低分子化合物的癌症以及纤维化抑制效果的制作方法
本发明涉及恶性肿瘤或者纤维化的治疗药。
背景技术:
作为人的死亡原因,恶性肿瘤、心脏疾病、脑血管疾病等为例的疾病居在上位。其中,由于恶性肿瘤的发生机制复杂,可以说是预防以及治疗尤其困难的疾病。
近年来,在恶性肿瘤的研究领域中,癌症干细胞的存在被发现后,树立新的治疗策略正在被人们所重视。癌症干细胞可分化成为癌细胞。在癌症患者当中存在去除癌细胞后经过一段时间有复发的可能,据说,这些是由于极少数的存活的癌症干细胞引起的。
癌症干细胞的特点在于,以往的众多抗癌剂很难对癌症干细胞发挥效果。关于此点,九州大学的中山教授等报告了如下的研究结果:通过使模型小鼠缺损Fbxw7,癌症干细胞可被伊马替尼(Imatinib)(抗癌剤)杀灭(非专利文献1)。但是,能直接抑制癌症干细胞的增殖的化合物至今没有得到。
另一方面,成为恶性肿瘤的原因的症状可举出组织的纤维化。比如,肝脏的纤维化进展会导致肝硬化、肝癌。另外,纤维化也会发生在肺、肾脏、心脏、皮肤等。非专利文献2中,关于纤维化治疗,记载了吡非尼酮(Pirfenidone)的临床试验结果。
另外,本申请的发明人等关于低分子化合物作了三个报告(非专利文献3、4、专利文献1)。非专利文献3、4中记载有显示肝癌细胞增殖的抑制效果以及Wnt/β-catenin信号抑制效果的低分子化合物。并且,非专利文献4的低分子化合物增强了CD44表达。非专利文献3、4中未记载具有什么样的结构以及功能的化合物对肝癌细胞显示增殖抑制效果。
专利文献1中记载了PN-1-2、PN-3-4、PN-3-13、HC-1以及IC-2抑制间充质干细胞的Wnt/β-catenin信号,使间充质干细胞分化诱导成肝细胞。该文献中没有关于癌细胞或者癌症干细胞的增殖抑制的记载。
现有技术文献
专利文献
专利文献1:WO2012/141038
非专利文献
非专利文献1:Takeishi et al.,Cancer Cell.2013 Mar 18;23(3):347-61.
非专利文献2:Noble et al.,Lancet.2011 May 21;377(9779):1760-9.
非专利文献3:Sakabe et al.,肝脏,53卷,Supplement 1,2012,A226,WS-54
非专利文献4:Seto et al.,肝脏,54卷,Supplement 1,2013,P-12
技术实现要素:
发明要解决的问题
如上所述,恶性肿瘤的治疗策略正在逐渐进展。但直到如今恶性肿瘤还是占据人的死亡原因的上位,仅靠以往的治疗策略是不充分的。
在恶性肿瘤的治疗领域中,已知根据各个骨架具有的特点,所给药的低分子化合物具有的药理作用会有很大变化。并且,在这个领域是不可靠性高的领域,可以说在开发新型的治疗方法时,难以预想能否得到所期望的药理作用。因此,新鉴定一种对恶性肿瘤具有治疗效果的低分子化合物,不是一件容易的事情。
进而,新鉴定一种不仅可以抑制恶性肿瘤细胞,还可以抑制癌症干细胞的增殖的低分子化合物,更不是一件容易的事情。
此外,如上所述,不断有关于纤维化治疗的研究成果的报告。但是对纤维化治疗有效的治疗药少,另外根据患者的情况也存在副作用的问题。因此,仅靠以往的抗纤维化剂是不充分的。
本发明鉴于以上情况进行的,其目的是提供一种针对恶性肿瘤、癌症干细胞或者纤维化的新型治疗药。
用于解决问题的方案
本申请的发明人等进行了深入研究,结果如后述的实施例所记载,成功地鉴定了具有抗恶性肿瘤效果的低分子化合物。并且,该低分子化合物不仅对肿瘤细胞有效,对癌症干细胞的增殖也具有抑制效果。癌症干细胞有可能成为恶性肿瘤的复发或者转移的原因是已知的,因此,作为恶性肿瘤的治疗药的有效成分,能抑制癌症干细胞的增殖的低分子化合物可以说是非常优异的化合物。更何况,还具有纤维化的抑制作用。本发明人等根据这些见解完成了本发明。
即,根据本发明的一个实施方式,提供一种恶性肿瘤的治疗药,其含有选自由以下结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物:
式中,
R1、R2、R4、R5以及R6相同或不同,表示H、卤素、硝基、氰基、OH、取代或未取代的烷基、取代或未取代的烯基、取代或未取代的烷氧基、芳基或者杂芳基;
R3以及R7相同或不同,表示H、取代或未取代的烷基或者取代或未取代的烯基;
环A表示取代或未取代的芳基或者取代或未取代的杂芳基;
R8以及R9相同或不同,表示取代或未取代的烷基或者取代或未取代的烯基;
m以及q相同或不同,表示1~4中的任一整数;
n表示1~3中的任一整数;
p以及r相同或不同,表示1~5中的任一整数。
另外,根据本发明的一个实施方式,提供一种癌症干细胞的治疗药,其含有选自由上述结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物:
式中,
R1、R2、R4、R5以及R6相同或不同,表示H、卤素、硝基、氰基、OH、取代或未取代的烷基、取代或未取代的烯基、取代或未取代的烷氧基、芳基或者杂芳基;
R3以及R7相同或不同,表示H、取代或未取代的烷基或者取代或未取代的烯基;
环A表示取代或未取代的芳基或者取代或未取代的杂芳基;
R8以及R9相同或不同,表示取代或未取代的烷基或者取代或未取代的烯基;
m以及q相同或不同,表示1~4中的任一整数;
n表示1~3中的任一整数;
p以及r相同或不同,表示1~5中的任一整数。
另外,根据本发明的一个实施方式,提供一种纤维化的治疗药,其含有选自由上述结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物:
式中,
R1、R2、R4、R5以及R6相同或不同,表示H、卤素、硝基、氰基、OH、取代或未取代的烷基、取代或未取代的烯基、取代或未取代的烷氧基、芳基或者杂芳基;
R3以及R7相同或不同,表示H、取代或未取代的烷基或者取代或未取代的烯基;
环A表示取代或未取代的芳基或者取代或未取代的杂芳基;
R8以及R9相同或不同,表示取代或未取代的烷基或者取代或未取代的烯基;
m以及q相同或不同,表示1~4中的任一整数;
n表示1~3中的任一整数;
p以及r相同或不同,表示1~5中的任一整数。
此外,作为本发明优选的实施方式,上述恶性肿瘤、癌症干细胞或者纤维化的治疗药中,上述R1、R2、R4、R5以及R6相同或不同,表示H、卤素、硝基、氰基、OH、烷基、卤代烷基、羟基烷基、烷基氨基、烷氧基、卤代烷氧基、羟基烷氧基、或者烷氧基氨基、被烷氧基取代的烷氧基、被烷氧基苯基取代的烷氧基、三烷基硅氧基烷基或者烷基二苯基硅氧基烷基,所述R3以及R7表示H,所述环A表示萘基、被5个卤素取代的苯基或者被1个甲基取代的呋喃基,所述R8以及R9相同或不同,表示烷基。
发明的效果
根据本发明,通过使用新型治疗药可以治疗恶性肿瘤、癌症干细胞或者纤维化。
附图说明
图1表示经过IC-2处理后的肝癌细胞的细胞存活率(浓度依赖性的方式)的调查结果。
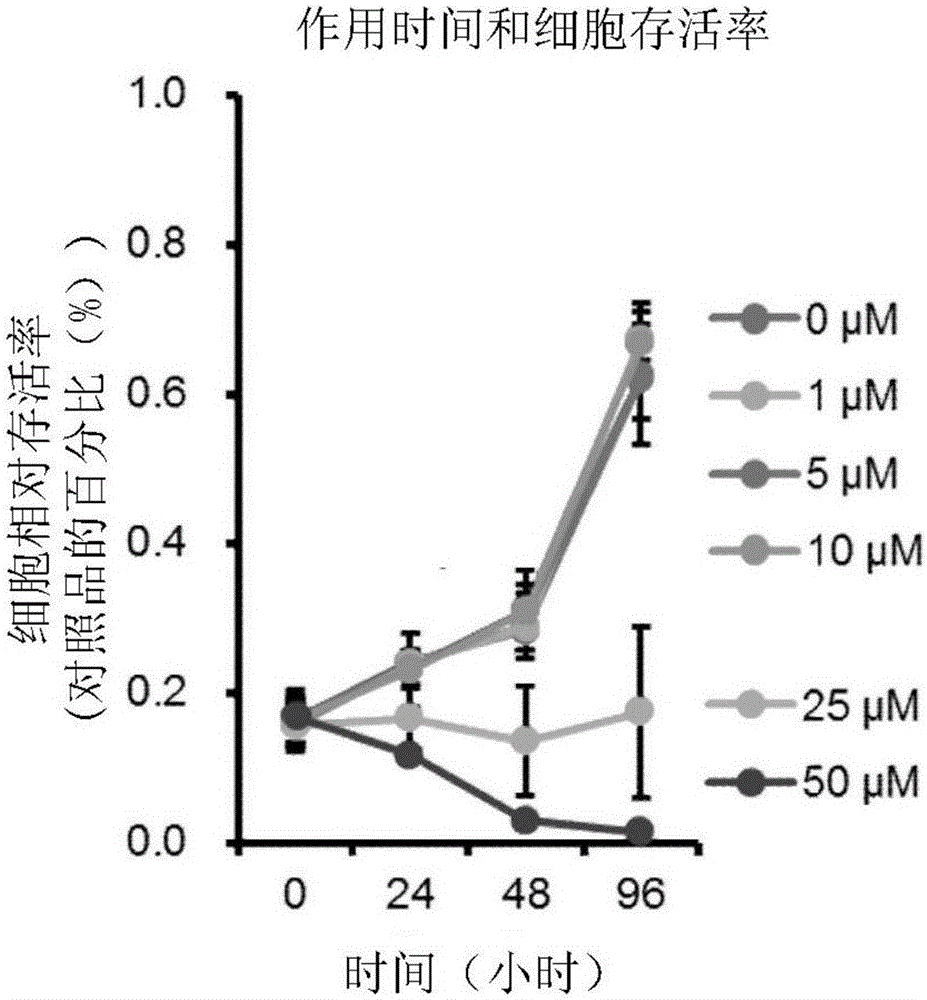
图2表示经过IC-2处理后的肝癌细胞的细胞存活率(时间依赖性的方式)的调查结果。
图3表示经过PN3-13处理后的肝癌细胞的细胞存活率(浓度依赖性的方式)的调查结果。
图4表示经过PN3-13处理后的肝癌细胞的细胞存活率(时间依赖性的方式)的调查结果。
图5表示经过HC-1处理后的肝癌细胞的细胞存活率(时间依赖性的方式)的调查结果。
图6表示将癌症干细胞用低分子化合物处理后的CD44阳性细胞数的调查结果。
图7表示肝癌模型小鼠的体重变化的调查结果。
图8表示肝癌模型小鼠的治疗结果。
图9表示低分子化合物对球形成能力的影响的调查结果。
图10表示将癌症干细胞用低分子化合物处理后的CD90阳性细胞数的调查结果。
图11表示将癌症干细胞用低分子化合物处理后的CD133阳性细胞数的调查结果。
图12表示将癌症干细胞用低分子化合物处理后的EpCAM阳性细胞数的调查结果。
图13表示萤光素酶检测结果。
图14表示大肠癌的抗肿瘤效果的调查结果。
图15表示大肠癌的抗肿瘤效果的调查结果。
图16表示大肠癌的抗肿瘤效果的调查结果。
图17表示大肠癌细胞的球形成能力的调查结果。
图18表示大肠癌的癌症干细胞的抑制效果的调查结果。
图19表示大肠癌的Wnt/β-catenin信号的抑制效果的调查结果。
图20表示大肠癌的Wnt/β-catenin信号的抑制效果的调查结果。
图21表示大肠癌的Wnt/β-catenin信号的抑制效果的调查结果。
图22表示鳞状细胞癌的抗肿瘤效果的调查结果。
图23表示鳞状细胞癌的抗肿瘤效果的调查结果。
图24表示鳞状细胞癌的抗肿瘤效果的调查结果。
图25表示鳞状细胞癌的抗肿瘤效果的调查结果。
图26表示鳞状细胞癌对癌症干细胞的抑制效果的调查结果。
图27表示大肠癌的Wnt/β-catenin信号的抑制效果的调查结果。
图28表示大肠癌的Wnt/β-catenin信号的抑制效果的调查结果。
图29表示大肠癌的Wnt/β-catenin信号的抑制效果的调查结果。
图30表示在实施例8中采用的IC-2衍生物的合成路线。
图31表示在实施例8中采用的IC-2衍生物的合成路线。
图32表示在实施例8中采用的IC-2衍生物的合成路线。
图33表示在实施例8中采用的IC-2衍生物的合成路线。
图34表示在实施例8中采用的IC-2衍生物的合成路线。
图35表示在实施例8中采用的IC-2衍生物的合成路线。
图36表示在实施例8中得到的IC-2衍生物的结构式和光谱数据。
图37表示在实施例8中得到的IC-2衍生物的结构式和光谱数据。
图38表示IC-2衍生物带来的Wnt/β-catenin信号的抑制效果的调查结果。
图39表示IC-2衍生物带来的Wnt/β-catenin信号的抑制效果的调查结果。
图40表示实施例的萤光素酶检测方案。
图41表示将肝星状细胞经过IC-2处理24小时后进行萤光素酶检测的结果。
图42表示将肝星状细胞经过IC-2+TGFβ处理24小时后进行萤光素酶检测的结果。
图43表示将肝星状细胞经过IC-2+Wnt3a处理24小时后进行萤光素酶检测的结果。
图44表示将肝星状细胞经过IC-2处理48小时后进行萤光素酶检测的结果。
图45表示将肝星状细胞经过IC-2+TGFβ处理48小时后进行萤光素酶检测的结果。
图46表示将肝星状细胞经过IC-2+Wnt3a处理48小时后进行萤光素酶检测的结果。
图47表示将肝星状细胞经过PN3-13+TGFβ处理24小时后进行萤光素酶检测的结果。
图48表示将肝星状细胞经过PN3-13+TGFβ处理48小时后进行萤光素酶检测的结果。
图49表示将肝星状细胞经过HC-1处理24小时后进行萤光素酶检测的结果。
图50表示将肝星状细胞经过HC-1+Wnt3a处理24小时后进行萤光素酶检测的结果。
图51表示将肝星状细胞经过HC-1+TGFβ处理24小时后进行萤光素酶检测的结果。
图52表示将肝星状细胞经过HC-1处理48小时后进行萤光素酶检测的结果。
图53表示将肝星状细胞经过HC-1+Wnt3a处理24小时后进行萤光素酶检测的结果。
图54表示将肝星状细胞经过HC-1+TGFβ处理48小时后进行萤光素酶检测的结果。
图55表示实施例的实时荧光定量PCR方案。
图56表示将肝星状细胞经过IC-2+TGFβ处理后的TGFβ的表达量的调查结果。
图57表示将肝星状细胞经过IC-2+TGFβ处理后的COL1A1的表达量的调查结果。
图58表示将肝星状细胞经过IC-2+TGFβ处理后的α-SMA的表达量的调查结果。
图59表示将肝星状细胞经过PN3-13+TGFβ处理后的TGFβ的表达量的调查结果。
图60表示将肝星状细胞经过PN3-13+TGFβ处理后的COL1A1的表达量的调查结果。
图61表示将肝星状细胞经过PN3-13+TGFβ处理后的α-SMA的表达量的调查结果。
图62表示将肝星状细胞经过HC-1+TGFβ处理后的TGFβ的表达量的调查结果。
图63表示将肝星状细胞经过HC-1+TGFβ处理后的COL1A1的表达量的调查结果。
图64表示将肝星状细胞经过HC-1+TGFβ处理后的α-SMA的表达量的调查结果。
图65表示给药IC-2后的肝损伤模型小鼠的羟脯氨酸量的调查结果。
图66表示给药IC-2后的肝损伤模型小鼠的纤维化区域的定量结果。
具体实施方式
以下对本发明的实施方式进行详细说明。另外,对于相同的内容,为避免重复,适当地省略说明。
本发明的一个实施方式是含有选自由结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的恶性肿瘤的治疗药。利用该治疗药可以治疗恶性肿瘤。
本发明的一个实施方式是含有选自由结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的癌症干细胞的治疗药。利用该治疗药可以治疗癌症干细胞。
本发明的一个实施方式是含有选自由结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的恶性肿瘤或者癌症干细胞的增殖抑制剂。利用该治疗药可以抑制恶性肿瘤細胞或者癌症干细胞的增殖。
本发明的一个实施方式是含有选自由结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的恶性肿瘤的复发抑制剂。利用该抑制剂可以抑制恶性肿瘤的复发。
本发明的一个实施方式是含有选自由结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的纤维化的治疗药。利用该治疗药可以治疗纤维化。
本发明的一个实施方式是含有选自由结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的伴随纤维化的疾病的治疗药。利用该治疗药可以治疗伴随纤维化的疾病。
结构式(1)、(2)以及(5)中,R1、R2、R4、R5以及R6相同或不同,表示H、卤素、硝基、氰基、OH、取代或未取代的烷基、取代或未取代的烯基、取代或未取代的烷氧基、芳基或者杂芳基。另外,R3以及R7表示H、取代或未取代的烷基或者取代或未取代的烯基。另外,环A表示取代或未取代的芳基或者取代或未取代的杂芳基。另外,R8以及R9相同或不同,表示取代或未取代的烷基或者取代或未取代的烯基。另外,m以及q相同或不同,表示1~4中的任一整数。另外,n表示1~3中的任一整数。另外,p以及r相同或不同,表示1~5中的任一整数。
上述R1、R2、R4、R5以及R6优选相同或不同,表示H、卤素、硝基、氰基、OH、烷基、卤代烷基、羟基烷基、烷基氨基、烷氧基、卤代烷氧基、羟基烷氧基、烷氧基氨基、或者被烷氧基取代的烷氧基、被烷氧基苯基取代的烷氧基、三烷基硅氧基烷基或者烷基二苯基硅氧基烷基。另外,所述R3以及R7优选H。另外,所述环A优选萘基、被5个卤素取代的苯基或者被1个甲基取代的呋喃基。在此情况的结构式(2)的化合物可以利用例如WO2012/141038记载的方法合成。另外,所述R8以及R9优选相同或不同,表示烷基。
本发明的一个实施方式中“卤素”是指F、Cl、Br或者I。
本发明的一个实施方式中“烷基”以及“烯基”在没有特别说明的情况下是指直链或支链状的烃链。
本发明的一个实施方式中“C1~C6”表示碳原子数为1、2、3、4、5或者6的烃。即,“C1~C6烷基”是碳原子数为1、2、3、4、5或者6的烷基。作为C1~C6烷基包括,例如,甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、正己基等。
本发明的一个实施方式中“烯基”包括,例如,乙烯基、1-丙烯基、2-丙烯基、2-甲基-1-丙烯基、1-丁烯基、2-丁烯基、3-丁烯基、3-甲基-2-丁烯基、1-戊烯基、2-戊烯基、3-戊烯基、4-戊烯基、4-甲基-3-戊烯基、1-己烯基、3-己烯基或者5-己烯基等。
本发明的一个实施方式中“烷氧基”包括,例如,甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、异丁氧基、仲丁氧基、叔丁氧基、戊氧基、异戊氧基、己氧基等。
本发明的一个实施方式中“取代或未取代”是指,未取代或者在可取代的位置上具有1、2、3、4或者5个取代基。另外,具有多个取代基时,这些取代基可相同或者不同。另外,取代的位置可以是1、2、3、4、5、6、7、8或者9位。在此,作为取代基可举例为H、卤素、硝基、氰基、OH、烷基、卤代烷基、羟基烷基、烷基氨基、环烷基、烯基、卤代烯基、羟基烯基、烯基氨基、环烯基、炔基、卤代炔基、羟基炔基、炔基氨基、烷氧基、卤代烷氧基、羟基烷氧基、烷氧基氨基、烷氧基苯基、三烷基硅氧基、烷基二苯基硅氧基、芳基或者杂芳基。
本发明的一个实施方式中“卤代烷基”是指,被一个以上的卤素取代的C1~C6烷基。该卤素的个数可以是,例如1、2、3、4、5、6或者13个,也可以是在此举例的两个数之间的范围内。或者,卤素在2个以上时,各个卤素的种类可相同或不同。卤代烷基包括,例如,氯甲基、二氯甲基、三氯甲基、氟甲基、二氟甲基、三氟甲基、溴甲基、二溴甲基、三溴甲基、氯乙基、二氯乙基、三氯乙基、氟乙基、二氟乙基、三氟乙基等。
本发明的一个实施方式中“羟基烷基”是指,被一个以上的羟基取代的C1~C6烷基。该羟基的个数可以是,例如1、2、3、4、5、6或者13个,也可以是在此举例的两个数之间的范围内。羟基烷基包括,例如,羟甲基、1-羟乙基、2-羟乙基、2-羟正丙基或2,3-二羟基正丙基等。
本发明的一个实施方式中“烷基氨基”是指,被一个以上的氨基取代的C1~C6烷基。该氨基的个数可以是,例如1、2、3、4、5、6或者13个,也可以是在此举例的两个数之间的范围内。烷基氨基包括,例如,甲基氨基或乙基氨基。
本发明的一个实施方式中“卤代烷氧基”与卤代烷基的烷基被烷氧基取代的物质等价。卤代烷氧基包括,例如,氟甲氧基、二氟甲氧基、三氟甲氧基、1-氟乙氧基、2-氟乙氧基、2-氯乙氧基、2-溴乙氧基、(1,1-二氟)乙氧基、(1,2-二氟)乙氧基、(2,2,2-三氟)乙氧基、(1,1,2,2-四氟)乙氧基、(1,1,2,2,2-五氟)乙氧基、1-氟正丙氧基、1,1-二氟正丙氧基、2,2-二氟正丙氧基、3-氟正丙氧基、(3,3,3-三氟)正丙氧基、(2,2,3,3,3-五氟)正丙氧基、4-氟正丁氧基、(4,4,4-三氟)正丁氧基、5-氟正戊氧基、(5,5,5-三氟)正戊氧基、6-氟正己氧基、(6,6,6-三氟)正己氧基、2-氟环丙氧基或2-氟环丁氧基等。
本发明的一个实施方式中“羟基烷氧基”与羟基烷基的烷基被烷氧基取代的物质等价。卤代烷氧基包括,例如,2-羟基乙氧基、2-羟基正丙氧基、3-羟基正丙氧基、2,3-二羟基正丙氧基或2-羟基环丙基等。
本发明的一个实施方式中“烷氧基氨基”与烷基氨基的烷基被烷氧基取代的物质等价。烷氧基氨基包括,例如,甲氧基氨基或乙氧基氨基等。
本发明的一个实施方式中“芳基”是指,C6~14的单环、双环或三环式芳烃环的基团。芳基包括,例如,苯基、萘基(1-萘基、2-萘基)、四氢萘基、茚基或芴基等。尤其优选萘基或被5个卤素取代的苯基。另外,芳基包括,例如,C5~8环烯基和在双键部位缩合的环基。
本发明的一个实施方式中“杂芳基”可以举出,环内具有5至14个的环原子以及共有π电子体系且含有1~4个选自N、S以及O的杂原子的取代基。杂芳基包括,例如,噻吩基、苯并噻吩基、呋喃基、苯并呋喃基、二苯并呋喃基、吡咯基、咪唑基、吡唑基、吡啶基、吡嗪基、嘧啶基、哒嗪基、四唑基、恶唑基、噻唑基或异恶唑基等。尤其优选被一个甲基取代的呋喃基。
本发明的一个实施方式中,结构式(1)所示的化合物的结构优选R1、R2以及R3分别是H、R4在4位且表示H、F、Cl、硝基、OH、CH2OH、甲氧基、甲氧基甲氧基或叔丁基二甲基硅氧基甲基。特别是,结构式(1)所示的化合物的结构从恶性肿瘤或者纤维化的治疗效果的观点考虑,越接近于结构式(3)所示的化合物的结构越优选。本发明的一个实施方式中,结构式(2)所示的化合物的结构从恶性肿瘤或者纤维化的治疗效果的观点考虑,越接近于结构式(4)所示的化合物的结构越优选。本发明的一个实施方式中,结构式(5)所示的化合物的结构从恶性肿瘤或者纤维化的治疗效果的观点考虑,越接近于结构式(6)所示的化合物的结构越优选。另外,结构式(3)、(4)以及(6)所示的化合物有时分别称为IC-2、PN3-13以及HC-1。在本说明书中PN3-13和PN-3-13所表示的意义相同。
本发明的一个实施方式中“盐”没有特别的限定,例如包括由任意的酸性(如,羧基)基团形成的阴离子盐或者由任意的碱性(如,氨基)基团形成的阳离子盐。盐类包括无机盐以及有机盐,包括[Berge,Bighley以及Monkhouse、J.Pharm.Sci.,1977,66,1-19]中记载的盐。例如,可举出金属盐、铵盐、与有机碱的盐、与无机酸的盐、与有机酸的盐、与碱性或酸性氨基酸的盐等。金属盐可举出,例如,碱金属盐(钠盐或钾盐等)、碱土金属盐(钙盐、镁盐或钡盐等)或者铝盐等。与有机碱的盐可举出,例如,与三甲胺、三乙胺、吡啶、甲基吡啶、2,6-二甲基吡啶、乙醇胺、二乙醇胺、三乙醇胺、环己胺、二环己胺或者N,N'-二苄基乙二胺等的盐。与无机酸的盐可举出,例如,与盐酸、氢溴酸、硝酸、硫酸或者磷酸等的盐。与有机酸的盐可举出,例如,与甲酸、乙酸、三氟乙酸、苯二甲酸、富马酸、草酸、酒石酸、马来酸、柠檬酸、琥珀酸、苹果酸、甲磺酸、苯磺酸或者对甲苯磺酸等的盐。与碱性氨基酸的盐可举出,例如,与精氨酸、赖氨酸或者鸟氨酸等的盐,与酸性氨基酸的盐可举出,例如,与天冬氨酸或者谷氨酸等的盐。
本发明的一个实施方式中“溶剂合物”是指由溶质和溶剂形成的化合物。关于溶剂合物比如可以参考[J.Honig et al.,The Van Nostrand Chemist’s Dictionary P650(1953)]。溶剂为水时所形成的溶剂合物是水合物。该溶剂优选对溶质的生物活性无影响的溶剂。作为这种优选的溶剂没有特别的限制,可举出水、乙醇以及乙酸。最优选的溶剂是水。本发明的化合物或者其盐可以在接触空气或者重结晶时吸收水分,根据情况具有吸湿水或成为水合物。在本发明的一个实施方式中“异构体”含有分子式相同但结构不同的分子。包括镜像异构体(Enantiomer)、几何(顺/反)异构体或者相互不成镜像的具有一个以上不对称中心的异构体(Diastereomer)。本发明的一个实施方式中“前药”是指前体的化合物,包括对受验体给药该化合物时,通过代谢过程或者各种化学反应引起化学变化而形成本发明的化合物或其盐或者其溶剂合物的化合物。关于前药比如可参考[T.Higuchi and V.Stella,“Pro-Drugs as Novel Delivery Systems”,A.C.S.Symposium Series,Volume 14]。
本发明的一个实施方式中“恶性肿瘤”包括,例如,正常的细胞引起突然变异而产生的肿瘤。恶性肿瘤可以从全身所有的脏器或者组织中产生。该恶性肿瘤包括,例如肺癌、食道癌、胃癌、肝癌、胰腺癌、肾癌、肾上腺癌、胆道癌、乳腺癌、结肠癌、小肠癌、卵巢癌、子宫癌、膀胱癌、前列腺癌、输尿管癌、肾盂癌、输尿管癌、阴茎癌、睾丸癌、脑癌、中枢神经系统癌、外周神经系统、头颈癌、脑胶质瘤、多形性成胶质细胞瘤、皮肤癌、黑色素瘤、甲状腺癌、唾液腺癌、恶性淋巴瘤、癌、肉瘤以及血液恶性肿瘤中的一种以上。上述肝癌可以是,例如,上皮性肿瘤或者非上皮性肿瘤,也可以是肝细胞癌或者胆管细胞癌。上述皮肤癌包括,例如,基底细胞癌、鳞状细胞癌或者恶性黑色素瘤。
本发明的一个实施方式中“癌症干细胞”包括癌细胞的起源细胞。该癌症干细胞包括表达癌症干细胞标记物的细胞。癌症干细胞标记物包括,例如,CD44、CD90、CD133或者EpCAM。
已知纤维化的典型症状是通过如下方式形成的:以胶原等为构成因素的结缔组织增生并代替正常组织,导致组织硬化而丧失正常功能。例如,可产生在肝、肺、肾、心脏或者皮肤等的每个组织。此外,例如,当大量的纤维化发生在肝组织会导致肝硬化,进而有可能成为肝癌。此外,在除了肝组织,随着纤维化的进展在各组织中有可能产生恶性肿瘤。纤维化包括伴随纤维化的疾病。伴随纤维化的疾病包括,例如,伴随纤维化的上述各组织的纤维化、硬化或者恶性肿瘤等。
本发明的一个实施方式中,“治疗”包括可发挥患者的疾病或者伴随疾病的一种以上的症状的症状改善效果、抑制效果或者预防效果。在本发明的一个实施方式中“治疗药”可以是,包括活性成分,以及包括在药理学上可容许的一个或一个以上载体的医药组合物。该医药组合物可以通过,例如,混合活性成分和上述载体,在制剂学的技术领域中已知的任意方法来制备。另外,治疗剂只要是用于治疗用途的物质在使用方式上没有特别的限制,可以单独使用活性成分,也可以是活性成分和任选成分的混合物。另外,上述载体的状态没有特别的限制,例如,可以是固体或液体(例如,缓冲液)。应当指出的是,恶性肿瘤的治疗药包括,例如,用于预防恶性肿瘤的药物(预防药物)、恶性肿瘤的复发抑制剂或者恶性肿瘤细胞增殖抑制剂。癌症干细胞的治疗药包括,例如,将癌症干细胞作为标的的治疗药,由癌症干细胞引起的恶性肿瘤的治疗药或者癌症干细胞的抑制剂。
治疗药的给药途径优选使用针对治疗有效的物质,例如,可以是静脉、皮下、肌肉、腹膜或者口服给药等。给药方式可以是,例如,注射剂、胶囊、片剂或者颗粒等。注射用水溶液中可以混合,例如,生理盐水、糖(例如,海藻糖)、NaCl或者NaOH等。另外,治疗药中可以混合,例如,缓冲剂(例如,磷酸盐缓冲液)或者稳定剂等。
给药量没有特别的限制,例如,可以是每次给药0.001、1、10、100或者1000mg/kg体重,也可以是任意两个值之间的范围。给药间隔没有特别的限制,例如,可以是每1、7、14、21或者28天给药1或2次,也可以是任意两个值之间的范围。给药量、给药间隔以及给药方式可根据患者的年龄或体重、症状或者靶器官等适当地选择。另外,治疗药优选含有治疗有效量或者能发挥所期望的功效的有效量的活性成分。
恶性肿瘤的治疗效果可通过图像检查、内窥镜检查、生研或者通过检测恶性肿瘤的标记物的检测来评价。另外,癌症干细胞的治疗效果可以通过图像检查、内窥镜检查、生研或者癌症干细胞的标记物的检测来评价。此外,纤维化的治疗效果可以通过图像检查、内窥镜检查、生研或者纤维化的标记物的检测来评价。每个治疗效果可由以下方式判断:患者或者患者来源的样品(例如,组织、细胞、细胞群或者血液)中的标记物的量在治疗药给药后显著下降时,可以判断有治疗效果。此时,治疗药给药后的标记物的量与给药前(或者是对照品)相比可以减少至0.7、0.5、0.3或者0.1倍以下。或者,患者来源样品中的标记物的阳性细胞数与治疗药给药后相比显著下降时,可以判断有治疗效果。此时,治疗药给药后的标记物的阳性细胞数与给药前(或者是对照品)相比可以减少至0.7、0.5、0.3或者0.1倍以下。另外,在後述的实施例2中使用将CD44阳性Huh7细胞移植到皮下的小鼠来评价治疗效果,但本申请的发明人等通过以下实验确认到以下事实:用通常的HuH7细胞来代替上述CD44阳性Huh7细胞时,也显示出IC-2对恶性肿瘤的治疗效果。
此外,悪性腫瘍的治疗效果可由以下方式判断:将患者来源的受验细胞的增殖速率在治疗药物给药后显著降低时,可以判断有治疗效果。此时,治疗药给药后的受验细胞的增殖速率与给药前(或者是对照品)相比可以减少至0.7、0.5、0.3或者0.1倍以下。应当指出,本发明的一个实施方式中“显著”例如可以使将在统计学的有意差利用Student t检验(单尾或双尾)来评价时,p<0.05或者p<0.01的状态。或者,也可以是产生实质性差异的状态。
本发明的一个实施方式中“患者”包括人或除人以外的哺乳动物(例如,小鼠、豚鼠、仓鼠、大鼠、小鼠、兔、猪、羊、山羊、牛、马、猫、狗、狨、猴或黑猩猩等一种或多种)。另外,患者可以是被判断或者诊断为患有恶性肿瘤或者纤维化的患者。另外,患者可以是需要对恶性肿瘤或者纤维化治疗的患者。或者,患者可以是被判断或者诊断为与健康人相比组织中的癌症干细胞数显著多的患者。此外,判断或者诊断可以通过图像检查、内窥镜检查、生研或者各种标记物的检测来评价。
本发明的一个实施方式中“细胞增殖被抑制的状态”包括受验细胞的增殖速率在与药剂处理前相比显著降低的状态。增殖速率可以测定一定期间内的细胞的增殖量来评价。增殖量的测定,例如,可以以吸光度为指标来测定,也可以用目视来进行。另外,增值量的测定可以通过以患者或者患者来源的样品中的恶性肿瘤的标记物的量为指标来测定。本发明的一个实施方式中“癌症干细胞的抑制”包括,例如,癌症干细胞的增殖的抑制、功能的抑制(例如,球形成的抑制、标记物表达的抑制)。
本发明的一个实施方式是含有选自由结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的恶性肿瘤、癌症干细胞或者纤维化的标记物抑制剂。本发明的一个实施方式是含有选自由结构式(1)、(2)以及(5)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的恶性肿瘤或者癌症干细胞的球形成抑制剂。该球形成抑制剂可用于恶性肿瘤或者癌症干细胞的治疗。
本发明的一个实施方式是一种治疗方法,该方法包括将包含在上述任意实施方式中的治疗药中的低分子化合物给药给患者的工序。本发明的一个实施方式是,包含在上述任意实施方式的治疗药中的低分子化合物的,用于治疗药的制造的使用。本发明的一个实施方式是一种增殖的抑制方法,该方法包括将包含在上述实施方式的增殖抑制剂中的低分子化合物给药给患者的工序。本发明的一个实施方式是,包含在上述任意实施方式的治疗药中的低分子化合物的,用于增殖抑制剂的制造的使用。本发明的一个实施方式是一种恶性肿瘤的复发抑制剂的治疗方法,该方法包括将包含在上述任意实施方式中的复发抑制中的低分子化合物给药给患者的工序。本发明的一个实施方式是,包含在上述任意实施方式的复发抑制剂中的低分子化合物的,用于恶性肿瘤的复发抑制剂的制造的使用。
本发明的一个实施方式是,如结构式(1)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物(在此,结构式(1)的R1、R2以及R3分别是H、R4在4位且表示F、Cl、硝基、OH、CH2OH、甲氧基、甲氧基甲氧基或叔丁基二甲基硅氧基甲基)。使用该化合物、其盐或者所述化合物及其盐的溶剂合物可以治疗恶性肿瘤、癌症干细胞以及纤维化。
本发明的一个实施方式是,包含如结构式(1)所示的化合物群中的一种以上的化合物、其盐、或者所述化合物及其盐的溶剂合物的Wnt/β-catenin信号抑制剂(在此,结构式(1)的R1、R2以及R3分别是H、R4在4位且表示F、Cl、硝基、OH、CH2OH、甲氧基、甲氧基甲氧基或叔丁基二甲基硅氧基甲基)。使用该化合物、其盐、或者所述化合物及其盐的溶剂合物可以抑制Wnt/β-catenin信号。Wnt/β-catenin信号抑制剂可利用于,例如,in vitro或者in vivo的各种用途。Wnt/β-catenin信号抑制剂可使用在,例如,根据Wnt/β-catenin信号抑制效果来改善疾病的治疗。
在本发明引用的所有的出版物、公报类(专利或专利申请),将其全部作为参考引用。
本说明书中的“或”在可采用文章中列举事项的“至少一个以上”时使用。“或者”也相同。本说明书中明确记载有“在两个值的范围内”时,该范围包括两个值本身。本说明书中“A~B”是指A以上B以下。
以上,对本发明的实施方式进行了说明,但这些仅为本发明的举例说明,可采用上述以外的各种构成。另外,也可采用合并上述实施方式中记载的构成。
实施例
以下,进一步对本发明的实施例进行说明,但本发明并不限定于此。
<实施例1>癌细胞以及癌症干细胞的抑制实验
1.1使用的试药例
·DMEM:Dulbecco’s Modified Eagle’s Medium粉末(Nissui,广岛,日本)、10mL 10%NaHCO3、5mL 100×glucose、10mL 50×glutamine、10%胎牛血清(FBS)(EQUITECH-BIO,Texas,USA)
·PBS(-):137mM NaCl、8.10mM Na2HPO4·12H2O、2.68mM KCl、1.47mM KH2PO4
·胰蛋白酶-EDTA溶液(Trypsin/EDTA Solution):Nacalai Tesque、0.25%胰蛋白酶、1mM EDTA
·低分子化合物IC-2(结构式(3))、PN3-13(结构式(4))、HC-1(结构式(6))以及ICG-001是根据WO2012/141038中记载的方法合成的。
1.2细胞的培养
将肝癌细胞株HuH7细胞在10cm的细胞培养皿(TPP Techno Plastic Products AG,Trasadingen,Switzerland)上利用DMEM在5%CO2、37℃以及100%的湿度条件下进行培养。达到70~90%融合的状态下加入胰蛋白酶-EDTA溶液200μL剥离细胞,以1000rpm在室温下离心5分钟后回收细胞,将1dish的细胞分成4dish进行传代。
1.3癌细胞的抑制效果(WST assay)
回收达到70~90%融合的HuH7细胞,在96孔板(FALCON)的各孔中按照以下方式播种:在浓度依赖性抗肿瘤效果检测中各孔播种1×104个细胞,在时间依赖性抗肿瘤效果检测中各孔播种5×103个细胞。24小时后用IC-2、PN3-13或者HC-1处理细胞,在37℃下进行孵化。作为对照品用了DMSO。
各低分子化合物的浓度如下。IC-2:0、1、5、10、25、50μM,PN3-13:0、1、5、10、25、50μM以及HC-1:0、1、5、10、25、50μM。在浓度依赖性抗肿瘤效果检测中用药剂处理4天后,在时间依赖性抗肿瘤效果检测中用药剂处理0、1、2、4天后,添加10%TetraColor ONE(SEIKAGAKU CORPORATION,东京,日本)100μL,在37℃下孵化45分钟,利用96孔用酶标仪(Micro Plate Reader)(TECAN,Zurich,Switzerland)测定吸光度(测定波长450nm/参考波长600nm)。测定结果采用与仅试剂的吸光度之差,作为仅细胞的吸光度,将其作为活细胞的数量。另外,各个低分子化合物的IC50是由IC50=10{LOG(A/B)×(50-C)/(D-C)+LOG(B)}求出的。A表示包括50%的高浓度,B表示包括50%的低浓度,C表示B的抑制率,D表示A的抑制率。
综上所述,利用IC-2处理,HuH7细胞的增殖显著地被抑制(图1、2)。并且,利用PN3-13处理时,HuH7细胞的增殖也被显著地抑制(图3、4)。另外,利用HC-1处理时,HuH7细胞的增殖也被显著地抑制(图5)。
1.5癌症干细胞的抑制效果(FACS分析)
按以下步骤,用癌症干细胞的标记物CD44作为指标来调查由低分子化合物带来的癌症干细胞抑制效果。回收达到70~90%融合的HuH7细胞,在10cm培养皿(TPP Techno Plastic Products AG,Trasadingen,Switzerland)上播种各1.5×106个细胞。15小时之后分别用六氯苯(Hexachlorophen)(15μM),ICG-001(15μM)、PKF118-310(5μM)、IC-2(50μM)、PN3-13(10μM)或者5-FU(0.5μM)处理,在37℃下进行孵化。对照品使用了各低分子化合物以及作为抗癌剂溶剂的DMSO。用药剂处理2天后从培养皿回收细胞,以1000rpm在4℃下进行5分钟离心,除去上清液,用1mL的0.5%FBS/2mM EDTA/PBS洗涤2次。在5%BSA/0.5%FBS/2mM EDTA/PBS 500μL中悬浮,在4℃下进行15分钟封闭。
添加抗人CD44单克隆抗体(Cell Signaling Technology Inc.,Danvers,MA,USA)5μL后,使之悬浮,4℃下在暗处进行第1次抗体反应10分钟。之后,用PBS 1mL洗涤三次。添加1.0μg的山羊抗小鼠IgG Alexa 488标记(Life Technologies Corp.,Carlsbad,CA,USA)后,使之悬浮,4℃下在暗处进行第2次抗体反应。用PBS 1mL洗涤3次后,再用5%FBS/2mM EDTA/DMEM洗涤1次。之后,在5%FBS/2mM EDTA/PBS 500μL中悬浮后,通过40μm的网状管(Becton,Dickinson and Company,Franklin,Lakes NJ,USA)。用Beckman Coulter-Moflo XDP(Beckman Coulter Inc.,Fullerton,CA,USA)进行分析。此外,有意差利用Student t检验(双尾)来评价,图中的*表示p<0.05,**表示p<0.01(通用于实施例中的全部图)。在n=5的条件下进行。
综上所述,利用IC-2或者PN3-13时,癌症干细胞的细胞数量与对照品相比显著地减少(图6)。利用代表性抗癌剂5-FU时,癌症干细胞的细胞数量反而增加。
<实施例2>利用肝癌模型小鼠的恶性肿瘤的治疗效果的评价
将CD44阳性HuH7细胞移植到皮下的小鼠分成3组(DMSO组:5只,5-FU组:4只,IC-2组:4只),将30mg/kg 5-FU、50mg/kg IC-2以及作为对照品使用各种药剂的溶剂DMSO,每3天进行腹膜给药。每3天测定各小鼠的体重、肿瘤的长径、短径,肿瘤的体积由以下公式来算出。肿瘤的体积=长径×(短径)2×0.5。肿瘤的体积是,将第0天(day 0)的体积进行标准化作成图表。为了充分评价5-FU的效果,5-FU的给药量设定为在论文中一般采用的25mg/kg的2倍量。IC-2的给药量是从在in vitro显示Wnt/β-catenin信号抑制效果的浓度算出所对应的浓度,设定为该浓度的2倍量。
综上所述,给药IC-2以及5-FU时均未发现体重的变化(图7)。这说明,IC-2以及5-FU均可安全地给药。并且,IC-2显示了明显高于5-FU的恶性肿瘤的治疗效果(图8)。
<实施例3>对球形成能力的影响
回收HuH7细胞后,用抗人CD44单克隆抗体(Cell Signaling Technology Inc.,Danvers,MA,USA)以及山羊抗小鼠IgG Alexa 488进行染色,用Beckman Coulter-Moflo XDP(Beckman Coulter Inc.,Fullerton,CA,USA)分离成CD44阴性划分和CD44阳性划分。将划分的细胞以5×104cells/well的量分别播种到24孔超低附着培养板(24-well Ultra-Low attachment multiwell plate,Corning,NY,USA)中,在37℃、5%CO2的存在下,用含有20ng/mL重组人表皮细胞生长因子(recombinant human epidermal growth factor)、20ng/mL人碱性成纤维细胞生长因子(human basic fibroblast growth factor)、1×B27、L-谷氨酰胺(L-glutamine)的DMEM/营养混合物(Nutrient Mixture)F-12Ham(Sigma-Aldrich,St.Louis,MO,USA)进行培养。播种24小时之后,将1%DSMO、50μM IC-2、10μM PN-3-13分别加到孔中,经过药剂处理1周后算出15个视野/孔(well)内的100μm以上的球数。
综上所述,IC-2以及PN-3-13抑制了CD44阴性以及CD44阳性的HuH7细胞的球形成(图9)。这说明,IC-2以及PN-3-13抑制了癌细胞以及癌症干细胞的功能。
<实施例4>癌症干细胞的FACS分析
4.1CD90(癌症干细胞标记物)
将肝癌细胞株HLF以1.5×106cells/dish的方式播种到10cm培养皿中,在37℃、5%CO2的存在下进行培养24小时后添加1%DSMO、0.5μM 5-FU、50μM IC-2进行孵化。用药剂处理2天后回收细胞,用0.5%BSA/0.5%FBS/2mM EDTA/PBS进行封闭处理。在500μL的细胞悬浮液中加入5μL APC小鼠抗人CD90抗体(BD Biosciences,San Jose,CA,USA)使之悬浮,在4℃进行孵化10分钟,由此进行第1次抗体反应。抗体反应后,用0.5%FBS/2mM EDTA/PBS洗涤2次,用5%FBS/2mM EDTA/DEME洗涤1次,在5%FBS/2mM EDTA/PBS 500μL中悬浮后,添加1μg/mL碘化丙啶(propidium iodide),通过40μm网状管。CD90表达细胞用Beckman Coulter-Moflo XDP进行分析。5-FU的浓度是根据WST化验24小时的IC50的浓度设定的。IC-2的浓度是根据显示Wnt/β-catenin信号抑制效果的浓度来设定的。
综上所述,CD90表达细胞的比率为,DMSO为21.8%、5-FU为24.1%、IC-2为13.5%(图10)。即,IC-2显示较高的肝癌干细胞的抑制效果。
4.2CD133(癌症干细胞标记物)
将肝癌细胞株HepG2以1.5×106cells/dish的量播种到10cm培养名,在37℃、5%CO2的存在下培养。24小时后添加1%DSMO、0.5μM 5-FU、50μM IC-2进行孵化、药剂处理2天后回收,将该回收的细胞用0.5%BSA/0.5%FBS/2mM EDTA/PBS进行封闭处理。在500μL细胞悬浮液中加入50μL抗人CD133单克隆抗体(Miltenyi Biotec)使之悬浮,在4℃下孵化10分钟,由此进行第1次抗体反应。第1抗体反应后,用PBS 1mL洗涤3次,加入1.0μg山羊抗小鼠IgG Alexa 488标记(Life Technologies Corp.,Carls bad,CA,USA)后,使之悬浮,4℃下在暗处进行10分钟第2次抗体反应。用5%FBS/2mM EDTA/PBS洗涤2次,再用5%FBS/2mM EDTA/DEME洗涤1次,用5%FBS/2mM EDTA/PBS 500μL悬浮后,加入1μg/mL碘化丙啶,通过40μm网状管。CD133表达细胞是用Beckman Coulter-Moflo XDP进行分析的。
综上所述,CD133表达细胞的比率为,DMSO为57.1%、5-FU为33.3%、IC-2为1.45%(图11)。即,IC-2显示较高的肝癌干细胞的抑制效果。
4.3EpCAM(癌症干细胞标记物)
将肝癌细胞株HepG2以1.5×106cells/dish的量播种到10cm培养名中,在37℃、5%CO2的存在下培养。24小时后加入1%DSMO、0.5μM 5-FU、50μM IC-2进行孵化、药剂处理2天后回收,将该回收的细胞用0.5%BSA/0.5%FBS/2mM EDTA/PBS进行封闭处理。500μL细胞悬浮液中加入0.3μL抗人EpCAM单克隆抗体(Cell Signaling Technology Inc.,Danvers,MA,USA)使之悬浮,在4℃进行10分钟孵化,由此进行第1次抗体反应。第1次抗体反应后,用PBS 1mL洗涤3次,加入1.0μg山羊抗小鼠IgG Alexa 488标记(Life Technologies Corp.,Carls bad,CA,USA)后,使之悬浮,4℃下在暗处进行10分钟第2次抗体反应。用5%FBS/2mM EDTA/PBS洗涤2次,再用5%FBS/2mM EDTA/DEME洗涤1次,用5%FBS/2mM EDTA/PBS 500μL悬浮后,加入1μg/mL碘化丙啶,通过40μm网状管。EpCAM表达细胞是用Beckman Coulter-Moflo XDP进行分析的。
综上所述,EpCAM表达细胞的比率为,DMSO为76.2%、5-FU为81.4%、IC-2为51.1%(图12)。即,IC-2显示较高的肝癌干细胞的抑制效果。
<实施例5>Wnt/β-catenin信号抑制效果的评价
5.1CF4-CMVpro-Luc稳定细胞株的构建
在5×106个HuH7细胞中加入30μg的pTCF4-CMVpro-Luc,用Cellject Pro(Thermo,Massachusetts,USA)导入质粒(Plasmid)(250V、1500μF)。转移基因24小时后添加2μg/mL嘌呤霉素(puromycin)进行筛选,培养液每隔3~5天交换。导入pTCF4-CMVpro-Luc的HuH7细胞分别在筛选开始的20天后发现有菌落的形成。将蓝枪头的顶端切断而作成的克隆环,在该环的边缘上抹上凡士林包围形成的菌落,用PBS稀释5倍的0.25%胰蛋白酶/1mM EDTA溶液30μL剥离并回收细胞,加入70μL的DMEM在96孔板上播种。
其次,回收达到70~90%融合的HuH7细胞,在24孔板(FALCON)的各孔中分别播种3.5×105个细胞。经过24小时后在15mL锥形离心管中回收细胞,在1000rpm、室温下离心5分钟,用1×PBS(-)1mL洗涤,在1000rpm、室温下再次进行5分钟离心。将细胞块悬浮到蛋白酶K缓冲液(Proteinase K Buffer)375μL中,加入十二烷基硫酸钠(sodium dodecyl sulfate,SDS)25μL、蛋白酶K(Proteinase K)(20mg/mL)4μL,在55℃下孵化一晚,消化细胞蛋白质。其次,加入400mL酚/氯仿用漩涡震荡混合器混合,在15000rpm、4℃下离心5分钟,回收上清液加入2倍量的100%乙醇,在-80℃下孵化30分钟。在15000rpm、4℃下离心分离20分钟后,舍去上清液加入70%乙醇500μL。在15000rpm、4℃下再次离心10分钟,最后舍去上清液,将沉淀物用20μL的超纯水溶解,用1μL分光光度计(LMS,东京,日本)测定DNA的量,使之调整至1ng/μL。之后,进行了聚合酶链反应(Polymerase Chain Reaction,PCR)。作为内标物(Internal Control)使用了针对甘油醛-3-磷酸脱氢酶(Glyceraldehyde-3-phosphate dehydrogenase,GAPDH)区域的引物(Primer)。并且,使用了针对荧光素酶(Luciferase)区域的引物。
PCR反应使用了rTaq(TOYOBO,大阪,日本),以10μL的规模,各基因的扩增分别在95℃进行2分钟,之后在95℃进行30秒,在60℃进行30秒,在72℃进行30秒,循环25次,在72℃进行5分钟(GAPDH),在95℃进行2分钟,在95℃进行30秒,在55℃进行30秒,在72℃进行30秒,循环30次,在72℃进行5分钟(luciferase基因)。
其次,进行了萤光素酶检测。回收达到70~90%融合的HuH7细胞,在24孔板(FALCON)的各孔中分别播种5×104个细胞。细胞播种17小时后,在24孔板的各孔中分别加入20%被动裂解液(Passive Lysis Buffer,PLB)(PROMEGA、Wisconsin、USA)100μL,在室温下震荡30分钟。在3.5mltube(SARSTEDT,Numbrecht,German)中分别加入荧光素酶分析试剂(Luciferase Assay Reagent,LAR)(PROMEGA)50μL。在24孔板中分别加入用PLB溶解的细胞10μL,用MiniLumat LB 9506(Berthold Technologies,Bad Wildbad,German)测定荧光素酶活性(luciferase activity)。
5.2萤光素酶检测
回收达到70~90%融合的pTCF4-CMVpro-Luc稳定表达HuH7细胞,在96孔板(FALCON)的各孔中分别播种1×104个细胞。细胞播种12小时后,用如图13所示浓度的IC-2、PN-3-13或者5-FU处理,在37℃下进行孵化。对照品用了DMSO。用低分子化合物处理2天后,在96孔白色板(Corning Inc.,New York,USA)的各孔中分别加入Steady-Glo(PROMEGA,Wisconsin,USA)100μL,在室温下孵化5分钟。之后,用荧光酶标仪infinite F500(TECAN,Zurich,Switzerland)测定荧光霉素的活性。
综上所述,IC-2以及PN-3-13显示出Wnt/β-catenin信号的抑制效果(图13)。5-FU未显示Wnt/β-catenin信号的抑制效果。
<实施例6>对大肠癌的抗肿瘤效果
6.1细胞培养
将大肠癌细胞(DLD-1,HCT 116,colo 205)在10cm细胞培养皿(TPP Techno Plastic Products AG,Trasadingen,Switzerland)上用DMEM,在5%CO2、37℃、100%湿度的条件下进行培养。达到70~90%融合时,用PBS(-)洗涤,相对于PBS(-)2mL添加胰蛋白酶-EDTA 300μL,在37℃下孵化5分钟后剥离细胞,用DMEM 5mL回收细胞。将回收的细胞在1000rpm条件下离心3分钟,除去上清液,在DMEM中悬浮后按1:4传代。
6.2抗肿瘤效果
将大肠癌细胞(DLD-1,HCT 116或者colo 205)在96孔板(TPP)上分别播种5×105个细胞。24小时后,用IC-2、PN3-13或者HC-1按照如图14~16所示的的浓度处理。用低分子化合物处理48小时后,加入10%TetraColor ONE(SEIKAGAKU CORPORATION,东京,日本)100μL,在37℃下孵化,用96孔板酶标仪(TECAN,Zurich,Switzerland)测定吸光度(测定波长450nm/参考波长600nm)。
综上所述,IC-2、PN3-13以及HC-1显示大肠癌细胞的抑制效果(图14~16)。
6.3球形成能力的评价
对DLD-1以细胞表面的标记物CD44的表达作为指标,划分成上位5%的群和下位5%的群组1次抗体使用抗CD44抗体(Anti-human CD44 antibody)(Abcam Ltd.,Cambridge,UK),使用山羊IgG Alexa 488抗小鼠(Life Technologies Corp.,Carlsbad,CA,USA)后,通过MoFlo XDP(Beckman Coulter Inc.,Fullerton,CA)解析·分取各组。回收的细胞用24孔超低附着培养板(Corning Inc.,Corning,NY),在添加有重组人碱性成纤维细胞生长因子(recombinant human basic fibroblast growth factor)(bFGF:Corefront Corporation,东京,日本)20ng/mL以及重组人表皮细胞生长因子(Sigma Life Science,St.Louis,United States)20ng/mL、B-27(life technologies)、L-谷氨酰胺(invitrogen,Life Technologies Corp.,Carlsbad,CA)200mmol/L、以及0.6%甲基纤维素(methyl cellulose)(Sigma Life Science)的无添加血清DMEM/F12培养基(Sigma Life Science)500μl下,在各孔中散布5,000个细胞来评价球形成能力的差异。第三天追加上述组成的培养基500ml,第7天进行评价。用位相差顕微镜,将各孔拍摄10个视野的照片,球的总数量用免费软件Image J计算,比较各群的球的总数。
在DLD-1,更强地表达CD44的细胞群的各孔的球形成数显著高于CD44的表达弱的细胞群、(图17)。由此可知,强烈表达CD44的群显示球形成能力,且显示含有更多的具有癌症干细胞性的细胞。
6.4癌症干细胞的抑制效果
将DLD-1在10cm培养皿上播种1×106个细胞。24小时后,用5-FU:0.5、5μM或者IC-2:50μM处理。并且,48小时后回收细胞。1次抗体使用抗CD44抗体(Abcam Ltd.,Cambridge,UK),使用山羊IgG Alexa 488抗小鼠(Life Technologies Corp.,Carlsbad,CA,USA)。其后,通过MoFlo XDP(Beckman Coulter Inc.,Fullerton,CA)进行分析。
综上所述,通过IC-2处理,CD44high细胞(强烈表达CD44的细胞)的比率相对于对照品显著减少(图18)。即,IC-2显示癌症干细胞的抑制效果。另外,用5-FU反而增加。
6.5Wnt/β-catenin信号的抑制效果
将大肠癌细胞(DLD-1,HCT 116,colo 205)在24孔板中分别播种3.34×104个细胞。24小时后用1μL脂质体2000(lipofectamine 2000)(invitrogen,Life Technologies Corp.,Carlsbad,CA)、50ng pTCF4-CMVpro-FLuc以及2.5ng pCMVpro-Rluc进行转染(transfection)。4小时后,用IC-2、PN3-13或者HC-1按照如图19~21所示的浓度处理。用低分子化合物处理48小时后,将20%被动裂解液(PROMEGA,Wisconsin,USA)在24孔板的各孔中分别加入100μL,在常温下震荡15分钟后,在-30℃静置一晚。第二天,通过Dual-LuciferaseR Reporter Assay System(PROMEGA),用MiniLumat LB 9506(Berthold Technologies,Bad Wildbad,German)测定荧光素酶活性。
综上所述,IC-2、PN3-13以及HC-1显示大肠癌细胞的Wnt/β-catenin信号抑制效果(图19~21)。
<实施例7>鳞状细胞癌的抗肿瘤效果
7.1细胞培养
将鳞状细胞癌的细胞株HSC2在10cm细胞培养皿(TPP Techno Plastic Products AG,Trasadingen,Switzerland)中,用DMEM在5%CO2、37℃、100%湿度的条件下培养。达到70~90%融合时,用PBS(-)洗涤后,相对于PBS(-)2mL添加胰蛋白酶-EDTA 300μL,在37℃孵化5分钟分后剥离细胞,用DMEM 5mL回收细胞。回收的细胞在1000rpm的条件下离心3分钟,除去上清液在DMEM中悬浮,按1:4传代。
7.2抗肿瘤效果抗
将HSC2在96孔板(TPP)上分别播种2.5×103个细胞。24小时后,用IC-2、PN3-13或者HC-1按照图22~24所示的浓度以及时间进行处理。用低分子化合物处理48小时后,添加10%TetraColor ONE(SEIKAGAKU CORPORATION,东京,日本)100μL,在37℃孵化,用96孔板酶标仪(TECAN,Zurich,Switzerland)测定吸光度(测定波长450nm/参考波长600nm)。另外,将鳞状细胞癌的细胞株HSC3也与上述相同的方法进行培养后,用IC-2调查抗肿瘤效果。
综上所述,IC-2、PN3-13以及HC-1显示鳞状细胞癌细胞的增殖抑制效果(图22~25)。
7.3癌症干细胞的抑制效果
将HSC2在10cm培养皿上分别播种5×105个细胞。24小时后,用5-FU:0.5μM、IC-2:25μM、PN-3-13:7.5μM或者HC-1:50μM进行处理。另外,还制备了未用低分子化合物处理的细胞(0μM)。此外,48小时后回收各细胞。作为1次抗体使用抗CD44抗体(Abcam Ltd.,Cambridge,UK),还使用了山羊IgG Alexa 488抗小鼠(Life Technologies Corp.,Carlsbad,CA,USA)。之后,通过BD bioscience FACS Aria细胞分选仪(BD Biosciences,CA,USA)进行分析。应当指出,各个低分子化合物的浓度是根据WST化验48小时的IC50の的浓度为基准设定的。
综上所述,CD44表达细胞的比率为,未用低分子化合物处理时83.9%,相对于此,用IC-2时减少至71.4%、用PN-3-13时减少至68.4%以及用HC-1时减少至49.8%(图26)。即,IC-2、PN3-13以及HC-1显示对癌症干细胞的抑制效果。另外,在其中HC-1的效果显著。此外,用5-FU时为83.3%,没有发现变化。
7.4Wnt/β-catenin信号抑制效果
将HSC2在24孔板上分别播种5×104个细胞。24小时后,用1μL脂质体2000(invitrogen,Life Technologies Corp.,Carlsbad,CA)、50ng pTCF4-CMVpro-FLuc以及2.5ng pCMVpro-Rluc进行转染。4小时后,用IC-2、PN3-13或者HC-1按照如图27~29所示的浓度进行处理。用低分子化合物处理48小时后,在24孔板上分别加入20%被动裂解液(PROMEGA,Wisconsin,USA)100μL,在常温下震荡15分钟后,在-30℃静置一晚。第二天,通过双荧光素酶报告系统(Dual-LuciferaseR Reporter Assay System)(PROMEGA),用MiniLumat LB 9506(Berthold Technologies、Bad Wildbad、German)测定荧光素酶活性。
综上所述,IC-2、PN3-13以及HC-1显示鳞状细胞癌细胞的Wnt/β-catenin信号的抑制效果(图27~29)。
<实施例8>IC-2衍生物的评价
8.1合成
IC-2衍生物是按照如图30~35所示的合成路线来合成的。合成的详细说明如下。合成的IC-2衍生物的结构式以及光谱数据如图36以及37所示。
化合物1
混合1-萘甲醛(1-naphtaldehyde)(1.6g,10mmol)和2,2-二甲氧基乙胺(2,2-dietoxyethanamine)1.3g,10mmol),在100℃搅拌30分钟~1小时。冷却后,反应混合物中加入EtOH(25mL),搅拌使之均匀,逐次少量添加NaBH4(0.38g,10mmol),之后在室温下搅拌1小时~一晚。反应结束后,通过减压浓缩蒸发EtOH,在得到的残渣中加入水(适量),将生成物用AcOEt萃取。分离的有机层用饱和盐水洗净,用Na2SO4干燥后,进行过滤以及减压浓缩。得到的残渣用硅胶柱色谱法(AcOEt/hexane=5/1)纯化得到化合物1(2.3g,8.5mmol,85%)的无色透明液体。
化合物1-R
用4-取代苯甲醛(benzaldehyde)(取代基R=Ome、Cl或者F)代替1-萘甲醛,通过与化合物1相同的步骤进行操作。
化合物2b
Fmoc-L-Phe-OH(0.54g,2.0mmol)的干燥DMF(7mL)溶液中加入HATU(0.76g,2.0mmol)和二异丙基乙胺(diisopropylethylamine,DIEA)(0.26g,2.0mmol),在室温搅拌30分钟后,在该反应混合物中添加1(0.54g,2.0mmol),在室温下搅拌一晚。反应结束后加水(20mL),用AcOEt萃取生成物。分离的有机层用饱和盐水洗涤2次,用Na2SO4干燥后,进行过滤以及减压浓缩。得到的残渣用硅胶柱色谱法(AcOEt/hexane=1/2)纯化得到2b(1.2g,1.9mmol,95%)的无色固体。
化合物2b-R
用化合物1-R(R=OMe、Cl又はF。在以下实施例8中相同)代替1,通过与2b相同的步骤进行操作。
化合物3b
2b(1.1g,1.7mmol)的CH2Cl2(20mL)溶液中加入二乙胺(diethylamine,DEA)(10mL),在室温下搅拌3小时。反应结束后,通过减压浓缩蒸发CH2Cl2以及多余的DEA,得到的残渣用硅胶柱色谱法(AcOEt/EtOH=5/1)纯化得到3b(0.55g,1.3mmol,76%)的无色透明的粘性液体。
化合物3b-R
用2b-R代替2b,通过与3b相同的步骤进行操作。
化合物4b
Fmoc-β-Ala-OH(2.5g,8.0mmol)的干燥DMF(15mL)溶液中加入HATU(3.3g,8.7mmol)和DIEA(1.1g,8.5mmol),在室温搅拌30分钟后,在其反应混合物中加入3b(3.3g,7.8mmol),在室温下搅拌一晚。反应结束后加水(30mL),用AcOEt萃取生成物。分离的有机层用饱和盐水洗涤2次,用Na2SO4干燥后,进行过滤以及减压浓缩。得到的残渣用硅胶柱色谱法(AcOEt/hexane=3/1)纯化得到4b(5.1g,7.1mmol,91%)的无色固体。
化合物4b-R
用3b-R代替3b,通过与4b相同的步骤进行操作。
化合物6b
4b(2.8g,3.9mmol)的CH2Cl2(10mL)溶液中加入DEA(6mL),在室温下搅拌3~4小时。通过减压浓缩蒸发CH2Cl2以及多余的DEA,得到的残渣中加入CH2Cl2(适量)使之成为均匀的溶液后,再次进行减压浓缩。进行2次该操作后得到的残渣中加入CH2Cl2(10mL)后,搅拌使之均匀,加入异氰酸苯(benzyl isocyanate)(0.78g,5.9mmol)在室温下搅拌一晚。反应结束后,通过减压浓缩蒸发CH2Cl2,得到的残渣用硅胶柱色谱法(AcOEt/EtOH=30/1)纯化得到6b(1.5g,2.4mmol,62%)的无色固体。
化合物6b-R
用4b–R代替4b,通过与6b相同的步骤进行操作。
化合物8b
4b(1.6g,2.3mmol)中加入蚁酸(10mL),在室温下搅拌一晚。反应结束后通过减压浓缩蒸发蚁酸,得到的残渣用硅胶柱色谱法(AcOEt/hexane=4/1)纯化得到8b(1.3g,2.1mmol,91%)的无色固体。
化合物8b-R
用4b-R代替4b,通过与8b相同的步骤进行操作。
化合物9b
8b(1.1g,1.8mmol)的CH2Cl2(5.5mL)溶液中加入二乙胺(diethylamine)(1.3g,18mmol,1.8mL),在室温下搅拌3小时。反应结束后通过减压浓缩蒸发CH2Cl2,得到的残渣用硅胶柱色谱法(AcOEt/EtOH=7/1)纯化得到9b(0.57g,1.4mmol,78%)的无色固体。
化合物9b-R
用8b-R代替8b,通过与9b相同的步骤进行操作。
IC-2(从6b合成)
6b(1.3g,2.1mmol)中加入蚁酸(8mL,0.21mol),在室温下搅拌一晚。反应结束后,通过减压浓缩蒸发蚁酸,得到的残渣用硅胶柱色谱法(AcOEt/EtOH=30/1)纯化得到IC-2(1.0g,1.9mmol,90%)的无色固体。
IC-2-R
用6b-R代替6b,通过与IC-2(从6b合成)相同的步骤进行操作。
IC-2(从9b合成)
9b(3.3g,8.3mmol)的CH2Cl2(10mL)溶液中加入异氰酸苯(benzyl isocyanate)(1.4g,11mmol),在室温下搅拌一晚。反应结束后,通过减压浓缩蒸发CH2Cl2,得到的残渣用硅胶柱色谱法(AcOEt/EtOH=30/1)纯化得到IC-2(3.7g,6.9mmol,83%)的无色固体。
4-(4-甲氧基苄基氧基)苯基乙酸(4-(4-Methoxybenxyloxy)phenylacetic acid)
甲基-4-羟基苯(methyl 4-hydroxyphenylacetate)(2.7g,16mmol)的干燥DMF(20mL)溶液中加入K2CO3(4.4g,32mmol)和4-甲氧基氯苄(4-methoxybenzyl chloride)(1.3g,8mmol),在室温下搅拌24小时。反应混合物中投入冰水(30mL)用EtOAc萃取生成物。分离的有机层用饱和盐水洗涤,用Na2SO4干燥后,进行过滤以及减压浓缩。得到的残渣中加入MeOH(24mL)和THF(8mL)搅拌使之均匀,缓慢加入NaOH水溶液(0.96g,24mmol,6mL),在室温下搅拌2小时。通过减压浓缩蒸发有机溶剂,加水(50mL)后用1M-硫酸使之呈酸性,用乙酸乙酯和THF萃取生成物。有机层用饱和盐水洗涤2次,用Na2SO4干燥,进行过滤以及减压浓缩。得到的残渣通过重结晶(EtOAc-THF)得到纯化4-(4-甲氧基苄基氧基)苯基乙酸(1.8g,6.8mmol,85%)。
4-甲氧基甲氧基苯乙酸(4-Methoxymethoxyphenylacetic acid)
甲基-4-羟基苯(Methyl 4-hydroxyphenylacetate)(2.5g,15mmol)的CH2Cl2(15mL)溶液中加入DIEA(3.9g,30mmol),边用冰浴冷却边加入氯甲基甲基醚(Chloromethyl Methyl Ether)(1.8g,23mmol),在该温度下搅拌10分钟后,返回到室温,搅拌一晚。通过减压浓缩蒸发CH2Cl2以及多余的氯甲基甲基醚后,加入MeOH(25mL)后搅拌呈均匀时加入KOH水溶液(3.0g,45mmol,5mL),在室温下搅拌1.5小时。反应混合物中加水(20mL),将水层分离后加入饱和NH4Cl水溶液(20mL),用稀硫酸将pH调至4。在其中加入EtOAc,分离有机层后,用饱和盐水洗涤以及用Na2SO4干燥。通过过滤、减压浓缩以及减压干燥得到4-甲氧基甲氧基苯基乙酸(2.2g,11mmol,76%)。
苄基4-羟基苯(Benzyl 4-hydroxyphenylacetate)
在Ar下,用冰浴冷却的4-羟基苯基乙酸(4-hydroxyphenylacetic acid)(3.0g,20mmol)的干燥DMF(20mL)溶液中加入NaH(60%的油分散液,0.88g,22mmol),在其温度下搅拌30分钟后,将溴苄(benzyl bromide)(6.8g,40mmol)经过30分钟分批加入。在冰浴冷却下搅拌3小时后,在室温下搅拌一晚。在反应混合物中加水(20mL)和EtOAc(20mL),充分搅拌后分离有机层,用5%-NaHCO3水溶液以及饱和盐水洗涤,之后用Na2SO4干燥。通过过滤以及减压浓缩后,得到的固体中加入正己烷后进行抽滤,得到的固体通过减压干燥得到苄基4-羟基苯(3.4g,14mmol,70%)。
4-(叔丁基二甲硅氧基甲基)苯乙酸(4-(tert-Butyldimethylsiloxymethyl)phenylacetic acid)
在Ar下,用冰浴冷却的4-羟甲基苯乙酸(4-Hydroxymethylphenylacetic acid)(1.7g,10mmol)的干燥DMF(10mL)溶液中加入NaH(60%的油分散液,0.44g,11mmol),在其温度下搅拌30分钟后,将溴苄(3.4g,20mmol)经过30分钟分批加入。冰浴冷却下搅拌2小时后在室温下搅拌一晚。在反应混合物中加水(20mL)和AcOEt(20mL)充分搅拌后分离有机层,用5%-NaHCO3水溶液和饱和盐水洗涤,并用Na2SO4干燥。过滤后,通过减压浓缩蒸发AcOEt,进一步通过减压蒸馏蒸发多余的溴苄。得到的残渣中加入干燥DMF(10mL),搅拌均匀后,加入叔丁基二甲基氯硅烷(tert-butyldimethylsilyl chloride)(2.0g,13mmol)和咪唑(imidazole)(1.4g,20mmol),在室温下搅拌2小时。在反应混合物中加水(20mL)用AcOEt萃取生成物。分离的有机层用饱和盐水洗涤,并用Na2SO4干燥。通过过滤以及减压浓缩后,得到的残渣中加入EtOH(15mL)搅拌呈均匀时加入5%-Pd/C(1.1g),系统内用H2置换。在室温下搅拌4小时后,通过将两张滤纸重叠设置的方式过滤除去Pd/C,滤液通过减压浓缩后,将残渣通过硅胶柱色谱法(AcOEt/hexane=1/2)得到4-(叔丁基二甲硅氧基甲基)苯乙酸(0.95g,3.4mmol,34%)。
IC-2-OMOM
4-甲氧基甲氧基苯乙酸(0.59g,3mmol)的甲苯(10mL)溶液中加入叠氮磷酸二苯酯(diphenylphosphoryl azide)(0.83g,3mmol)和Et3N(0.36g,3.6mmol),在80℃下搅拌2小时。冷却后,加入正己烷(15mL),搅拌一段时间后将上清液用倾析(decantation)收集。在残渣中再次加入正己烷(7mL),搅拌一段时间后将上清液用倾析收集,并再次进行了相同的操作。将收集的上清液减压浓缩,在残渣中加入CH2Cl2(8mL)呈均匀时加入9b(0.40g,1mmol)。将混合物在室温下搅拌一晚后,通过减压浓缩蒸发有机溶剂,残渣通过硅胶柱色谱法(AcOEt)得到IC-2-OMOM(0.50g,0.85mmol,84%)。
IC-2-NO2
通过与IC-2-OMOM相同的步骤,用4-硝基苯乙酸(4-nitrophenylacetic acid)进行操作。但是,在途中省略了加入正己烷收集上清液的操作,冷却后直接在反应混合物中加入CH2Cl2以及9b。硅胶柱色谱法:AcOEt/EtOH=8/1。收率:24%。
IC-2-OPMB
通过与IC-2-OMOM相同的步骤,用4-(4-甲氧基苄氧基)苯乙酸(4-(4-Methoxybenzyloxy)phenylacetic acid)进行操作。但是,仅用甲苯无法使4-(4-甲氧基苄氧基)苯乙酸成为均匀的溶液,还加入干燥THF(5mL)进行了操作。硅胶柱色谱法:AcOEt/EtOH=30/1。收率:93%。
IC-2-MOTBS
通过与IC-2-OMOM相同的步骤,用4-(叔丁基二甲基硅氧基)苯乙酸(4-(tert-butyldimethylsiloxymethyl)phenylacetic acid)进行操作。硅胶柱色谱法:AcOEt。收率:66%。
IC-2-OH
IC-2-OMOM(0.44g,0.74mmol)的CH2Cl2(3mL)溶液中加入CF3CO2H(1.1mL,14mmol),在室温下搅拌1小时。将反应混合物缓慢加入到5%-NaHCO3水溶液(40mL)中,生成物用AcOEt萃取。分离的有机层用饱和盐水洗涤,并用Na2SO4干燥。通过过滤以及减压浓缩后得到的残渣用硅胶柱色谱法(AcOEt/EtOH=30/1)纯化得到IC-2-OH(0.16g,0.29mmol,39%)。
IC-2-MOH
IC-2-MOTBS(0.51g,0.75mmol)的干燥THF(8mL)溶液中,在冰水冷却的条件下加入TBAF的THF溶液(1M,1.5mL,1.5mmol),在该温度下搅拌10分钟,在室温下搅拌1.5小时。在反应混合物中加水(20mL)后,生成物用AcOEt萃取。分离的有机层用饱和盐水洗涤,并用Na2SO4干燥。通过过滤以及减压浓缩后得到的残渣用硅胶柱色谱法(AcOEt/EtOH=10/1)纯化得到IC-2-MOH(0.30g,0.53mmol,71%)。
8.2Wnt/β-catenin信号的抑制效果
回收达到70~90%融合的pTCF4-CMVpro-Luc稳定表达HuH7细胞,在96孔板(FALCON)的各孔中分别播种1×104个细胞。24小时后用如图38以及39所示浓度的IC-2衍生物(IC-2-OMe、IC-2-F、IC-2-Cl或者IC-2-NO2)处理,在37℃进行孵化。作为对照品用DMSO。用低分子化合物处理4天后,将Steady-Glo(PROMEGA,Wisconsin,USA)在96孔白色板(Corning Inc.,New York,USA)的各孔中分别加入100μL,在室温下孵化5分钟。之后,用荧光酶标仪infinite F500(TECAN,Zurich,Switzerland)测定荧光霉素活性。
综上所述,IC-2衍生物显示Wnt/β-catenin信号的抑制效果(图38以及39)。
<实施例9>纤维化的抑制
9.1细胞培养条件
人肝星状细胞株LX-2细胞是在10%FBS DMEM中维持培养的。将在试验中使用的细胞在1%FBS DMEM中悬浮,进行播种。细胞密度约为2.0×105cell/cm2的条件下进行。
9.2所使用的低分子化合物的浓度
IC-2:0、15、20、25μM,PN-3-13:0、5.5、6.0、6.5μM,HC-1:0、12、16、20μM。
9.3萤光素酶检测
用萤光素酶检测调查各低分子化合物的Wnt/β-catenin信号的抑制效果。萤光素酶检测是按照如图40以及在以下9.3.1~9.3.2项所示的条件下进行的。图40是用低分子化合物+TGFβ处理时的方案,但是在单独用低分子化合物或者用低分子化合物+Wnt3a处理时也进行了相同的实验。另外,TGFβ和Wnt3a是为了提高Wnt/β-catenin信号的强度而添加的。
9.3.1样品的回收
将5×被动裂解液用MQ稀释5倍。以100μl/孔的方式添加后震荡20分钟。在-30℃下,以O/N进行冻结。
9.3.2测定
将前一天冻结的样品、LARII、Stop&Glo Buffer、Stop&Glo Substrate在室温下孵化大约一小时(除了样品进行遮光)。准备样品数量+1个样品份的Stop&Glo液(Stop&Glo Buffer 49μl,Stop&Glo Substrate 1μl)。在3.5ml离心管中分注50μl的LARII,添加样品10μl,设置到测定器。完成第一回的测定后添加50μl的Stop&Glo液,再次设置到测定器进行测定。
9.3.3结果
用IC-2处理24小时后萤光素酶检测的结果如图41~43所示。单独用IC-2处理、或者用IC-2+TGFβ或IC-2+Wnt3a处理的任一种方式处理时,LX-2细胞的Wnt/β-catenin信号都会被抑制。
用IC-2处理48小时后的萤光素酶检测的结果如图44~46所示。单独用IC-2,或者用IC-2+TGFβ处理时,LX-2细胞的Wnt/β-catenin信号被抑制。
用PN3-13处理24、48小时后的萤光素酶检测的结果如图47、48所示。用PN3-13+TGFβ处理时,根据浓度,LX-2细胞的Wnt/β-catenin信号被抑制。
用HC-1处理24小时后的萤光素酶检测的结果如图49~51所示。单独用HC-1、或者用HC-1+TGFβ处理时,LX-2细胞的Wnt/β-catenin信号被抑制。
用HC-1处理48小时后的萤光素酶检测的结果如图52~54所示。单独用HC-1、或者用HC-1+TGFβ、HC-1+Wnt3a中的任一种处理方式时,LX-2细胞的Wnt/β-catenin信号都会被抑制。
9.4实时荧光定量PCR
用实时荧光定量PCR调查各低分子化合物的纤维化抑制效果。实时荧光定量PCR是按照如图55、以及以下9.4.1~9.4.2项所示的条件进行的。另外,TGFβ、α-SMA、COL1A1是纤维化标记物。
9.4.1样品的制备
MQ:5.2μL/样品、MgCl2:0.8μl/样品、10μM引物F:0.5μl、10μM引物R:0.5μl、LightCycler FastStart DNA Master SYBER GreenI(1a+1b):1.0μM/样品作为pre-Mix制备,在各孔中分注8μL。添加样品2μL。
9.4.2PCR反应温度
·GAPDH
95℃:10min→[95℃:10sec→60℃:10sec→72℃:10sec]×35循环
·α-SMA
95℃:10min→[95℃:10sec→56℃:5sec→72℃:10sec]×40循环
·COL1A1
95℃:10min→[95℃:10sec→58℃:5sec→72℃:10sec]×40循环
·TGFβ
95℃:10min→[95℃:1sec→56℃:5sec→72℃:10sec]×40循环
9.4.3结果
用IC-2+TGFβ处理24、48小时后的实时荧光定量PCR的结果示于图56~58。随着IC-2浓度的增加,纤维化标记物的表达量减少。尤其,α-SMA的表达量显著减少。
用PN3-13+TGFβ处理24、48小时后的实时荧光定量PCR的结果示于图59~61。除了24小时处理时的TGFβ以外,随着PN3-13浓度的增加,纤维化标记物的表达量减少。尤其,α-SMA的表达量显著减少。
用HC-1+TGFβ处理24、48小时后的实时荧光定量PCR的结果示于图62~64。随着HC-1浓度的增加,纤维化标记物的表达量减少。尤其,α-SMA的表达量显著减少。
<实施例10>利用肝损伤模型小鼠的纤维化治疗效果的评价
10.1实验动物以及饲养条件
采用健康的7周龄的C57BL/6雄性小鼠(日本SLC,静冈,日本)预备饲养1星期。整个预备饲养以及实验期间都是在室温22±1℃、湿度50±5%的动物室内饲养,使小鼠自由摄取饲料以及水。
10.2肝纤维化的诱导方法以及药物的给药方法
将四氯化碳(CCl4:和光纯药工业,大阪,日本),以0.2ml/kg、每周3次、4、6以及8星期的条件下,用微量注射器(伊藤制作所,静冈,日本)在腹腔内给药。使用的四氯化碳是用玉米油(和光纯药工业)溶解的浓度为10%的溶液。将该四氯化碳溶液给药4星期后,将小鼠分成媒剂(Vehicle)给药组、甘草甜素给药组、ICG-001给药组以及IC-2给药组,将四氯化碳和同时按照以下方法制备的药液,以每星期3次、4星期的条件下,用微量注射器在腹腔内给药。另外,四氯化碳和药液每天交替给药。
将甘草甜素(东京化成工业,东京,日本)溶解到生理盐水中,用4M NaOH溶液调成pH7.0,制备30mg/ml的浓度。为了使IC-2以及ICG-001(AdooQ BioScience,California,USA)分别成40mg/ml以及10mg/ml的浓度,将其溶解于Wellsolve(celeste,东京,日本)中,再用60℃的热水浴中加热10分钟,使之完全溶解。溶解有这些药剂的Wellsolve溶液中加入9倍量的生理盐水。其次,采取所需量的甘草甜素、IC-2以及ICG-001,使浓度成为甘草甜素为150mg/kg,IC-2为10.6、21.2mg/kg以及ICG-001为5mg/kg的药液,将IC-2以及ICG-001制备成wellsolve和生理盐水以1:9比率混合的溶液,将甘草甜素制备成加入生理盐水至液量成200μl的溶液。另外,作为媒剂使用将wellsolve和生理盐水以1:9比率混合的溶液。此外,IC-2的给药量是根据在in vitro的有效浓度而设定的。
10.3肝脏的切除
将四氯化碳给药4星期后,使用安装有27G注射针的1mL的一次性注射器(Disposable syringes)对5只小鼠,相对于每1g腹腔给药1μl的量给药全身麻醉药戊巴比妥(Somnopentyl)(共立制药,东京,日本),进行麻醉诱导。麻醉诱导后,使用27G注射针以及1mL注射器从下腔静脉全血采集,切除整个肝脏。另外,四氯化碳给药8星期后对各组8只小鼠实施如同上述的操作。从切除的肝脏中的1cm见方左右的组织片用于组织学解析,浸泡在4%多聚甲醛(nacalai tesque,京都,日本)。剩余的肝组织片用手术器械弄成碎片,用液态氮瞬间冻结后,将其保存在-80℃的超低温储罐中,直到将其用于实验。
10.4羟脯氨酸的测定
通过上述方法切除的冻结肝组织块用手术器械切成湿重为50mg,加入500μl的超纯水,用POLYTRON均质器(Polytron homogenizer)破碎。将破碎液先在液态氮冻结后在室温下解冻,用超声波细胞破碎仪BioRuptur(Cosmo Bio,东京,日本)在冰水中15秒、冷却15秒为1次循环的方式,共实施30循环、15分钟的超声波处理。该破碎液100μl中加入等量的12N浓盐酸,在设定成120℃的孵化器块(Block incubator)(IKA、Staufen、Germany)中进行16小时的水解反应处理。剩余的破碎液用于测定溶液中的蛋白质的量。水解反应处理后,在室温下冷却后,用移液管吹打水解产物成细腻状态,在室温、3000rpm的条件下进行5分钟离心操作。离心操作后将10μl的上清液加到1.5mL离心管中,用冷却蒸发器(佐久间制作所,东京日本)去除盐酸。然后,用羟脯氨酸测定试剂盒(Hydroxyproline quantification kit)(BioVision,California,USA)进行羟脯氨酸的测定。去除盐酸的水解溶解物中加入100μl的氯胺-T溶液,用涡旋振荡混合器VORTEX充分搅拌后静置25分钟。之后,将加入100μl DMAB溶液,在60℃的热水浴中反应90分钟。在室温下冷却后,将反应溶液移到96孔板,用酶标仪(TECAN,Zurich,Switzerland)测定560nm的吸光度。根据校正曲线,从得到的吸光度算出羟脯氨酸的量。
破碎液中的蛋白质量是用蛋白质检测用浓缩燃料试剂(Bio-Rad,California,USA),通过Bradford法测定的。破碎液在15000rpm、4℃下进行10分钟离心,将得到的上清液1μl加到96孔板。在其中加入用超纯水稀释至5倍的蛋白质测定燃料试剂浓缩(Protein assay Dye Reagent concentrate)200μl,搅拌后静置15分钟,用酶标仪(TECAN)测定595nm的吸光度。根据校正曲线,从得到的吸光度算出相对于每1μl破碎液的蛋白质量,求出相对于蛋白质量的羟脯氨酸的量来评价纤维化程度。
10.5天狼星红染色
由上述的方法切除的肝组织块用4%多聚甲醛在室温下固定16小时,用石蜡包埋后用切片机(Microtome)作成组织块,用天狼猩红染色试剂盒(Picosirius Red Stain Kit)(Polysciences,Pennsylvania,USA)
按照所附的操作方法,用色天狼星红染色胶原纤维染。之后,用倒置荧光相差显微镜BZ-9000(KEYENCE,东京,日本)在亮视野下,拍摄每组织块10张100倍放大像,测定各拍摄图像中的相对于组织面积染成红色纤维的面积,来定量纤维化阳性面积比。
10.6结果
羟脯氨酸量的测定结果示于图65。与Vehicle给药组相比,甘草甜素给药组(阳性对照品)的羟脯氨酸的量降低,在IC-2给药组发现大幅度的降低。
用天狼星红染色的纤维化区域的定量结果示于图66。可以认定,与Vehicle组相比IC-2给药组的纤维化区域减少。
<考察>
如上所述,通过利用IC-2等可抑制癌细胞以及癌症干细胞的增殖。癌症干细胞可以成为恶性肿瘤的复发或者转移的原因是已知的。因此,作为恶性肿瘤的治疗药的有效成分,可抑制癌症干细胞增殖的IC-2等可以说是非常优异的化合物。并且,IC-2等还具有抑制作为癌症的发生原因的纤维化的作用。
以上是利用本发明的实施例进行说明的。这些实施例仅属于例示,可以有各种变形例,并且本领域的技术人员也可以理解这些各种变形例也在本发明的范围内。
- 还没有人留言评论。精彩留言会获得点赞!
- 能够与配体偶联的单甲基缬氨酸化合物的制造方法与工艺
- 烟梗纤维微波降解制备烟用料液方法及应用与流程
- 利用特异性挥发物筛选昆虫相关嗅觉蛋白的方法与制造工艺
- 小分子共价抑制剂的计算机筛选方法及其在筛选S-腺苷甲硫氨酸脱羧酶的共价抑制剂的应用与制造工艺
- 一种合成抗原的分子量检测方法及应用与制造工艺
- 一种用于MALDI‑TOF‑MS分析小分子的氧化锌沸石复合材料及其制备方法与制造工艺
- 用于原位收集根系挥发物的试验装置及方法与制造工艺
- 一种核壳型纳米香精胶囊的制备方法与制造工艺
- 小分子共价抑制剂在制备抑制S-腺苷甲硫氨酸脱羧酶的药物上的应用及其筛选方法与制造工艺
- Linderolide H在制备治疗鼻炎药物中的应用的制造方法与工艺