IKKε/TBK1抑制剂与β肾上腺素能激动剂或交感神经系统激活剂的组合的制作方法
本发明是根据由美国国立卫生研究院授予的DK060591在政府支持下进行的。政府享有本发明的某些权利。
领域
本文提供了治疗肥胖症和肥胖症相关病状的方法,所述方法包括施用IKKε/TBK1抑制剂与β肾上腺素能激动剂或交感神经系统激活剂的组合以及包含这类组合的药物组合物。
背景技术:
肥胖症会在肝和脂肪组织中产生一种慢性、低度发炎状态,伴随巨噬细胞浸润和使胰岛素作用衰减的炎性细胞因子和趋化因子的局部分泌,从而导致胰岛素抵抗和2型糖尿病的后续发展(Wellen和Hotamisligil,2005;Hotamisligil,2006;Lumeng等人,2007;Shoelson等人,2007;所述参考文献以引用的方式整体并入本文)。众多研究表明在若干群体当中炎症与胰岛素抵抗之间存在强相关性(Hotamisligil,2006;所述参考文献以引用的方式整体并入本文)。另外,炎症途径的基因切除或药理学抑制可以将肥胖症与胰岛素抵抗解除关联(Hotamisligil,2006;Shoelson等人,2007;所述参考文献以引用的方式整体并入本文)。
转录因子NFκB及其炎症程序在肥胖的肝和脂肪组织中的胰岛素抵抗的发展中发挥了重要作用(Yuan等人,2001;Arkan等人,2005;Wunderlich等人,2008;Chiang等人,2009;所述参考文献以引用的方式整体并入本文)。NFκB由IκB激酶(IKK)家族激活,所述家族具有四个成员:IKKα、IKKβ、IKKε以及TBK1。IKKα和IKKβ与支架配偶体NEMO发生作用以激活NFκB(Hacker和Karin,2006;所述参考文献以引用的方式整体并入本文)。虽然IKKβ的药理学抑制或基因切除定义了这种激酶在胰岛素抵抗中的作用(Yuan等人,2001;Arkan等人,2005;所述参考文献以引用的方式整体并入本文),但是并不确定非经典激酶IKKε和TBK1的作用。
肥胖症是由食物摄取增加,而能量消耗减少引起的复杂的代谢疾病。肥胖症还会增加发展成2型糖尿病、心脏病、中风、关节炎以及某些癌症的风险。有大量证据表明,脂肪组织在肥胖症状态下会对儿茶酚胺诸如肾上腺素变得不太敏感,并且这种降低的敏感性反过来会减少能量消耗。然而,并不完全理解这个过程的细节。
发明概要
本文提供了治疗肥胖症和肥胖症相关病状的方法,所述方法包括施用IKKε/TBK1抑制剂与β肾上腺素能激动剂或交感神经系统激活剂的组合以及包含这类组合的药物组合物。
在一些实施方案中,本发明提供治疗患有与肥胖症、胰岛素抵抗或肝性脂肪变性相关联的病状的受试者的方法,所述方法包括:向患有与肥胖症、胰岛素抵抗或肝性脂肪变性相关联的病状的受试者施用:(i)IKKε和/或TBK1抑制剂,以及(ii)β肾上腺素能激动剂或交感神经系统激活剂。在一些实施方案中,施用会引起受试者体内的体脂肪的减少。在一些实施方案中,受试者已经具有或处于经历肥胖症、糖尿病或胰岛素抵抗的风险。在一些实施方案中,糖尿病是II型糖尿病。在一些实施方案中,治疗导致葡萄糖代谢增加、体脂肪减少、体脂肪增加缺乏、胰岛素受体信号传导增加、胰岛素受体磷酸化水平降低、肝中慢性炎症减少或得到预防、脂肪组织中慢性炎症减少或得到预防、肝性脂肪变性减少或得到预防、代谢能量消耗得到促进、循环游离脂肪酸减少或胆固醇减少。在一些实施方案中,受试者患有肝性脂肪变性(脂肪肝疾病)。在一些实施方案中,受试者患有脂肪性肝炎。在一些实施方案中,受试者是超重的或肥胖的。在一些实施方案中,受试者是人。
在一些实施方案中,方法还包括以下步骤,所述步骤包括测试受试者的选自由以下组成的组的疾病或病状:受损的胰岛素信号传导、肥胖症、糖尿病、胰岛素抵抗、代谢综合征、肝性脂肪变性、慢性肝脏炎症以及脂肪组织中的慢性炎症。在一些实施方案中,方法还包括基于所述测试评定治疗的效果的步骤。在一些实施方案中,方法还包括基于所述评定来调节治疗。在一些实施方案中,调节治疗包括以下各项中的一个或多个:改变IKKε/TBK1抑制剂的剂量、换成不同的IKKε/TBK1抑制剂、改变β肾上腺素能激动剂或交感神经系统激活剂的剂量、换成不同的β肾上腺素能激动剂或交感神经系统激活剂、增加附加治疗。
在一些实施方案中,方法包括施用共同配制成单一药物组合物的IKKε和/或TBK1抑制剂和β肾上腺素能激动剂或交感神经系统激活剂。在其他实施方案中,IKKε和/或TBK1抑制剂和β肾上腺素能激动剂或交感神经系统激活剂是分开的药物组合物,并且是共同施用的(例如,在1小时内、在20分钟内、在15分钟内、在5分钟内、在1分钟内、同时等)。
在一些实施方案中,本发明提供药物组合物,所述药物组合物包含:(i)IKKε和/或TBK1抑制剂,以及(ii)β肾上腺素能激动剂或交感神经系统激活剂。在一些实施方案中,IKKε和/或TBK1抑制剂包含小分子。在一些实施方案中,IKKε和/或TBK1抑制剂包含具有式I的结构:
其中R1是氢、烷基、苯基、羧基、羟基、烷氧基或氨基,所述R1可以是未取代的或被一个烷基取代;m是0、1或2;并且R2是烷基、烷氧基、卤素、硝基、羟基、羧基、亚丁二烯基(–CH=CH–CH=CH–),所述R2与任何邻近的碳原子或氨基形成苯环,所述R2可以是未取代的或被至少一个烷基取代;以及其生理学上可接受的盐。在一些实施方案中,IKKε和/或TBK1抑制剂包含氨来呫诺。在一些实施方案中,β肾上腺素能激动剂或交感神经系统激活剂包含小分子。在一些实施方案中,β肾上腺素能激动剂或交感神经系统激活剂包含β2肾上腺素能受体激动剂。在一些实施方案中,小分子β肾上腺素能激动剂或交感神经系统激活剂是芬特明。
附图简述
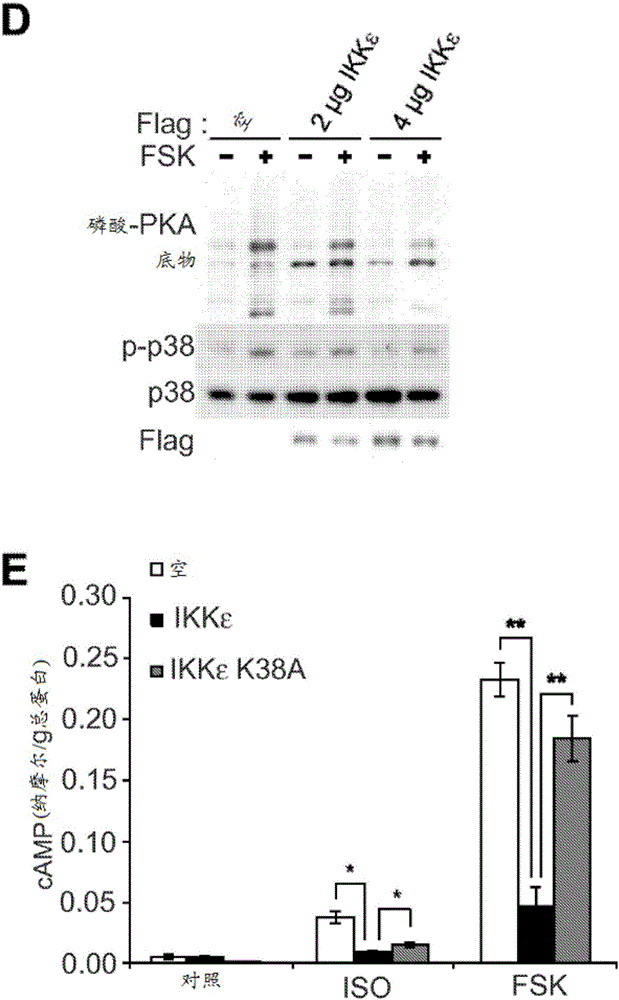
图1示出IKKε和TBK1过表达降低对3T3-L1脂肪细胞中β-肾上腺素能/cAMP途径的敏感性。(A)在用或未用10μM ISO(黑条)或10μM CL-316,243(CL,灰条)处理4小时之后,Ucp1表达在表达空载体、Flag-IKKε或Flag-IKKε K38A的3T3-L1脂肪细胞中的增加倍数。(B)用或未用10μM ISO或10μM CL处理的表达空载体(白条)、Flag-IKKε(黑条)或Flag-IKKε K38A(灰条)的3T3-L1脂肪细胞中的甘油释放。(C)图1B的全细胞溶解产物的免疫印迹。复制结果,重复三次。D.E.表示避光暴露,而L.E.表示光暴露。(D)用或未用50μM FSK处理15分钟的表达空载体或Flag-IKKε的3T3-L1脂肪细胞中的全细胞溶解产物的免疫印迹。(E)用或未用10μM ISO或50μM FSK处理15分钟的表达空载体、Flag-IKKε或Flag-IKKε K38A的3T3-L1脂肪细胞的cAMP水平。
图2示出用TNFα进行延长处理以依赖于IKKε和TBK1的活性的方式降低脂肪细胞对β-肾上腺素能刺激的敏感性。(A)用或未用不同浓度的如所指示的TNFα处理24小时,之后用或未用10μM ISO或50μM FSK处理的3T3-L1脂肪细胞中的甘油释放。(B)在50μM氨来呫诺(Am)预处理存在或不存在下,用或未用100ng/ml TNFα处理24小时,之后用或未用10μM ISO或50μM FSK处理的3T3-L1脂肪细胞的cAMP水平。(C)在1μM CAY10576(CAY)预处理存在或不存在下,用或未用100ng/ml TNFα处理24小时,之后用或未用50μM FSK处理的3T3-L1脂肪细胞的cAMP水平。(D)用或未用不同浓度的与图2A相同的TNFα处理24小时,之后用或未用10μM ISO或50μM FSK处理的3T3-L1脂肪细胞的全细胞溶解产物的免疫印迹。在多个实验中,结果是重复的。‘[’指示总HSL。‘n.s.’表示非特异性条带。箭头指示CGI-58。(E)在用浓度增加(0、10、50和200μM)的氨来呫诺持续30分钟的预处理存在或不存在下,用或未用50ng/ml TNFα或100μg/ml poly(I:C)处理24小时,之后用或未用10μM ISO处理15分钟的3T3-L1脂肪细胞的全细胞溶解产物的免疫印迹。
图3示出IKKε和TBK1通过激活PDE3B来降低cAMP水平。(A)用或未用50μM FSK、250μM IBMX或两者一起处理15分钟的表达空载体、Flag-IKKε或Flag-TBK1的3T3-L1脂肪细胞的cAMP水平。(B)用或未用10μM ISO或50μM FSK,连同或不连同10μM扎达维林(Zarda)一起处理15分钟的表达空载体、Flag-IKKε或Flag-TBK1的3T3-L1脂肪细胞的cAMP水平。(C)使用从HEK293T细胞免疫沉淀的HA-PDE3B或1μg MBP(髓磷脂碱性蛋白)作为如所指示的重组激酶的底物进行的体外激酶反应的32P磷酸图像。(D)用抗-HA抗体进行免疫沉淀,之后用或未用CIP处理的免疫沉淀物(上图)和共同表达HA-PDE3B与Flag-IKKε/TBK1或Flag-IKKε/TBK1K38A的Cos-1细胞的全细胞溶解产物(下图)的免疫印迹。D.E.表示避光暴露,而L.E.表示光暴露。(E)共同表达HA-PDE3B与Flag-TBK1或Flag-TBK1 K38A的HEK293T细胞的GST-14-3-3下拉产物的免疫印迹。丽春红S染色法显示GST-14-3-3下拉中使用的珠粒的量。
图4示出IKKε和TBK1使PDE3B在丝氨酸318处磷酸化,从而导致14-3-3β的结合。(A)质谱实验中PDE3B通过IKKε或TBK1磷酸化的位点(P-位点)的概括。(B)共同表达HA-PDE3B或HA-PDE3B S318A与Flag-TBK1的HEK293T细胞的GST-14-3-3下拉产物的免疫印迹。丽春红S染色法显示GST-14-3-3下拉中使用的珠粒的量。(C)硝酸纤维素膜上的GST-14-3-3覆盖(上图)和共同表达HA-PDE3B或HA-PDE3B S318A与Flag-TBK1的HEK293T细胞的全细胞溶解产物的免疫印迹(IB)(下图)。(D)用或未用100ng/ml TNFα处理16小时,之后用或未用25μM FSK处理15分钟的表达空载体、HA-PDE3B或HA-PDE3B S318A的3T3-L1脂肪细胞的cAMP水平。
图5示出IKKε/TBK1抑制剂氨来呫诺在饮食诱导的肥胖小鼠中的白脂肪组织中敏化受β-肾上腺素能激动剂刺激的脂解。(A)在用氨来呫诺或媒介物对照处理4天的ND-或HFD-喂食的小鼠中进行CL-316,243注射之后15分钟的血清FFA(左图)和甘油(右图)水平的增加倍数。(B)在用氨来呫诺或媒介物预处理1小时之后的离体附睾(左图)和腹股沟(右图)WAT中的甘油释放。CL-316,243处理从时间零开始。(C)在60分钟CL-316,243处理之后的图5B的腹股沟WAT溶解产物的免疫印迹。(D)在用氨来呫诺或媒介物对照处理4天之后的HFD-喂食小鼠中进行CL-316,243(CL)或盐水对照注射之后20分钟的附睾WAT中的cAMP水平。(E)在用氨来呫诺或媒介物对照处理4天之后的HFD-喂食小鼠中进行CL-316,243或盐水对照注射之后5分钟的附睾WAT的免疫印迹。(F)每个治疗组中的小鼠的相对氧消耗。
定义
术语“核因子κ-B激酶亚基ε的抑制剂”、“I-κ-B激酶ε”、“IKKε”和“IKKi”在本文中可互换使用来指代由IKBKE基因编码的酶。
详述
本文提供了治疗肥胖症和肥胖症相关病状的方法,所述方法包括施用IKKε/TBK1抑制剂与β肾上腺素能激动剂或交感神经系统激活剂的组合以及包含这类组合的药物组合物。在一些实施方案中,本发明提供氨来呫诺或其他IKKi/TBK1抑制剂与β-肾上腺素能激活剂(例如,β2或β3)或与激活交感神经系统的试剂(例如,安非他明,芬特明)的组合。
在一些实施方案中,本发明提供一种减少受试者体内的体脂肪或预防体脂肪增加的方法,所述方法包括:向正经历超重或身体肥胖或处于超重或身体肥胖风险的受试者施用组合物,所述组合物是治疗有效剂量的(i)IKKε/TBK1抑制剂以及(ii)(A)β肾上腺素能激动剂或(B)交感神经系统激活剂。在一些实施方案中,所述施用导致受试者体内体脂肪的增加减少或得到预防。在一些实施方案中,受试者正经历或处于经历病状诸如糖尿病和胰岛素抵抗的风险。在一些实施方案中,施用药物组合物带来以下结果,诸如:葡萄糖代谢增加、胰岛素受体信号传导增加、胰岛素受体磷酸化水平降低、肝中慢性炎症减少或得到预防、脂肪组织中的慢性炎症减少或得到预防、肝性脂肪变性减少或得到预防、代谢能量消耗得到促进、循环游离脂肪酸减少和/或胆固醇减少。
在一些实施方案中,IKKi抑制剂是TBK1/IKKi双重抑制剂。在一些实施方案中,TBK1/IKKi抑制剂是小分子。例如,在一些实施方案中,TBK1/IKKi双重抑制剂是2-氨基-4-(3'-氰基-4'-吡咯烷)苯基-嘧啶化合物或其衍生物或类似物(Li等人,2013年10月6日的Int J Cancer.,doi:10.1002/ijc.28507;所述参考文献以引用的方式整体并入本文)。在其他实施方案中,TBK1/IKKi双重抑制剂是A20、TAX1BP1(Parvatiyar等人,The Journal of Biological Chemistry,285,14999-15009(2010).;所述参考文献以引用的方式整体并入本文)或其衍生物或类似物。在一些实施方案中,TBK1/IKKi抑制剂是氨来呫诺、其衍生物或类似物、或其药学上可接受的盐。
例如,美国专利号4,143,042中描述了氨来呫诺、或2-氨基-7-异丙基-1-氮杂呫吨酮-3-羧酸;2-氨基-7-异丙基-5-氧代-5H-色烯并[2,3-b]吡啶-3-羧酸,所述专利以引用的方式整体并入本文。在一些实施方案中,化合物具有式I的结构:
其中R1是氢、烷基、苯基、羧基、羟基、烷氧基或氨基,所述R1可以是未取代的或被一个烷基取代;m是0、1或2;并且R2是烷基、烷氧基、卤素、硝基、羟基、羧基、亚丁二烯基(–CH=CH–CH=CH–),所述R2与任何邻近的碳原子或氨基形成苯环,所述R2可以是未取代的或被至少一个烷基取代;以及其生理学上可接受的盐。上述式中的每一个中指定的取代基可以在氮杂呫吨酮环的任选的位置或在6-、7-、8-或9-位置中的多个位置处取代。
在式(I)中,由R1和R2表示的烷基可以是任何具有1至6个碳原子的直链、支链或环烷基。烷基的典型实例可以是甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、环戊基、己基、环己基等。
由R1和R2表示的烷氧基可以例如是烷基部分中具有1至4个碳原子的烷氧基,诸如甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基等。
由R1表示的单-烷基取代的氨基可以是烷基部分中具有1至3个碳原子的氨基,诸如甲氨基、乙氨基、丙氨基或异丙氨基。由R2表示的卤素可以是氯、溴、碘或氟。
由R2表示的烷基取代的氨基包括单-或二-烷基取代的氨基,所述氨基的烷基部分是具有1至3个碳原子的烷基部分,例如,甲氨基、乙氨基、丙氨基、异丙氨基、二甲氨基、二乙氨基或二丙氨基。
具有通式(I)的化合物可以通过以下方式转化成对应的有机胺盐、碱金属盐或铵盐:根据常规方式使(I)与有机胺(例如,乙醇胺、二乙醇胺、dl-甲基麻黄素、1-(3,5-二羟基苯基)-L-异丙基氨基乙醇、异丙肾上腺素、右美沙芬、海群生(乙胺嗪)、二乙胺、三乙胺等)、碱金属氢氧化物(例如,氢氧化钠、氢氧化钾等)或氨反应,例如通过将它们混合在一起并在合适的溶剂中加热。
在一些实施方案中,提供作用于β肾上腺素受体的拟交感神经药(例如,小分子、肽、RNA等),所述拟交感神经药具有β阻断剂相反的作用。在一些实施方案中,提供β肾上腺素能受体激动剂。在一些实施方案中,β肾上腺素能受体激动剂模拟肾上腺素和去甲肾上腺素信号传导的作用。在一些实施方案中,β肾上腺素能受体激动剂激活β1、β2和β3受体中的一个或多个。
在一些实施方案中,提供选自多巴酚丁胺、异丙肾上腺素(β1和β2)、扎莫特罗、肾上腺素等的β1激动剂。其他合适的β1激动剂处在本发明的范围内。
在一些实施方案中,提供β2激动剂,所述β2激动剂选自沙丁胺醇(在美国为沙丁胺醇(albuterol))、左沙丁胺醇(在美国为左沙丁胺醇(Levalbuterol))、非诺特罗、福莫特罗、异丙肾上腺素(β1和β2)、间羟异丙肾上腺素、沙美特罗、特布他林、克仑特罗、异他林、吡布特罗、丙卡特罗、利托君、肾上腺素等。其他合适的β2激动剂处在本发明的范围内。
在一些实施方案中,任何合适的β3激动剂(例如,米拉贝隆)处在本发明的范围内。
在一些实施方案中,β肾上腺素能受体激动剂选自以下列表,包括但不限于沙丁胺醇(沙丁胺醇、喘乐宁)、左沙丁胺醇(左沙丁胺醇、左旋沙丁胺醇)、特布他林(博利康尼)、吡布特罗(Maxair)、丙卡特罗、克仑特罗、间羟异丙肾上腺素(Alupent)、非诺特罗、甲磺酸比托特罗、利托君、异丙肾上腺素、沙美特罗(Serevent Diskus)、福莫特罗(Foradil、信必可)、班布特罗、克仑特罗、茚达特罗(indacaterol)、阿布他明、苯呋洛尔、溴乙酰烯丙心安甲烧(bromoacetylalprenololmenthane)、溴沙特罗、西马特罗、西拉唑啉、地诺帕明、多培沙明、依替福林、海索那林、去甲乌药碱、异克舒令、马布特罗、甲氧那明、苄丙酚胺、奥昔非君、普瑞特罗、雷托巴胺、瑞普特罗、利米特罗、曲托喹酚、妥洛特罗、齐帕特罗、净特罗(zinterol)等。
在一些实施方案中,提供交感神经系统激活剂。这类试剂可以通过任何合适的机制(例如,作用于一种或多种神经递质(例如,去甲肾上腺素、5-羟色胺和多巴胺肾上腺素和/或肾上腺素),增加所述一种或多种神经递质的释放,或抑制所述一种或多种神经递质的重摄取,作为肾上腺素能受体激动剂来发挥作用等)来激活交感神经系统。合适的交感神经系统激活剂可以选自苯并二氮平(例如,安定(Valium)、氯硝西泮(Klonopin)、劳拉西泮(Ativan)、替马西泮(Restoril)、氟硝西泮(Rohypnol)、三唑仑(海乐神)、阿普唑仑(赞安诺)等)、安非他明(例如,安非他明(阿得拉)、甲基苯丙胺(Desoxyn)、哌甲酯(利他林)、芬特明、4-甲米雷司、芬美曲秦(Preludin)、甲卡丙酮、芬氟拉明(Pondimin、Fen-Phen)、右芬氟拉明(Redux)、假麻黄碱(舒达非特)、麻黄碱、苯丙醇胺(老版Triaminic)、去氧肾上腺素(舒达非特PE)等)、芬特明、托吡酯等。其他合适的交感神经系统激活剂处在本发明的范围内。
在一些实施方案中,本发明可用于治疗或预防超重和肥胖症。最广泛接受的对肥胖症的临床定义是世界卫生组织(WHO)基于BMI确定的标准。在这个约定下,对于成人来说,1级超重(通常且简单地称为超重)是BMI处于25-29.9kg/m2。2级超重(通常称为肥胖症)是BMI处于30-39.9kg/m2。3级超重(通常称为严重或病态肥胖症)是BMI大于或等于40kg/m2。外科文献经常使用不同的分类来识别特别严重的肥胖症。在这个背景下,BMI大于40kg/m2被描述为严重肥胖症,BMI处于40-50kg/m2被称为病态肥胖症,并且BMI大于50kg/m2被称为超级肥胖。对儿童肥胖症的定义涉及针对年龄相当和性别相当对照受试者,BMI分别大于第85百分位(通常用于定义超重)或第95百分位(通常用于定义肥胖症)。肥胖症的继发性原因包括但不限于甲状腺功能减退、库欣综合征、胰岛素瘤、下丘脑性肥胖、多囊卵巢综合征、遗传综合征(例如,普-威综合征、阿耳斯特雷姆综合征、巴德-别德尔综合征、科恩综合征、博耶逊综合征、弗勒利希综合征)、生长激素缺乏、口服避孕药使用、药剂诱导的肥胖症(例如,吩噻嗪、丙戊酸钠、卡马西平、三环抗抑郁药、锂、糖皮质激素、醋酸甲地孕酮、噻唑烷二酮、磺酰脲、胰岛素、肾上腺素能拮抗剂、5-羟色胺拮抗剂[尤其是赛庚啶]),饮食失调(尤其是重复暴食症、神经性贪食症、夜食症)、性腺功能减退症、假性甲状旁腺功能减退症以及与管喂相关的肥胖症。在一些实施方案中,本文描述的药物组合物和治疗可用于治疗前述继发性原因引起的肥胖症中的一种或多种。
在一些实施方案中,对受试者进行测试以例如通过以下方式评定疾病或病状(例如,肥胖症和/或相关病症,包括但不限于胰岛素抵抗、糖尿病、脂肪变性、非酒精性脂肪型肝炎以及动脉粥样硬化)的存在、不存在或级别:测定或测量生物标志物、代谢物、身体症状、适应症等以确定肥胖症和/或相关病症(包括但不限于胰岛素抵抗、糖尿病、脂肪变性、非酒精性脂肪型肝炎以及动脉粥样硬化)的风险或存在,并且因此基于测试结果来用本文描述的药物组合治疗受试者。在一些实施方案中,对患者进行测试、治疗,并且之后再次对所述患者进行测试以监测治疗反应。在一些实施方案中,可以进行测试和治疗的循环,而不限制测试和治疗的模式(例如,测试/治疗、测试/治疗/测试、测试/治疗/测试/治疗、测试/治疗/测试/治疗/测试、测试/治疗/治疗/测试/治疗/治疗等)、周期性或每个测试和治疗阶段之间的间隔持续时间。
通常预期根据所提供的技术的组合物和/或药物组合被配制用于向哺乳动物施用,并且尤其是向患有对这类化合物的施用有反应的病状的人施用。因此,在以药物组合物或组合施用预期化合物的情况下,预期制剂可以是呈与药学上可接受的载体的混合物的形式。例如,预期化合物和组合可以作为药理学上可接受的盐口服施用,或者以生理盐水溶液(例如,缓冲至约7.2至7.5的pH)形式静脉内施用。常规缓冲液诸如磷酸盐、碳酸氢盐或柠檬酸盐可以用于这个目的。当然,本领域普通技术人员可以在本说明书的教导内修改制剂,以为特定施用途径提供众多制剂。具体而言,可以对预期化合物进行修改以使它们更易溶于水或其他媒介物,这例如可以通过本领域普通技术人员熟知的微小的修改(盐配制、酯化等)来容易地完成。本领域普通技术人员也熟知,为了控制本发明化合物的药物代谢动力学以用于在患者体内获得最大有益效果,修改特定化合物的施用途径和给药方案。
在某些药物剂型中,可以形成用于各种目的的预期化合物的前药形式,所述各种目的包括降低毒性、提高器官或靶细胞特异性等。在各种前药形式当中,本发明化合物的酰化(乙酰化或其他)衍生物、吡啶酯和各种盐形式是优选的。本领域普通技术人员将认识到如何容易地将本发明化合物修改成为前药形式,以促进活性化合物向宿主生物或患者体内的靶部位递送。合适时,本领域普通技术人员也会利用前药形式的有利的药物代谢动力学参数,以将本发明化合物递送至宿主生物或患者体内的靶部位,以使所述化合物的期望效果最大化。类似地,应了解到,预期化合物还可以代谢成其生物活性形式,并且因此特别涵盖本文的化合物的所有代谢物。此外,预期化合物(及其组合)可以结合又另一种试剂施用以用于治疗肥胖症和相关病症,包括但不限于胰岛素抵抗、糖尿病、脂肪变性、非酒精性脂肪型肝炎以及动脉粥样硬化。
对于向受试者施用,预期以药学有效量施用化合物和/或组合。普通技术人员认识到,药学有效量将根据所使用的治疗剂、受试者的年龄、病状和性别以及受试者的疾病程度来变化。一般而言,剂量不应该大到产生不良的副作用,诸如高粘滞综合征、肺水肿、充血性心力衰竭等。所述剂量还可以由个别临床医师来调节以实现所需治疗目标。
如本文所使用,涵盖实际用量的术语“药学有效量”将取决于施用途径、正治疗的受试者的类型以及考虑中的特定受试者的身体特征。用于确定这一量的这些因素及其关系对于医疗、兽医和其他相关领域的技术人员而言是熟知的。可以定制这个量和施用方法以实现最优功效,但是将取决于这类因素诸如体重、饮食、并行药物处理以及本领域技术人员将认识到的其他因素。
在一些实施方案中,提供单一药物组合物,所述单一药物组合物包含(i)IKKε/TBK1抑制剂和(ii)(A)β肾上腺素能激动剂或(B)交感神经系统激活剂。在其他实施方案中,施用分开的药物组合物,一种包含IKKε/TBK1抑制剂,而另一种包含β肾上腺素能激动剂和/或交感神经系统激活剂。可以共同地或分开地确定和/或调节分开的药物组合物的施用剂量和方案。
对剂量数量和频率进行选择以在基本上不具有有害作用的情况下发挥化合物的有效水平。当口服或静脉内施用时,剂量的范围通常会是0.001至10,000mg/kg/天或剂量(例如,0.01至1000mg/kg/天或剂量;0.1至100mg/kg/天或剂量,1至100mg/kg/天或剂量、或其中的量)。
在一些实施方案中,向受试者施用单一剂量。在其他实施方案中,在间隔数小时、数天、数周等的两个或更多个时间点内施用多个剂量。在一些实施方案中,在一个较长时间段(例如,长期(chronically)),例如在数月或数年(例如,1、2、3、4、5、6、7、8、9、10、11、12或更多个月或更多年)的时段内施用药物组合物。在这类实施方案中,在延长时段的持续时间内可以基于有规律的预定计划(例如,每天、每周等)来服用药物组合物。
施用药学有效量的方法包括而不限于肠胃外、口服、腹膜内、鼻内、局部、舌下、直肠以及阴道形式的施用。肠胃外施用途径包括例如皮下、静脉内、肌内、胸骨内注射和输液途径。在一些实施方案中,氨来呫诺、其衍生物或其药学上可接受的盐是口服施用的。
药物组合物优选地包含本发明技术的一种或多种化合物结合一种或多种药学上可接受的载体、稀释剂或赋形剂。药学上可接受的载体在本领域中是已知的,诸如在例如Remingtons Pharmaceutical Sciences,Mack Publishing Co.(A.R.Gennaro编辑,1985)中描述的那些,所述参考文献出于所有目的以引用的方式明确并入本文。
因此,在一些实施方案中,组合物(包含一种试剂或药物组合物)配制为片剂、胶囊、延时释放片剂、延时释放胶囊;延时释放球粒;缓释片剂、缓释胶囊;缓释球粒;快速释放片剂、快速释放胶囊;快速释放球粒;舌下片剂;凝胶胶囊;微型胶囊;经皮递送制剂;经皮凝胶;经皮贴片;无菌溶液;制备来用作肌内或皮下注射液、用作直接进入靶部位的注射液、或用于静脉内施用的无菌溶液;制备用于直肠施用的溶液;制备用于通过胃饲管或十二指肠饲管施用的溶液;用于直肠施用的栓剂;制备成溶液或酏剂的经口食用液体;局部乳膏;凝胶;洗剂;酊剂;糖浆剂;乳剂;或混悬剂。
在一些实施方案中,延时释放制剂是持续释放、持续作用、延长释放、受控释放、调节释放或连续释放机体,例如,所述组合物被配制成随时间的推移快速、缓慢或以任何适当的释放率溶解治疗剂。
在一些实施方案中,本发明技术的药物制剂物和/或制剂以颗粒的形式提供。如本文所使用的颗粒意指可以全部或部分由如本文所述的治疗剂组成的纳米颗粒或微颗粒(或在一些情况下为更大颗粒)。颗粒可以包含芯由包衣包裹的制剂物和/或制剂,所述包衣包括但不限于肠溶包衣。制剂物和/或制剂也可以分散在整个颗粒中。制剂物和/或制剂也可以被吸收到颗粒中。颗粒可以具有任何级数的释放动力学,包括零级释放、一级释放、二级释放、延迟释放、持续释放、即时释放以及其任何组合等。除了制剂物和/或制剂,所述颗粒可以包括任何常规用于药学和医学领域的那些材料,包括但不限于溶蚀的、不溶蚀的、生物可降解的或不能生物降解的材料或其组合。颗粒可以是包含呈溶液或半固体状态的制剂的微囊剂。颗粒在视觉上可以是任何形状。
不能生物降解的聚合物材料和可生物降解的聚合物材料两者均可以用于制造用于递送制剂物和/或制剂的颗粒。这类聚合物可以是天然的或合成的聚合物。聚合物基于释放需要的时间段来选择。特别感兴趣的生物粘附性聚合物包括H.S.Sawhney、C.P.Pathak和J.A.Hubell在Macromolecules,(1993)26:581-587中描述的可生物溶蚀水凝胶,所述参考文献的教导以引用的方式并入本文。这些聚合物包括聚透明质酸、酪蛋白、明胶、明胶蛋白、聚酐、聚丙烯酸、藻酸盐、壳聚糖、聚(甲基丙烯酸甲酯)、聚(甲基丙烯酸乙酯)、聚(甲基丙烯酸丁酯)、聚(甲基丙烯酸异丁酯)、聚(甲基丙烯酸己酯)、聚(甲基丙烯酸异癸酯)、聚(甲基丙烯酸月桂酯)、聚(甲基丙烯酸苯基酯)、聚(丙烯酸甲酯)、聚(丙烯酸异丙酯)、聚(丙烯酸异丁酯)以及聚(丙烯酸十八烷基酯)。
在一些实施方案中,药物组合物用缓冲剂配制。缓冲剂可以是任何药学上可接受的缓冲剂。缓冲液系统包括柠檬酸盐缓冲液、醋酸盐缓冲液、硼酸盐缓冲液以及磷酸盐缓冲液。缓冲液的实例包括柠檬酸、柠檬酸钠、醋酸钠、醋酸、磷酸钠和磷酸、抗坏血酸钠、酒石酸、马来酸、甘氨酸、乳酸钠、乳酸、抗坏血酸、咪唑、碳酸氢钠和碳酸、琥珀酸钠和琥珀酸、组氨酸以及苯甲酸钠和苯甲酸。
实验
肥胖症会产生涉及NFκB途径的慢性炎症状态,这会导致非经典IκB激酶IKKi和TBK1的持续升高。本发明的实施方案的发展过程中进行的实验证明这些激酶会使白脂肪组织中的β-肾上腺素能信号传导衰减。用这些激酶的特异性抑制剂处理3T3-L1脂肪细胞能恢复被TNFα和Poly(I:C)衰减的β-肾上腺素能信号传导和脂解。相反地,激酶的过表达会减少响应于异丙肾上腺素或毛喉素而发生的对Ucp1、脂解、cAMP水平以及激素敏感性脂肪酶的磷酸化的诱导。非经典IKK通过磷酸化和激活主要脂肪细胞磷酸二酯酶PDE3B来降低儿茶酚胺敏感性。通过用药物氨来呫诺治疗肥胖小鼠而对这些激酶进行的体内抑制逆转了肥胖症诱导的儿茶酚胺抵抗,并且响应于β-3肾上腺素能激动剂的注射而恢复PKA信号传导。这些研究表明通过减少脂肪细胞中cAMP的产生,IKKε和TBK1会导致肥胖症期间对能量消耗的阻遏。
实施例1
IKKε和TBK1过表达降低对3T3-L1脂肪细胞中β-肾上腺素能/cAMP途径的敏感性
脂肪组织的交感神经激活涉及通过刺激脂解和脂肪氧化来维持能量平衡(Coppack等人,1994;Langin,2006;Festuccia等人,2011;所述参考文献以引用的方式整体并入本文)。在白色和褐色脂肪组织中通过β-肾上腺素能激动剂或冷暴露而进行的β-肾上腺素能信号传导的激活通过环AMP(cAMP)起始级联事件,以Ucp1的转录上调而告终,这导致质子泄漏和能量消耗增加(Himms-Hagen等人,2000;Cao等人,2004;Yehuda-Shnaidman等人,2010;所述参考文献以引用的方式整体并入本文)。与野生型(WT)对照相比较,在高脂肪饮食(HFD)时,IKKε-缺陷型小鼠展现出增加的能量消耗,伴随白脂肪库中Ucp1的增加的表达(Chiang等人,2009;所述参考文献以引用的方式整体并入本文)。仅在HFD-喂食的小鼠中观察到IKKε-缺陷型小鼠的增加的能量消耗(Chiang等人,2009;所述参考文献以引用的方式整体并入本文),从而表明在肥胖症期间诱导IKKε之后,激酶会阻遏对营养过度产生的增加的适应性产热反应。这种效应进一步通过以下方式来分析:在3T3-L1脂肪细胞中过表达IKKε并且在用非选择性β-肾上腺素能激动剂即异丙肾上腺素(ISO)或β3-肾上腺素能激动剂CL-316,243处理之后检查Ucp1基因表达。通过归一化处理样品相对于对照样品中的相对Ucp1mRNA水平来计算Ucp1基因表达的倍数差异。用ISO或CL-316,243处理表达空载体的细胞分别导致Ucp1mRNA水平增加1.6倍或两倍(图1A)。当WT IKKε在这些细胞中过表达时,响应于ISO或CL-316,243而发生的对Ucp1基因表达的诱导是迟钝的。然而,IKKεK38A的激酶失活突变体的表达(Fitzgerald等人,2003;所述参考文献以引用的方式整体并入本文)不太有效,但仍然适度地阻遏Ucp1表达。
除了增加的Ucp1表达之外,IKKε敲除小鼠还展现出增加的脂解和脂肪氧化(Chiang等人,2009;所述参考文献以引用的方式整体并入本文),从而表明肥胖小鼠的脂肪组织的减少的脂解可能部分归因于IKKε和TBK1的增加的表达(Chiang等人,2009;所述参考文献以引用的方式整体并入本文)。针对非经典IKK通过以下方式将肥胖症依赖型增加模型化:在3T3-L1脂肪细胞中过表达IKKε,之后测定响应于ISO或CL-316,243而发生的甘油释放。虽然异丙肾上腺素和CL-316,243两者都增加了表达空载体的细胞的脂解,但是WT IKKε过表达超过40%会降低异丙肾上腺素和CL-316,243的脂解作用,并且还会减少基础甘油释放(图1B)。因IKKε过表达而发生的脂解的减少通过响应于ISO或CL-316,243而发生的HSL和脂滴包被蛋白的磷酸化的大幅减少来实现(图1C)。催化失活的激酶的表达不能有效地阻断脂解信号传导,但是通过过表达实现的蛋白质的水平低于WT激酶(图1B、图1C,图1)。TBK1的过表达减少了响应于异丙肾上腺素或腺苷酸环化酶激活子毛喉素而发生的HSL的磷酸化(图1)。当IKKε在用毛喉素刺激的3T3-L1脂肪细胞中过表达(图1D)时获得相同的结果,这通过用抗-磷酸-PKA底物基序抗体进行蛋白质印迹法来检测。
IKKε的过表达还会阻遏响应于毛喉素(图1D)或异丙肾上腺素(图1)而发生的p38(p-p38)的磷酸化,而IKKεK38A的过表达不具有作用(图1)。虽然甘油释放可能是HSL和脂滴包被蛋白磷酸化发生变化的结果,但是值得注意的是,我们并未直接测定甘油中间体的再酯化是否也会受到影响。在一起考虑时,这些数据表明与在肥胖症中观察到的类似,IKKε或TBK1的过表达会阻遏脂解信号传导。预期的是,激酶失活突变体的部分有效性反映了因其二聚作用而激活内源性IKKε或TBK1激酶(Larabi等人,2013;Tu等人,2013;所述参考文献以引用的方式整体并入本文)。
由于PKA信号传导响应于儿茶酚胺而负责Ucp1诱导(Klein等人,2000;Cao等人,2001;所述参考文献以引用的方式整体并入本文);在本发明的实施方案的发展过程中进行实验以研究IKKε和TBK1通过降低cAMP水平而诱导的脂肪细胞的β-肾上腺素能敏感性的降低。IKKε在3T3-L1脂肪细胞中过表达超过80%会减少因异丙肾上腺素和毛喉素而产生的cAMP水平的增加,而IKKεK38A的过表达却不会如此(图1E)。先前的研究已经显示对脂肪组织中的肾上腺素能刺激物的降低的敏感性可以归因于减少的β-肾上腺素能受体(Reynisdottir等人,1994;所述参考文献以引用的方式整体并入本文)或α2-肾上腺素能受体的增加的表达(Stich等人,2002;所述参考文献以引用的方式整体并入本文)。
实施例2
用TNFα进行延长处理以依赖于IKKε和TBK1的活性的方式降低脂肪细胞对β-肾上腺素能刺激的敏感性
肥胖症伴随促炎性巨噬细胞浸润到脂肪组织中;这些细胞分泌炎性细胞因子,诸如TNFα,所述炎性细胞因子会通过刺激分解代谢途径来产生胰岛素抵抗(Hotamisligil,2006;Lumeng等人,2007;Ye和Keller,2010;Ouchi等人,2011;所述参考文献以引用的方式整体并入本文)。虽然已知TNFα会增加脂肪细胞的脂解(Zhang等人,2002;Souza等人,2003;Green等人,2004;Plomgaard等人,2008;所述参考文献以引用的方式整体并入本文),但是也有证据表明肥胖症中存在可能用于阻遏能量消耗的抗炎反应(Gregor和Hotamisligil,2011;Saltiel,2012;Calay和Hotamisligil,2013;Reilly等人,2013;所述参考文献以引用的方式整体并入本文)。在本发明的实施方案的发展过程中使用TNFα来进行实验以在细胞培养物中将肥胖脂肪组织的炎性环境模型化,从而确定细胞因子在此背景下是否也有可能调控β-肾上腺素能信号传导。虽然用TNFα的短期处理会使通过毛喉素处理产生的cAMP的增加加强,但是这种效应在12小时之后消减。在24小时暴露之后,TNFα抑制因毛喉素而产生的第二信使的产生(图2)。因此,脂肪细胞中的促炎性细胞因子TNFα的分解代谢作用是瞬时的并且之后是抑制阶段。
用TNFα将3T3-L1脂肪细胞处理24小时以依赖于IKKβ的活性和NFκB途径的方式在活性部位处诱导IKKε的表达以及增加的TBK1磷酸化(Reilly等人,2013;所述参考文献以引用的方式整体并入本文)。在本发明的实施方案的发展过程中进行实验以确定通过TNFα的长期处理产生的对β-肾上腺素能敏感性的阻遏是否归因于非经典IKK的增加的活性。用TNFα的长期处理会阻遏响应于β-肾上腺素能刺激物而发生的对Ucp1基因表达的诱导(图2),而IKKε mRNA(Ikbke)的表达被上调。用TNFα将3T3-L1脂肪细胞处理24小时以剂量依赖型方式减少响应于异丙肾上腺素和毛喉素两者而发生的甘油释放(图2A)。TNFα处理还减少受异丙肾上腺素和毛喉素刺激的cAMP产生;即在很大程度上通过用选择性的但结构上不相关的IKKε和TBK1的抑制剂氨来呫诺(图2B)(Reilly等人,2013;所述参考文献以引用的方式整体并入本文)或CAY10576(图2C)(Bamborough等人,2006;所述参考文献以引用的方式整体并入本文)预孵育细胞就可挽救的效应。通过用TNFα处理细胞也会减少受异丙肾上腺素刺激的β-肾上腺素能信号传导(图2D),这通过HSL、脂滴包被蛋白和由PKA底物基序抗体识别的其他蛋白质的减少的磷酸化显现出来,而通过TNFα的处理,IKKε表达被并行上调并且TBK1磷酸化出现增加。用氨来呫诺预处理3T3-L1脂肪细胞还会阻断TNFα对受异丙肾上腺素刺激的β-肾上腺素能信号传导的抑制作用,这通过用抗-磷酸-PKA底物基序抗体、抗磷酸-HSL和抗-磷酸-脂滴包被蛋白抗体进行蛋白质印迹法来确定(图2E)。响应于异丙肾上腺素而发生的p38的磷酸化也以剂量依赖型方式通过氨来呫诺来大幅加强。先前的研究显示toll样受体3(TLR3)激动剂Poly(I:C)会导致IKKε和TBK1的直接激活(Hemmi等人,2004;Clark等人,2009;Clark等人,2011;所述参考文献以引用的方式整体并入本文)。类似于TNFα,同时用Poly(I:C)处理3T3-L1脂肪细胞会减少对响应于β-肾上腺素能刺激而发生的cAMP产生、脂解和磷酸化的刺激(图2),并且Poly(I:C)对脂肪组织对异丙肾上腺素刺激的敏感性的抑制作用通过氨来呫诺预处理得到部分恢复,但并不是在TNFα处理中所观察到的那种程度(图2E)。这些结果表明肥胖症相关联的炎症会导致IKKε和TBK1的激活,这会使脂肪细胞对β-肾上腺素能刺激的敏感性降低。
实施例3
IKKε和TBK1通过激活PDE3B来降低cAMP水平
cAMP水平还可以通过磷酸二酯酶来调控,所述磷酸二酯酶使第二信使裂解并且在过程中使cAMP-依赖性信号衰减。磷酸二酯酶3B(PDE3B)是脂肪细胞中表达的主要PDE同工型脂肪细胞(Zmuda-Trzebiatowska等人,2006;所述参考文献以引用的方式整体并入本文)。细胞内和体内的PDE3B的基因切除或药理学抑制揭示了酶在脂质和葡萄糖代谢中的重要作用(Choi等人,2006;Berger等人,2009;Degerman等人,2011;所述参考文献以引用的方式整体并入本文)。PDE3B通过胰岛素而在脂肪细胞中发生的磷酸化和激活被认为是由Akt介导的,并且cAMP自身通过促进PKA-依赖性磷酸化以及PDE3B的激活而充当其自身水平的负反馈调控因子(Degerman等人,2011;所述参考文献以引用的方式整体并入本文)。
在本发明的实施方案的发展过程中进行的实验证明在过表达IKKε的受毛喉素或异丙肾上腺素刺激的3T3-L1脂肪细胞中的cAMP产生受到削弱(图1E),因此进行附加实验来确定非经典IKK是否可能通过脂肪细胞中增加的PDE3B的活性来对肾上腺素能刺激去敏化。用非特异性磷酸二酯酶抑制剂IBMX在过表达IKKε或TBK1的3T3-L1的脂肪细胞中进行预处理挽救了响应于毛喉素而对cAMP产生具有的全部刺激(图3A)。有趣的是,选择性PDE3B和PDE4抑制剂扎达维林(Schudt等人,1991;所述参考文献以引用的方式整体并入本文)还会阻断IKKε和TBK1过表达响应于3T3-L1脂肪细胞中的异丙肾上腺素和毛喉素而对cAMP水平产生的抑制作用(图3B),从而表明PDE3B作为非经典IKK的靶标的重要作用。
接下来检查IKKε和TBK1是否直接将PDE3B磷酸化以调控cAMP水平。在体外用[γ-32P]ATP孵育重组TBK1、Akt和PKA,并且将PDE3B纯化作为底物。先后通过SDS-PAGE和放射自显影术来测定磷酸化。TBK1直接催化PDE3B的磷酸化;磷酸化如先前所报道还通过用Akt和PKA孵育来产生(Kitamura等人,1999;Palmer等人,2007;所述参考文献以引用的方式整体并入本文)(图3C)。IKKε还在体外催化这种磷酸化。在PDE3B用识别14-3-3结合基序的抗体印迹时也检测到了通过在体外用TBK1、IKKε和PKA孵育而产生的这种磷酸化的增加(图3)。当纯化的PDE3B在体外用相同量的重组TBK1和经典IKKβ激酶孵育时,几乎检测不到PDE3B因IKKβ而产生的磷酸化,从而表明PDE3B是非经典IKK的更好的靶标的特异性级别(图3)。相对于ATP而言,这种磷酸化是剂量依赖型的(图3)。
为了确定IKKε是否可以在细胞中将PDE3B磷酸化,使IKKε及其失活突变体K38A与HA-标记的PDE3B共同表达在HEK293T细胞中,之后用抗-HA抗体来进行免疫沉淀(IP)。细胞中IKKε的表达引起PDE3B的电泳迁移率的变化,而在表达IKKε K38A时不会检测到这种变化(图3)。在细胞中表达IKKε而非其激酶失活突变体K38A之后也检测到了PDE3B的磷酸化,这通过用识别14-3-3结合基序的抗体进行印迹来检测。为了确定这种分子位移是否依赖于PDE3B的磷酸化,使HA-PDE3B连同IKKε、TBK1或其激酶失活突变体一起共同表达在Cos-1细胞中,并且用或不用小牛肠磷酸酶(CIP)处理HA免疫沉淀物。两种野生型激酶的表达降低了PDE3B的电泳迁移率(图3D,将泳道3、7与泳道4、8进行比较),这可以通过用磷酸酶处理来逆转。激酶失活突变体都不具有作用(图3D,将泳道5、9与泳道6、10进行比较)。
先前的研究表明IKKε和TBK1通过包括其激酶结构域近侧的泛素样结构域(ULD)的序列结合至其相应的底物。这个结构域在IKK家族成员当中是高度保守的,并且IKKε与TBK1具有49%的同一性(Ikeda等人,2007;May等人,2004;所述参考文献以引用的方式整体并入本文)。为了证实PDE3B是IKKε和TBK1的真实的底物,从TBK1制备GST-ULD结构域融合蛋白并且用3T3-L1脂肪细胞溶解产物来孵育这个融合蛋白。所述融合蛋白特异性地将内源性PDE3B从这些溶解产物中沉淀出来(图3)。为了进一步探索这两种蛋白质的相互作用,使WT TBK1及其K38A突变体与HA-标记的PDE3B共同表达在HEK293T细胞中,并且用抗-HA抗体来免疫沉淀所述蛋白质。激酶失活TBK1优先与PDE3B发生免疫共沉淀,而几乎检测不到PDE3B与WT TBK1的相互作用(图3)。这些数据表明TBK1和IKKε与底物诸如PDE3B缔合,并且随后在磷酸化之后发生解离。
为了进一步测试PDE3B通过IKKε和TBK1发生的磷酸化在起始其与14-3-3β的相互作用中的作用,制备了GST-14-3-3β融合蛋白,所述融合蛋白用来自共同表达TBK1与PDE3B的HEK293T细胞的溶解产物来孵育。在通过TBK1而非通过其失活K38A突变体发生的磷酸化之后,优先通过GST-14-3-3β下拉PDE3B,而GST珠粒单独并未富集PDE3B和其磷酸化形式两者(图3E)。
实施例4
IKKε和TBK1使PDE3B在丝氨酸318处磷酸化,从而导致结合14-3-3β
为了评估PDE3B通过IKKε和TBK1发生的磷酸化的调控作用,我们将确定哪些位点被磷酸化。HA-PDE3B与IKKε和TBK1共同表达在Cos-1细胞中,并且通过IP发现磷酸化的PDE3B富集了抗-HA抗体。之后通过LC-MS/MS质谱法确定人PDE3B的磷酸化位点。这个分析揭示丝氨酸22、299、318、381、463、467和503都被两种激酶磷酸化;所述激酶之间不存在差异(图4A)。有趣的是,PDE3B的磷酸化特征与已知的Akt和PKA特征两者都不匹配(Lindh等人,2007)。然而,先前已经鉴别了脂肪细胞和肝细胞中的小鼠PDE3B在响应于胰岛素和毛喉素两者而在丝氨酸299和丝氨酸318(等同于小鼠PDE3B中的丝氨酸277和296的残基)上发生的磷酸化(Lindh等人,2007)。虽然已知若干丝氨酸残基响应于刺激会在PDE3B上发生磷酸化,但是丝氨酸318(人)具有最佳的表征。这个残基驻留在Akt和PKA两者的共有磷酸化序列内,并且一旦发生磷酸化,就还能用作共有14-3-3结合基序(Lindh等人,2007;Palmer等人,2007)。我们因此构建了PDE3B的Ser318Ala(S318A)突变体,并且通过GST-14-3-3覆盖测定来检查其与GST-14-3-3β融合蛋白的相互作用。有趣的是,虽然用TBK1进行了孵育,但是PDE3B的磷酸缺陷型S318A突变体并未特异地与GST-14-3-3β相互作用,而野生型蛋白质却有如此行为(图4B、图4C)。在GST下拉测定中,通过蛋白质印迹法仍然检测到了PDE3B S318A的分子位移(图4B),从而表明PDE3B仍然通过TBK1在其他位点上进行磷酸化,但对于14-3-3β结合来说并不重要。
为了检查PDE3B在丝氨酸318处发生的磷酸化的功能重要性,我们使WT PDE3B及其S318A突变体过表达在3T3-L1脂肪细胞中,并且测试细胞对TNFα的反应。细胞内WT PDE3B的过表达会减少受毛喉素刺激的cAMP产生以及HSL因TNFα而产生的磷酸化的衰减,而PDE3B S318A是无效的(图4D,图4—补充图1A,1B)。这些数据表明虽然IKKε和TBK1可以在若干位点上将PDE3B磷酸化,但是丝氨酸318因促进PDE3B与14-3-3β之间的相互作用而在磷酸二酯酶功能的调控中可能是特别重要的。更重要的是,这个残基是介导IKKε和TBK1对脂肪细胞对β-肾上腺素能刺激的敏感性的负面作用的主要位点。
实施例5
IKKε/TBK1抑制剂氨来呫诺在饮食诱导的肥胖小鼠中的白脂肪组织中敏化受β-肾上腺素能激动剂刺激的脂解
为了测试非经典IKK在维持体内能量平衡中的功能重要性,在本发明的实施方案的发展过程中进行实验以研究IKKε和TBK1的选择性抑制剂氨来呫诺的施用是否可以逆转啮齿类动物中饮食诱导的儿茶酚胺抵抗。向小鼠喂食高脂肪或正常饮食,用氨来呫诺通过口服强饲法处理所述小鼠,持续4天(在观察到体重减轻的时间点之前),并且之后给予β3-肾上腺素能激动剂CL-316,243的单次腹膜内(IP)注射。CL-316,243的注射刺激了媒介物和氨来呫诺两者处理的正常饮食(ND)的小鼠中的血清FFA和甘油水平的三倍增加。CL-316,243增加血清FFA的作用在HFD-喂食的媒介物处理的小鼠中出现显著衰减。然而,用氨来呫诺处理的HFD-喂食的小鼠以与正常饮食小鼠类似的方式响应,但是事实上所述正常饮食小鼠的体重与对照HFD-喂食的小鼠匹配(图5A)。与媒介物处理的HFD-喂食的小鼠相比较,氨来呫诺处理的HFD小鼠的血清甘油水平的增加倍数也显著更高。此外,用氨来呫诺离体预处理来自HFD小鼠的白脂肪组织还增强了甘油释放(图5B)。这种效应在腹股沟脂肪库中更为显著,在其中,与媒介物预处理的组织相比较,氨来呫诺预处理增加了响应于CL-316,243处理而发生的HSL、脂滴包被蛋白和由PKA底物基序抗体识别的其他蛋白质的磷酸化(图5C)。如先前针对其他抑制剂所报道(Clark等人,2009;Reilly等人,2013),氨来呫诺同时还增加TBK1因反馈抑制的缓解而在Ser172处发生的磷酸化。
为了检查氨来呫诺对TBK1和IKKε的抑制是否通过增加对cAMP产生的刺激来逆转对儿茶酚胺诱导的体内脂解的抵抗,在CL-316,243IP注射之后,我们测量了来自HFD小鼠的附睾脂肪组织中的cAMP水平。用氨来呫诺预处理的HFD小鼠体内的cAMP的水平在CL-316,243IP注射之后出现增加(图5D)。与此相一致的是,用氨来呫诺预处理的HFD-喂食的小鼠的HSL磷酸化在CL-316,243IP注射之后也出现增加(图5E)。
为了检查通过用氨来呫诺靶向非经典IKK而对肥胖脂肪组织中的儿茶酚胺抵抗产生的抑制是否会导致饮食诱导的肥胖小鼠体内的能量消耗的增加,测量了媒介物或氨来呫诺处理的HFD-喂食的小鼠在代谢笼中接受单次CL-316,243注射之后的氧消耗率。与媒介物处理的HFD-喂食的小鼠相比较,CL-316,243增加能量消耗的作用在氨来呫诺处理的HFD-喂食的小鼠中更为显著(图5F)。这些数据表明用选择性抑制剂氨来呫诺靶向非经典IKK改善了肥胖脂肪组织中的儿茶酚胺抵抗。
- 还没有人留言评论。精彩留言会获得点赞!
- 包含与β-3-肾上腺素能受体激动剂组合的去氨加压素的方法和组合物的制作方法
- 具有毒蕈碱受体拮抗体和β2肾上腺素能受体激动剂两种活性的2-氨基-1-羟乙基-8-羟基 ...的制作方法
- 靶向于肌动蛋白结合蛋白Transgelin-2的化合物及其新用图
- 一种α-2肾上腺素受体和σ受体配体的组合物的制作方法
- 天然植物皂苷功效组分的筛选方法及用图
- 通过玻璃体内和前房内途径用于治疗眼内压和眼部疾病的α-2肾上腺素能激动剂的制作方法
- 用于治疗组织创伤的α-肾上腺素能激动剂的制作方法
- 具有β2肾上腺素能受体激动剂和M3毒蕈碱受体拮抗剂两种活性的2-氨基-1-羟乙基-8-羟 ...的制作方法
- 具有毒蕈碱受体拮抗剂和β2肾上腺素能受体激动剂活性的化合物的制作方法
- 具有毒蕈碱受体拮抗剂和β2肾上腺素能受体激动剂活性的化合物的制作方法
- 作为去甲肾上腺素(ne)与血清素(5-h t)活性和单胺重摄取的调控剂以治疗血管舒缩症 ...的制作方法
- 一种治疗广泛性焦虑症的药物及其制备方法
- 用于治疗和诊断与五羟色胺-、肾上腺素-、去甲肾上腺素-、谷氨酸-和促肾上腺皮质素释 ...的制作方法
- 单胺再摄取抑制剂和钾通道激活剂的组合的制作方法
- 具有5-羟色胺能活性和/或去甲肾上腺素能活性的取代的苯乙胺的制作方法
- 去甲肾上腺素重摄取调节剂用于预防和治疗血管舒缩症的用途的制作方法
- 去甲肾上腺素重摄取调节剂用于预防和治疗血管舒缩症的用途的制作方法
- 产生治疗疼痛的止痛化合物的细胞系的制作方法
- 重酒石酸去甲肾上腺素药物组合物冻干粉针注射剂的制作方法
- 重酒石酸去甲肾上腺素注射液及其制剂工艺的制作方法