,并洗脱液收集在一起;
用离心干燥仪除去洗脱液中的溶剂,重新溶解在0.2%的甲酸溶液,获得o-糖肽。
以下通过实施例进一步阐述发明技术方案:
实施例1:条件一下的n-糖肽和o-糖肽分离
将糖蛋白溶解在0.3ml的缓冲液a中,加入酰基树脂并在室温下混合2小时,添加10μl1mnacnbh3后继续混合2小时。用300μl缓冲液b冲洗两次。对固相化的糖蛋白的半胱氨酸进行烷基化(先用含有5mmdtt,2m尿素,0.5m碳酸氢铵的溶液进行还原处理,然后加入10mm的碘乙酰胺并在20℃和避光环境中振荡0.5个小时)。用1m氯化钠,10%乙腈溶液冲洗后,用200mm1-羟基苯并三唑水合物和200mmedc.hcl(n-(3-二甲基氨基丙基)-n-乙基碳二亚胺盐酸盐)的乙醇溶液,在37℃恒温与固定在树脂上的糖蛋白唾液酸进行衍生化反应1小时,分别用100%乙醇,1mnacl和hplc纯净水将样品洗涤三次。加入胰蛋白酶在100mm碳酸氢铵和1m尿素溶液中(ph=8.0)降解固相化的糖蛋白。收集产生的多肽后,采用亲水色谱柱对糖肽(n-糖肽和o-糖肽的混合物)进行富集并用乙腈的水溶液(40%乙腈,60%水)清洗;用离心干燥仪除去溶剂后在-20℃保存。
将多肽溶解在300μl缓冲液b中,与glycocatch树脂混合后振荡1小时,然后在600rpm离心1min,收集上层清液,用300μl缓冲液b对树脂进行了二次重洗。所有的洗脱液汇集用于c18纯化后,用离心干燥仪除去溶剂并重新溶解在50μl的0.2%甲酸溶液进行n-糖肽分析。分别用1mnacl500μl、10%乙腈溶液500μl和纯水500μl洗涤树脂3次后,将400μl20mm碳酸氢铵含有100单位的切割酶的溶液和树脂混合后在37℃振荡8小时后,离心,上层清液以及用300μl10%乙腈溶液清洗3次的洗脱液混合在一起,用离心干燥仪除去溶剂。
实施例2:条件二下的n-糖肽和o-糖肽分离
将糖蛋白溶解在0.5ml的缓冲液a中,加入酰基树脂并在室温下混合4小时,添加25μl1mnacnbh3后继续混合4小时。用500μl的缓冲液b冲洗两次。对固相化的糖蛋白的半胱氨酸进行烷基化(先用含有12mmdtt,8m尿素,1m碳酸氢铵的溶液进行还原处理,然后加入16mm的碘乙酰胺并在37℃和避光环境中振荡1个小时)。用1m氯化钠,10%乙腈溶液冲洗后,用300mm1-羟基苯并三唑水合物和300mmedc.hcl(n-(3-二甲基氨基丙基)-n-乙基碳二亚胺盐酸盐)的乙醇溶液,在37℃恒温与固定在树脂上的糖蛋白唾液酸进行衍生化反应1小时,分别用100%乙醇,1mnacl和hplc纯净水将样品洗涤三次加入胰蛋白酶在100mm碳酸氢铵和1m尿素溶液中(ph=8.0)降解固相化的糖蛋白。收集产生的多肽后,采用亲水色谱柱对糖肽(n-糖肽和o-糖肽的混合物)进行富集并用乙腈的水溶液(40%乙腈,60%水)清洗;用离心干燥仪除去溶剂后在-20℃保存。
将多肽溶解在400μl缓冲液b中,与glycocatch树脂混合后振荡1小时,然后在800rpm离心3min,收集上层清液,用500μlpbs缓冲液树脂进行了二次重洗。所有的洗脱液汇集用于c18纯化后,用离心干燥仪除去溶剂并重新溶解在50μl的0.2%甲酸溶液进行n-糖肽分析。分别用1mnacl500μl、10%乙腈溶液500μl和纯水500μl洗涤树脂3次后,将450μl25mm碳酸氢铵含有100单位的切割酶的溶液和树脂混合后在37℃振荡12小时后,离心,上层清液以及用300μl10%乙腈溶液清洗3次的洗脱液混合在一起,用离心干燥仪除去溶剂。
实施例3:条件三下的n-糖肽和o-糖肽分离
将糖蛋白溶解在2ml的缓冲液a中,加入酰基树脂并在室温下混合6小时,添加30μl1mnacnbh3后继续混合6小时。用600μl缓冲液b冲洗两次。对固相化的糖蛋白的半胱氨酸进行烷基化(先用含有15mmdtt,10m尿素,2m碳酸氢铵的溶液进行还原处理,然后加入20mm的碘乙酰胺并在50℃和避光环境中振荡2个小时)。用1m氯化钠,10%乙腈溶液冲洗后,用250mm1-羟基苯并三唑水合物和250mmedc.hcl(n-(3-二甲基氨基丙基)-n-乙基碳二亚胺盐酸盐)的乙醇溶液,在37℃恒温与固定在树脂上的糖蛋白唾液酸进行衍生化反应1小时,分别用100%乙醇,1mnacl和hplc纯净水将样品洗涤三次加入胰蛋白酶在100mm碳酸氢铵和1m尿素溶液中(ph=8.0)降解固相化的糖蛋白。收集产生的多肽后,采用亲水色谱柱对糖肽(n-糖肽和o-糖肽的混合物)进行富集并用乙腈的水溶液(40%乙腈,60%水)清洗;用离心干燥仪除去溶剂后在-20℃保存。
将多肽溶解在500μl缓冲液b中,与glycocatch树脂混合后振荡1小时,然后在1200rpm离心5min,收集上层清液,用500μlpbs缓冲液树脂进行了二次重洗。所有的洗脱液汇集用于c18纯化后,用离心干燥仪除去溶剂并重新溶解在50μl的0.2%甲酸溶液进行n-糖肽分析。分别用1mnacl500μl、10%乙腈溶液500μl和纯水500μl洗涤树脂3次后,将500μl30mm碳酸氢铵含有100单位的切割酶的溶液和树脂混合后在37℃振荡15小时后,离心,上层清液以及用400μl10%乙腈溶液清洗3次的洗脱液混合在一起,用离心干燥仪除去溶剂。
实施例4:对提取的o-糖肽进行液相色谱-质谱分析和数据处理
将获得的样品注射10μl到uitimate3000lc/fusionorbitrap质谱中进行分析。每个样品重复3次。糖肽使用easy-sprayc18柱分离后在orbitrap质谱进行分析。碰撞能量设为27,自动增益控制目标为1.0×104。所获质谱数据用软件包byonic和byologic进行数据分析。
实施例5:血型糖蛋白a(gpa)的糖基化分析
glycocatch方法可以用于人类红细胞膜表面的主要糖蛋白gpa(uniprotid:p02724.2)的糖基化分析。先期研究已经证明gpa包含一个n-糖基化位点和多于15个的o-糖基化位点。采用上述的方法分析显示在n[45]dt处有一个单一的n-糖基化位点和22个o-糖基化位点(图2)。在gpa中鉴定出7个n-多糖结构,包括man9,复合多糖(h3n6,h5n5)和杂化唾液酸n-多糖。o-糖基化位点主要包括s[12]、t[31]、s[32]、s[33]、t[47]、t[58]、s[66]、t[77]、t[108]、s[111]和s[139]。galnac似乎是最丰富的o-多糖。
实施例6:甲型流感病毒神经氨酸苷酶(neuraminidase)的糖基化分析
从2009年1月到2010年8月持续了19个多月的2009年猪流感大流行属于h1n1流感病毒。该病毒是一种新的h1n1毒株,是由之前禽类、猪、人类流感病毒的三重重组,并进一步与欧亚猪流感病毒结合而产生的。神经氨酸苷酶是病毒的一个表面糖蛋白,在病毒侵入寄主细胞中发挥重要作用。其n295s突变与甲型流感的多重耐药有关。n295s变异对神经氨酸苷酶n-糖基化和o-糖基化的影响可以采用glycocatch方法进行研究。
通过采用上述分析步骤对野生型和n295s神经氨酸苷酶进行分析后,发现野生型和n295s所有的9个n-糖基化位点和n-多糖的丰度都相似。这是意料之中的,因为n295s并不在这9个n-糖基化位点附近。但是,n295s神经氨酸酶上的o-糖基化与野生型相比有显著不同(图3)。在野生型中主要含有h1n2和h1n2f1两种o-多糖,而n295s含有5种o-多糖,除h1n2和h1n2f1外,还包括s1h1n1,h2n2和h2n2f1(图3)。另外,野生型和n295s的o-糖基化的相对丰度是不同的。例如,野生型在s[31],t[148],s[413]等位点有最丰富的o-多糖,而n295s在s[95],t[148],t[215]处有最丰富的o-多糖。由于n295s上的突变,s[295]上成为一个o-糖基化位点。有趣的是,n295s中未检测到s[299]的o-糖基化。这可能是由于和s[295]之上的o-多糖的位阻竞争造成的。
由技术常识可知,本发明可以通过其它的不脱离其精神实质或必要特征的实施方案来实现。因此,上述公开的实施方案,就各方面而言,都只是举例说明,并不是仅有的。所有在本发明范围内或在等同于本发明的范围内的改变均被本发明包含。
- 还没有人留言评论。精彩留言会获得点赞!