脂质运载蛋白突变蛋白的控制释放制剂的制作方法
相关申请的交叉引用
本申请引用美国临时申请61/231,365,并要求所述申请的优先权的权益,所述申请于2009年8月5日提交至美国专利及商标局,出于各种目的,其内容通过引用全文并入本文。
发明领域
本申请涉及用于控制释放与可生物降解聚合物组合的脂质运载蛋白突变蛋白或其缀合物的药物组合物。本发明还涉及用于控制递送脂质运载蛋白突变蛋白及其缀合物的方法、用于生产控制释放制剂的方法以及由此生产的制剂。最后,本发明涉及本发明的用于控制递送脂质运载蛋白突变蛋白的制剂用于延长脂质运载蛋白突变蛋白的体内半衰期、提高脂质运载蛋白突变蛋白的生物利用度、或降低脂质运载蛋白突变蛋白给予受试者后的免疫原性的用途,以及涉及用于治疗疾病或障碍的方法,所述方法包括给予有其需要的受试者本发明的制剂。
发明背景
脂质运载蛋白家族成员(Pervaiz、S.和Brew、K.(1987)FASEB J.1、209-214)通常为小的分泌蛋白,其特征在于一系列不同的分子识别特性:主要是它们结合多种疏水分子(例如类视黄醇、脂肪酸、胆固醇、前列腺素、胆绿素、信息素、促味剂和添味剂)的能力、它们与特异细胞表面受体并形成大分子复合物的结合。尽管过去将它们主要分类为转运蛋白,但是现在明确了脂质运载蛋白完成多种生理功能。这些功能包括在视黄醇转运、嗅觉、信息素信号传递以及前列腺素生物合成中的作用。还表明脂质运载蛋白参与免疫应答的调节和细胞内环境稳定的介导(例如Flower,D.R.(1996)Biochem.J.318、1-14和Flower,D.R.等人(2000)Biochim.Biophys.Acta 1482、9-24综述)。
脂质运载蛋白之间的全序列保守性非常低,经常具有低于20%的序列同一性。形成强烈对比的是,其整体折叠模式高度保守。脂质运载蛋白结构的中心部分由单个八股反平行β折叠组成,所述β折叠自身闭合形成连续的氢键β桶。桶的一个末端被穿过其底部的N端肽段以及连接β折叠股的三个肽环立体封闭。β桶的另一个末端暴露于溶剂并包括靶标结合位点,其由四个肽环形成。在其它刚性脂质运载蛋白骨架中,正是这种环的多样性产生不同的结合模式,每一种模式能容纳不同大小、形状和化学特征的靶标(例如上文的Flower,D.R.(1996);上文的Flower,D.R.等人(2000),或Skerra,A.(2000)Biochim.Biophys.Acta 1482,337-350中的综述)。
脂质运载蛋白家族成员已成为关于具有确定的配体结合特性的蛋白质的研究对象。PCT公开文本WO 99/16873公开了在四个肽环区域具有突变氨基酸的脂质运载蛋白家族的多肽,所述四个肽环排列于包围结合口袋的圆柱状β桶结构的末端,并且与包含大菜粉蝶(Pieris brassicae)后胆色素结合蛋白的氨基酸位置28至45、58至69、86至99、和114至129的线性多肽序列中的那些区段对应。
PCT公开文本WO 00/75308公开了后胆色素结合蛋白的突变蛋白,其特异地结合洋地黄毒苷,而国际专利申请WO 03/029463和WO 03/029471分别涉及人中性粒细胞明胶酶结合的脂质运载蛋白(hNGAL)和载脂蛋白D的突变蛋白。为进一步提高和优化脂质运载蛋白变体的配体亲和力、特异性以及折叠稳定性,已经提出了使用脂质运载蛋白家族的不同成员的多种方法(Skerra,A.(2001)Rev.Mol.Biotechnol.74,257-275;Schlehuber、S.,和Skerra,A.(2002)Biophys.Chem.96、213-228),例如替换其它氨基酸残基。PCT公开文本WO 2006/56464公开了人中性粒细胞明胶酶结合的脂质运载蛋白的突变蛋白,其具有对低纳摩尔范围的CTLA-4结合亲和力。
PCT公开文本WO 2005/19256公开了对不同或相同靶标配体具有至少一个结合位点的泪脂质运载蛋白的突变蛋白,并且提供了用于生成此类人泪脂质运载蛋白的突变蛋白的方法。根据该PCT专利申请,使泪脂质运载蛋白的一级序列中的某些氨基酸延伸序列(特别是包含成熟人泪脂质运载蛋白的氨基酸7-14、24-36、41-49、53-66、69-77、79-84、87-98和103-110的环区域)进行突变,从而生成对给定配体具有高结合亲和力的突变蛋白。所得的突变蛋白对纳摩尔范围内,多数情况下<100nM的所选配体(KD)具有结合亲和力。
PCT公开文本WO 2008/015239公开了人泪脂质运载蛋白的突变蛋白,其具有非天然配体,包括IL-4受体、VEGF和VEGF受体的至少一个结合位点,并且提供了用于制备此类脂质运载蛋白突变蛋白的方法。该申请公开了可以对人泪脂质运载蛋白一级序列中的某些氨基酸延伸序列进行突变以生成突变蛋白,所述突变蛋白对给定靶标具有高结合亲和力。所报导的结合亲和力在纳摩尔范围内。所述突变位置在环区域中且在天然的成熟人泪脂质运载蛋白线性多肽序列的氨基酸序列位置26-34、56-58、80、83、104-106和108中任意处具有至少一个突变,并在天然的成熟人泪脂质运载蛋白线性多肽序列的氨基酸序列位置61和153中任意处具有至少一个突变。或者,所公开的突变蛋白在天然的成熟人泪脂质运载蛋白线性多肽序列的氨基酸序列位置34、80和104中任意处具有至少一个突变。
理想的是拥有这样的组合物和制剂,所述组合物和制剂能够控制递送这些脂质运载蛋白突变蛋白、提高体内半衰期、降低突变蛋白的免疫原性和/或提高突变蛋白的生物利用度,从而进一步改善脂质运载蛋白的突变蛋白在治疗应用中的适用性。
因此,本发明的目的是提供满足该需求的脂质运载蛋白突变蛋白缀合物和制剂。
发明概述
通过具有独立权利要求的特征的缀合物、药物制剂和控制释放制剂实现了该目的。
第一方面,本申请提供缀合物,所述缀合物包含脂质运载蛋白突变蛋白和选自蛋白质、蛋白质结构域、肽、脂肪酸、脂质、多糖和/或有机聚合物的部分。
在一个实施方式中,所述缀合物包含脂质运载蛋白突变蛋白和蛋白质、蛋白质结构域、肽、脂肪酸、脂质、多糖和/或有机聚合物部分,所述缀合物基本上由脂质运载蛋白突变蛋白和蛋白质、蛋白质结构域、肽、脂肪酸、脂质、多糖和/或有机聚合物部分组成,或所述缀合物由脂质运载蛋白突变蛋白和蛋白质、蛋白质结构域、肽、脂肪酸、脂质、多糖和/或有机聚合物部分组成。所述部分还可以包含前述化合物的组合,例如糖肽、脂质修饰的肽和蛋白质、糖脂、用有机基团修饰的多糖,例如烷基化的多糖等。
在本发明的一个实施方式中,使脂质运载蛋白突变蛋白和选自蛋白质、蛋白质结构域、肽、脂肪酸、脂质、多糖和/或有机聚合物及其组合的部分彼此共价地连接。此连接可以经由脂质运载蛋白突变蛋白或氨基酸侧链的N-或C-末端。在一个具体实施方式中,所述连接经由突变的氨基酸残基形成,所述突变的氨基酸残基例如半胱氨酸或赖氨酸残基。
发明详述
在本发明的缀合物中,所述部分可以促进控制递送脂质运载蛋白突变蛋白,延长脂质运载蛋白突变蛋白的体内半衰期,提高脂质运载蛋白突变蛋白的生物利用度和/或降低脂质运载蛋白突变蛋白的免疫原性。
延长血清半衰期的部分可以是亲水聚合物、脂肪酸分子(例如棕榈酸(Vajo&Duckworth 2000,Pharmacol.Rev.52,1-9)、免疫球蛋白的Fc部分、免疫球蛋白的CH3结构域、免疫球蛋白的CH4结构域、白蛋白或其片段、白蛋白结合肽、或白蛋白结合蛋白质、泛素、源自泛素的肽、转铁蛋白等等。
白蛋白结合蛋白质可以是细菌白蛋白结合蛋白质、抗体、抗体片段(包括结构域抗体,例如见US专利6,696,245)、或对白蛋白具有结合活性的脂质运载蛋白突变蛋白。相应地,用于延长本发明脂质运载蛋白突变蛋白的半衰期的适合的缀合配偶体包括白蛋白(Osborn,B.L.等人(2002)Pharmacokinetic and pharmacodynamic studies of a human serum albumin-interferon-alpha fusion protein in cynomolgus monkeys J.Pharmacol.Exp.Ther.303,540-548),或白蛋白结合蛋白质,例如细菌白蛋白结合结构域,例如一种链球菌蛋白G(T.和Skerra,A.(1998)Use of an albumin-binding domain for the selective immobilisation of recombinant capture antibody fragments on ELISA plates.J.Immunol.Methods 218,73-83)。可以用作缀合配偶体的白蛋白结合肽的其它实例例如具有Cys-Xaa1-Xaa2-Xaa3-Xaa4-Cys共有序列的那些,其中Xaa1是Asp、Asn、Ser、Thr或Trp;Xaa2是Asn、Gln、His、Ile、Leu或Lys;Xaa3是Ala、Asp、Phe、Trp或Tyr;Xaa4是Asp、Gly、Leu、Phe、Ser或Thr,如US专利申请2003/0069395或Dennis等人(Dennis,M.S.,Zhang,M.,Meng,Y.G.,Kadkhodayan,M.,Kirchhofer,D.,Combs,D.&Damico,L.A.(2002).,,Albumin binding as a general strategy for improving the pharmacokinetics of proteins.”J Biol Chem 277,35035-35043)所述。
在其它实施方式中,可以将白蛋白自身或白蛋白的生物活性片段用作本发明的脂质运载蛋白突变蛋白的缀合配偶体。术语“白蛋白”包括所有的哺乳动物白蛋白,例如人血清白蛋白或牛血清白蛋白或大鼠白蛋白。可以以重组方式生成白蛋白或其片段,如US专利5,728,553或欧洲专利申请EP 0330451和EP 0361991中所述。可以将重组的人白蛋白Novozymes Delta Ltd.(Nottingham,UK)缀合或融合至脂质运载蛋白突变蛋白,从而延长突变蛋白的半衰期。
如果白蛋白结合蛋白质是抗体片段,其可以是结构域抗体。涉及结构域抗体(dAbs)从而能够精确控制生物物理特性和体内半衰期,获得最佳安全性和有效产物特征。结构域抗体例如可从Domantis Ltd.(Cambridge,UK and MA,USA)商购获得。
将转铁蛋白用作延长本发明突变蛋白的血清半衰期的部分,可以以基因工程方式将所述突变蛋白融合至非糖基化的转铁蛋白的N或C末端或二者。非糖基化的转铁蛋白的半衰期为14-17天,而转铁蛋白融合蛋白将类似地具有延长的半衰期。转铁蛋白载体还具有高的生物利用度、生物学分布和循环稳定性。此技术可从BioRexis(BioRexis Pharmaceutical Corporation,PA,USA)商购获得。用作蛋白质稳定剂/半衰期延长配偶体的重组人转铁蛋白(DeltaFerrinTM)也可从Novozymes Delta Ltd.(Nottingham,UK)商购获得。
如果将免疫球蛋白的Fc部分用于延长本发明突变蛋白的血清半衰期的目的,可以使用可从Syntonix Pharmaceuticals,Inc(MA,USA)商购获得的SynFusionTM技术。使用此Fc-融合技术能够产生长效作用的生物药剂,并且可以例如由连接至抗体的Fc区域的两个拷贝的突变蛋白组成,以改善药代动力学、溶解性和生产效率。
又一个延长本发明突变蛋白的半衰期的选择是将长的非结构化的柔性富甘氨酸序列(例如具有约20至80个连续甘氨酸残基的多甘氨酸)融合至本发明的突变蛋白的N或C末端。此方法例如公开于WO2007/038619,还具有术语“rPEG”(重组PEG)。
本文使用的术语“亲水聚合物”是指任何水溶性的线性、分支、分叉、支叉、枝状、多臂或星形聚合物,包括但不限于聚乙二醇和聚乙二醇/聚丙二醇共聚物,聚氧乙烯化丙三醇和类似聚合物。优选地,所述聚合物的分子量范围为约300道尔顿至约70,000道尔顿,更优选约500道尔顿至约50,000道尔顿,更优选约5,000道尔顿至约30,000道尔顿。
用于本发明的亲水聚合物通常具有至少一个引入的反应性基团,以通过氨基、羧基、巯基、磷酸酯或羟基官能团连接至目标生物活性分子。可以根据标准操作程序制备用于本申请的亲水聚合物(例如聚乙二醇),其中其一端以甲氧基封端,另一端被活化以便于缀合至生物活性分子的活性基团。例如,US专利6,113,906描述了使用“U形”(即分支的)形式的聚乙二醇上的琥珀酰亚胺琥珀酸酯或琥珀酰亚胺碳酸酯反应性基团与蛋白质的氨基反应。US专利5,650,234描述了使用聚乙二醇的N-羟基苯并三唑碳酸酯、2-羟基嘧啶碳酸酯和N-羟基-2-吡咯烷酮碳酸酯衍生物与蛋白质的氨基反应,从而形成稳定的氨基甲酸酯键。US专利5,672,661描述了使用丙酸和丁酸取代的聚乙二醇的琥珀酰亚胺酯与蛋白质的氨基反应,从而形成稳定的酰胺键。US专利5,446,090描述了使用聚乙二醇的乙烯基-砜衍生物,从而与蛋白质的巯基形成稳定的硫醚键。US专利5,880,255描述使用聚乙二醇的tresyl(2,2,2-三氟代乙烷-磺酰基)衍生物与蛋白质的氨基反应,从而形成简单且稳定的仲胺连接。US专利5,252,714描述了使用聚乙二醇的丙醛衍生物与蛋白质的氨基反应,形成稳定的仲胺键。可以有意地将由此类亲水聚合物连接至生物活性分子形成的键设计成稳定的或不稳定的(即可逆的)。此外,可以根据标准操作程序制备用于本申请的亲水聚合物,使其具有两个相似的(例如同质双功能的)或不相似的(例如异质双功能的)官能团,所述官能团可用于促进缀合至生物活性分子的活性基团。例如,WO 01/26692A1描述了使用异质双功能的聚乙二醇衍生物来进行蛋白质修饰。这些专利的全部内容都通过引用并入本文。
因此,用于本发明的缀合物的示例性亲水聚合物包括但不限于聚亚烷基二醇、聚氧乙烯化多元醇、羟乙基淀粉、多羟基酸、聚乳酸、聚乙醇酸、及其共聚物以及其线性、分支和活化的衍生物。
聚亚烷基二醇可以是取代的、未取代的、线性或分支的。它们还可以是活化的聚亚烷基衍生物。聚亚烷基二醇包括但不限于聚乙二醇、聚丙二醇、和聚乙二醇/聚丙二醇共聚物。
在本发明的一个实施方式中,所述亲水聚合物是聚乙二醇(PEG)。
PEG是指线性或分支的中性聚醚,其化学式为HO-(CH2CH2O)n-H。PEG高度溶解于水和许多有机溶剂(例如二氯甲烷、乙醇、甲苯、丙酮和氯仿),并且易于以多种大小(分子量)和官能化的结构(例如氨基-、羧基-、巯基-封端的)得到。已发现PEG无毒,已被FDA批准用于药物(肠胃外药物、局部药物、栓剂、鼻部喷剂)、食品和化妆品中。在溶液中,PEG是高度水合的聚合物,其中每个单体(环氧乙烷单元)可以结合多达三个水分子。并且,据信PEG具有影响数层更松散地结合的水合水分子的结构的能力。水中单个表面结合链的行为的分子模拟显示,该聚合物显示出具有极大程度的区段柔性。因此,推测聚合物在水性环境中占有极大的流体动力学体积。这些发现用于解释PEG显著有效地排除其它聚合物(天然的和合成的)存在的原因。对其它聚合物的排除是支持PEG排斥蛋白质,与其它合成聚合物形成两相体系的能力的主要驱动力,并且使得该聚合物为非免疫原性而不产生抗原。当PEG共价地连接至蛋白质时,它通常将聚合物的许多有利性质转移给所得的缀合物。由于上述多个有利特性,PEG非常适合用于蛋白质修饰。
本文使用的术语“PEG”包括任何PEG聚合物,包括PEG的氨基-反应性衍生物(“PEG反应物”)。已知多种用于蛋白质缀合的PEG反应物。典型的PEG反应物为一端以醚键封端(例如O-甲基)且另一端用反应性基团官能化的线性PEG聚合物。其它PEG反应物是分支的或枝状的,同样具有非反应性末端和反应性官能团的组合以连接至蛋白质。或者,可以使用具有相似或不相似的反应性官能团的同质或异质双官能PEG反应物以连接至蛋白质。
PEG反应物的实例包括但不限于,醛、N-羟基琥珀酰亚胺碳酸酯、N-羟基琥珀酰亚胺丙酸酯、对硝基苯基-碳酸酯、N-马来酰亚胺、N-琥珀酰亚胺、硫醇或苯并三唑-碳酸酯封端的一类或其它氨基-反应性的活化的PEG类。
PEG聚合物的分子量可以在例如300至70,000Da的范围内。可以通过接头基团将反应性官能团与PEG链分开。任选地,聚合物在PEG和接头之间具有可降解的内部的键。相应地,在本发明的一个实施方式中,可以以亲电方式活化PEG聚合物的反应性基团以与蛋白质亲核试剂反应。亲电基团的实例是n-羟基琥珀酰亚胺碳酸酯、tresyl和醛官能团。具有这些官能团的PEG反应物会反应形成与蛋白质的氨基的共价键。用于PEG缀合至蛋白质氨基的优选PEG反应物是丙酸接头mPEG-SPA的mPEG琥珀酰亚胺活性酯。另一个优选的PEG反应物是单甲氧基PEG-醛(mPEG-Ald)。
上文已提及适合的聚乙二醇(PEG)分子。其它实例描述于WO 99/64016、US专利6,177,074或涉及干扰素的US专利6,403,564中,或者已针对其它蛋白质例如PEG-修饰的天冬酰胺酶、PEG-腺苷脱氨酶(PEG-ADA)或PEG-超氧化物歧化酶进行了描述(例如参见Fuertges et al.(1990)The Clinical Efficacy of Poly(Ethylene Glycol)-Modified Proteins J.Control.Release 11,139-148)。其它实例例如可见于US专利4,179,337、US专利5,446,090和US专利5,880,255。本领域已知,这种连接可以使得分子量明显增加和使得改性治疗性蛋白质的血液清除率降低(例如参加US专利5,320,840),以及使得免疫原性减弱、从可生物降解聚合物药物递送体系的释放持续时间延长、在可生物降解药物递送体系中可实现的药物载量提高(相对于未聚乙二醇化的药物)、和药物突释降低(相对于未聚乙二醇化的药物)(例如参见PCT公布WO 02/036169)。这些专利和专利申请的内容通过引用全文并入本文。
所述聚氧乙烯化多元醇包括但不限于聚氧乙烯化丙三醇、聚氧乙烯化的山梨糖醇、聚氧乙烯化葡萄糖及其衍生物,例如酯。
活化的衍生物可以包括氨基-反应性衍生物,所述氨基-反应性衍生物包括选自醛、硫醇、N-羟基琥珀酰亚胺、琥珀酰亚胺、马来酰亚胺、PNP-碳酸酯和苯并三唑封端的亲水聚合物。在一个具体实施方式中,亲水聚合物的活化的衍生物是马来酰亚胺-衍生的PEG,其然后与突变蛋白的半胱氨酸残基的自由硫醇基团反应,其中这些半胱氨酸侧链可以天然的存在于蛋白质中或可以通过突变人工引入。为了使这些硫醇基团可用于偶联化学,可以使蛋白质经历还原步骤,以还原形成胱氨酸桥的半胱氨酸残基。在一个实施方式中,此步骤使用还原剂,例如三(2-羧乙基)盐酸膦(TCEP)或β-巯基乙醇。
聚合物的分子量范围可以是约300至约70000道尔顿,包括例如聚亚烷基二醇,例如分子量为约5、7、10、12、15、20、25、30、35、40、45、50、55、60、65或70kDa的聚乙二醇。
并且,例如如在US专利6,500,930或6,620,413所述,出于血清半衰期延长的目的,可以将碳水化合物低聚物和聚合物例如淀粉或羟乙基淀粉(HES)缀合至本发明的突变蛋白。
在本发明的缀合物中,脂质运载蛋白突变蛋白可以选自属于蛋白质的脂质运载蛋白家族的蛋白质的任何突变蛋白。已在上文提及了示例性脂质运载蛋白,并且包括但不限于视黄醇结合蛋白质(RBP)、后胆色素结合蛋白(BBP)、载脂蛋白D(APO D)、中性粒细胞明胶酶结合的脂质运载蛋白(NGAL)、泪脂质运载蛋白(TLPC)、α2-微球蛋白相关蛋白质(A2m)、24p3/uterocalin(24p3)、von Ebners腺蛋白质1(VEGP 1)、von Ebners腺蛋白质2(VEGP 2)和主要变应原Can f1前体(ALL-1)。优选的脂质运载蛋白突变蛋白包括人中性粒细胞明胶酶结合的脂质运载蛋白(hNGAL)、人泪脂质运载蛋白(hTLPC)、人载脂蛋白D(APO D)和大菜粉蝶后胆色素结合蛋白的突变蛋白。
人泪前白蛋白,目前称为泪脂质运载蛋白(TLPC或Tlc;SWISS-PROT数据库登记号P31025)最开始被描述为人泪液的主要蛋白质(约为总蛋白质含量的三分之一),但是近来已在数种其它分泌组织中被鉴定到,所述分泌组织包括前列腺、鼻粘膜和气道粘膜。已在大鼠、猪、狗和马中发现了同源的蛋白质。
人中性粒细胞明胶酶结合的脂质运载蛋白(hNGAL,也称作Lcn2,SWISS-PROT数据库登记号P80188)为富含于人血浆中的178个氨基酸的糖蛋白。人Lcn2的动物同源物为大鼠α2-微球蛋白相关蛋白质(A2m;SWISS-PROT数据库登记号P31052)和小鼠24p3/uterocalin(24p3;SWISS-PROT数据库登记号P11672)。
人载脂蛋白D(Apo-D;SWISS-PROT数据库登记号P05090)为与其它载脂蛋白序列无显著相似性的高密度脂蛋白的组分。其与血浆视黄醇结合蛋白质和载体蛋白质的α2微球蛋白超家族(也称作脂质运载蛋白)的其它成员具有高度同源性。
后胆色素结合蛋白(BBP;SWISS-PROT数据库登记号P09464)为富含于大菜粉蝶中的蓝色色素蛋白。
在本发明的一个实施方式中,所述脂质运载蛋白突变蛋白与下述蛋白的氨基酸序列具有至少50%、60%、70%、75%、80%、85%、90%或95%序列同源性:人泪脂质运载蛋白、人中性粒细胞明胶酶结合的脂质运载蛋白、大鼠α2-微球蛋白相关蛋白质、小鼠24p3/uterocalin、后胆色素结合蛋白、人载脂蛋白D、视黄醇结合蛋白质、von Ebners腺蛋白质1(VEGP 1)、von Ebners腺蛋白质2(VEGP 2)或主要变应原Can f1前体(ALL-1)。
在另一个实施方式中,所述脂质运载蛋白突变蛋白可以与下述蛋白的氨基酸序列具有至少50%、60%、70%、75%、80%、85%、90%或95%序列同一性:人泪脂质运载蛋白、人中性粒细胞明胶酶结合的脂质运载蛋白、大鼠α2-微球蛋白相关蛋白质、小鼠24p3/uterocalin、后胆色素结合蛋白、人载脂蛋白D、视黄醇结合蛋白质、von Ebners腺蛋白质1(VEGP 1)、von Ebners腺蛋白质2(VEGP 2)或主要变应原Can f1前体(ALL-1)。
术语“同源性”以其通常的含义在本文中使用,并且包括相同氨基酸以及认为是在两个互相比较的蛋白质线性氨基酸序列等价位置的保守替换氨基酸(例如天冬氨酸残基交换谷氨酸残基)。“同一性”或“序列同一性”表示量度它们的相似性或关系的序列特性。本发明所用的术语“序列同一性”或“同一性”表示本发明多肽序列与有关序列(同源性)比对之后,形成配对的相同残基相对于这两个序列较长一个的残基数量的百分比。通过将相同残基的数量除以残基总数再使其结果乘以100来量度同一性。
本文中,例如可以使用程序BLASTP,blastp 2.2.5版(2002年11月16日;参见Altschul、S.F.等人(1997)Nucl.Acids Res.25、3389-3402)测定序列同源性或序列同一性百分比。在此实施方式中,同源性百分比基于包括前肽序列的完整多肽序列比对(matrix:BLOSUM 62;gap costs:11.1;cutoff value set to 10-3),在配对比较中使用人泪脂质运载蛋白作为参考。其计算为,以BLASTP程序输出的结果表示的“阳性氨基酸”(同源氨基酸)除以程序选择用于比对的氨基酸总数的百分比。在这方面应注意,所选择的氨基酸的总数可以与人脂质运载蛋白2的长度不同。
在本发明的缀合物中,所述脂质运载蛋白突变蛋白以可检测的亲和力结合给定的非天然靶标。所述靶标可以是蛋白质、蛋白质结构域、蛋白质片段或肽。
在本发明的一个实施方式中,被泪脂质运载蛋白突变蛋白结合的靶标为选自下述的蛋白质或其片段:血管内皮生长因子(VEGF)、血管内皮生长因子受体2(VEGF-R2)、干扰素4受体α链(IL-4Rα)、细胞毒性T淋巴细胞抗原-4(CTLA-4)、受体酪氨酸激酶c-Met或其片段。还包括VEGF-R2、IL-4Rα、CTLA-4或c-Met的胞外区域或结构域作为配体。所述配体通常为哺乳动物来源的。在一个实施方式中,这些配体为人来源的,但是作为示例性实例,它们还可以是小鼠、大鼠、猪、马、犬、猫、牛、绒猴或猕猴来源的等等。
在本发明的缀合物中,所述脂质运载蛋白突变蛋白可以在包围天然脂质运载蛋白结合口袋的四个肽环AB、CD、EF和GH中的任意序列位置包含至少一个突变氨基酸残基。这些环形成所述脂质运载蛋白的已知结合位点(因此被称为开放末端)。在人泪脂质运载蛋白(hTLPC;SWISS-PROT数据库登记号P31025)中,AB环包含天然的成熟人泪脂质运载蛋白的线性多肽序列的位置24-36,CD环包含位置53-66,EF环包含位置69-77,GH环包含位置103-110。本文使用的这四个环的定义根据Flower(上文的Flower,D.R.(1996)和上文的Flower,D.R.等人(2000))。在大菜粉蝶后胆色素结合蛋白中,这四个肽环排列于包围结合口袋的圆柱形β桶结构的末端,对应于包含大菜粉蝶后胆色素结合蛋白的线性多肽序列的氨基酸位置28-45、58-69、86-99和114-129的线性多肽序列中的区段。
在其它实施方式中,所述脂质运载蛋白突变蛋白在包围天然的脂质运载蛋白结合口袋的四个肽环AB、CD、EF和GH中的任意序列位置具有至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40或41个突变氨基酸残基。
在其它实施方式中,所述脂质运载蛋白突变蛋白在对应于天然的成熟人泪脂质运载蛋白(SWISS-PROT数据库登记号P31025)的线性多肽序列的序列位置24-36、53-66、79-84和103-110的任意序列位置具有至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40或41个突变的氨基酸残基。在优选实施方式中,所述脂质运载蛋白突变蛋白在对应于天然的成熟人泪脂质运载蛋白的线性多肽序列的序列位置26-34、56-58、80、83、104-106和108的任意序列位置具有至少2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17或18突变氨基酸残基。技术人员使用本领域熟知的技术和程序可以容易地确定蛋白质的脂质运载蛋白家族其他成员的相应序列位置。
本发明的脂质运载蛋白突变蛋白可以包括突变氨基酸序列位置以外的野生型(天然的)氨基酸序列。另一方面,本文公开的脂质运载蛋白突变蛋白还可含有进行突变的环区域中序列位置以外的氨基酸突变,只要这些突变不干扰突变蛋白的结合活性和折叠即可。使用已建立的标准方法(Sambrook,J.等人(1989)Molecular Cloning:A Laboratory Manual,2nd Ed.,Cold Spring Harbor Laboratory Press,Cold Spring Harbor,NY)可以非常容易地在DNA水平上实现此类突变。氨基酸序列的可能改变为插入或缺失以及氨基酸替换。此类替换可以是保守的,即氨基酸残基被化学上相似的氨基酸残基替代。保守替换的实例为以下组成员的替代:1)丙氨酸、丝氨酸和苏氨酸;2)天冬氨酸和谷氨酸;3)天冬酰胺和谷氨酰胺;4)精氨酸和赖氨酸;5)异亮氨酸、亮氨酸、甲硫氨酸和缬氨酸;和6)苯丙氨酸、酪氨酸和色氨酸。另一方面,也可以在氨基酸序列中引入非保守改变。此外,除了替代单一的氨基酸残基,还可以插入或缺失泪脂质运载蛋白一级结构的一个或多个连续的氨基酸,只要这些缺失或插入产生稳定的折叠/功能突变蛋白(例如参见,其中产生N和C末端截短的突变蛋白的实验部分)。
氨基酸序列的此类修饰包括单一氨基酸位置的定点突变,以便通过掺入特定限制性酶的切割位点而简化突变脂质运载蛋白基因或其部分的亚克隆。此外,可掺入这些突变以进一步提高脂质运载蛋白突变蛋白与给定靶标的亲和力。并且,可以引入突变,以便调节突变蛋白的某些特征,例如以改善折叠稳定性、血清稳定性、蛋白质抗性或水溶解性,或者如果必要的话以降低聚集倾向。例如,天然存在的半胱氨酸残基可以突变成其它氨基酸,以防止二硫键的形成。然而,还可以有意地将其它氨基酸序列位置突变成半胱氨酸,以便引入新的反应性基团,例如用于缀合至本申请的部分或用于形成非天然存在的二硫键。
本申请还包括如上文定义的截短的脂质运载蛋白突变蛋白,所述突变蛋白中缺失了一个或多个N末端和/或C末端氨基酸,例如成熟脂质运载蛋白的序列的前四个N末端氨基酸残基和/或后两个C末端氨基酸残基。
在本发明缀合物的一个具体实施方式中,所述脂质运载蛋白突变蛋白包含SEQ ID No.1–110中任一项所述的氨基酸序列、基本上由SEQ ID No.1–110中任一项所述的氨基酸序列组成、或由SEQ ID No.1–110中任一项所述的氨基酸序列组成。所述脂质运载蛋白突变蛋白可以与SEQ ID No.1-110中任一项的脂质运载蛋白突变蛋白具有70、75、80、85、88、90、92、94、96或98%序列同一性。
上述部分之一缀合至脂质运载蛋白突变蛋白可以经由所述脂质运载蛋白突变蛋白的N末端、C末端或氨基酸侧链进行。为促进缀合,所述部分可以包含反应性基团,例如上文描述的那些。适合的氨基酸侧链可以天然存在于各脂质运载蛋白的氨基酸序列中,或者可以通过突变引入。在经由突变引入适合的结合位点的情况下,一种可选方法是在适当的位置用半胱氨酸残基替代氨基酸。在一个实施方式中,该突变包括下述的至少一种:针对人泪脂质运载蛋白(SWISS PROT数据库条目P31025)的线性多肽序列的Thr 40→Cys、Glu 73→Cys、Arg 90→Cys、Asp 95→Cys或Glu 131→Cys替换,或者蛋白质的脂质运载蛋白家族其它成员的相应替换。之后可以将在这些位置任意处新产生的半胱氨酸残基用于将突变蛋白缀合至延长突变蛋白的血清半衰期的部分上,所述部分例如PEG或其活化的衍生物。
在这些任意序列位置引入半胱氨酸的人泪脂质运载蛋白突变蛋白的一个示例性实例是VEGF结合人泪脂质运载蛋白突变蛋白S236.1-A22(SEQ ID NO:22)。本领域已知此类突变的其它实例,且包括下述文献公开的那些:PCT公开文本WO 99/16873、WO 00/75308、WO 03/029471、WO 03/029462、WO03/029463、WO 2005/019254、WO 2005/019255、WO 2005/019256、WO2006/56464、和WO 2008/015239,所述文献的内容通过引用全文并入本文。
在另一个实施方式中,为了提供用于将上述部分之一缀合至本发明的突变蛋白的适合的氨基酸侧链,可以通过突变引入人工氨基酸。通常,可以将此类人工氨基酸设计成更有反应性,因此促进期望部分的缀合。可以经由人工tRNA引入的这样的人工氨基酸的一个实例是对乙酰基-苯基丙氨酸。
在一些实施方式中,可以将脂质运载蛋白突变蛋白及其缀合的部分用于形成融合蛋白。在此类实施例中,以其N末端或C末端将脂质运载蛋白突变蛋白融合至蛋白质、蛋白质结构域或肽部分。
在其它实施方式中,可以将所述脂质运载蛋白突变蛋白融合至另一个蛋白质、蛋白质结构域或肽,例如具有相同或不同结合特异性的第二脂质运载蛋白突变蛋白(这造成“Duocalins”的形成,参见Schlehuber,S.,and Skerra,A.(2001),Duocalins,engineered ligand-binding proteins with dual specificity derived from the lipocalin fold.Biol.Chem.382,1335-1342),并且可以将所得的包含所述脂质运载蛋白突变蛋白的融合蛋白缀合至上述部分。
本领域已知用于产生脂质运载蛋白突变蛋白的技术(例如参见上文的Flower,D.R.(1996);上文的Flower,D.R.等人(2000),上文的Skerra,A.(2000),上文的Skerra,A.(2001);上文的Schlehuber,S.,和上文的Skerra,A.(2002);以及PCT公开文本WO 99/16873、WO 00/75308、WO 03/029471、WO 03/029462、WO 03/029463、WO 2005/019254、WO 2005/019255、WO2005/019256、WO 2006/56464和WO 2008/015239)。这些专利申请每一个的内容都通过引用全文并入本文。
本文可互换地使用的术语“非天然配体”、“非天然结合配偶体”或“非天然靶标”是指这样的化合物:所述化合物在生理条件下不结合各天然的脂质运载蛋白。所述靶标(配体)可以是任何游离的或缀合形式的化合物,其显示具有下述物质的特征:免疫半抗原、激素例如类固醇激素或其任意生物聚合物或片段,例如蛋白质或蛋白质结构域、肽、脱氧寡核苷酸、核酸、寡糖或多糖或其缀合物、脂质或类似的大分子。
包含于本发明缀合物中的脂质运载蛋白突变蛋白保留了它们以可检测的亲和力结合期望靶标的能力,所述可检测的亲和力例如具有至少200nM的解离常数。当前在一些实施方式中优选这样的脂质运载蛋白突变蛋白,其以对给定靶标的至少100、20、1nM或甚至更低的解离常数结合期望靶标。可以通过多种方法测量突变蛋白与期望靶标的结合亲和力,所述方法例如荧光滴定法、竞争ELISA或表面等离子共振(BIAcore)。
本发明还涉及用于控制释放脂质运载蛋白突变蛋白的药物制剂,所述制剂包含与聚合物、脂质或脂质体以及任选的药学上可接受的赋形剂组合的脂质运载蛋白突变蛋白或其缀合物。
术语“控制释放”或“持续释放”是指对根据本发明的药物递送制剂递送的脂质运载蛋白突变蛋白分子的速率和/或数量的控制。控制释放可以是连续的或不连续的、和/或线性或非线性的。这可以通过一种或多种类型的聚合物组成、药物载量、包含赋形剂或降解促进剂,单独、组合或顺序使用以产生期望效果而得以实现。“零序”或“线性释放”通常理解为,表示在期望的时间段期间,作为量/单位时间的函数,释放的生物活性分子的量随时间变化保持相对恒定。“多相”通常理解为表示释放发生一次以上的“突释”。
在一个实施方式中,脂质体包封脂质运载蛋白突变蛋白或其缀合物。在一个具体实施方式中,所述脂质体分散或乳化于水性基础介质中。
在一个实施方式中,所述聚合物为可生物降解聚合物。在一个这样的实施方式中,所述可生物降解聚合物选自聚丙交酯、聚乙交酯、聚(丙交酯-共-乙交酯)、聚乳酸、聚乙醇酸、聚(乳酸-共-乙醇酸)、聚己内酯、聚碳酸酯、聚酯酰胺、聚酸酐、聚(氨基酸)、聚原酸酯、聚缩醛(polyacetyls)、聚氰基丙烯酸酯、聚醚酯、聚二噁烷酮、聚亚烷基烷基酯(polyalkylen alkylate)、聚乙二醇和聚丙交酯或聚(丙交酯-共-乙交酯)的共聚物、可生物降解的聚氨酯和某些类型的蛋白质和多糖聚合物、以及其共混物和共聚物。在另一个实施方式中,所述可生物降解聚合物选自多羟基酸、聚乳酸、聚丙交酯、聚乙交酯、聚乙醇酸、和其共聚物以及其衍生物。所述可生物降解聚合物还可以包括聚酸酐、聚原酸酯和多糖聚合物,或者由聚酸酐、聚原酸酯和多糖聚合物组成。一种适合的聚酸酐共聚物是双(对羧基苯氧基)丙烷和癸二酸的共聚物。其它适合的可生物降解的亲水聚合物包括生物聚合物,例如上述蛋白质和多糖聚合物,包括但不限于透明质酸、壳聚糖、明胶、胶原、淀粉、糊精和纤维素。这些生物大分子可以交联,以形成三维水凝胶。
在一个实施方式中,所述可生物降解聚合物为聚-(D,L-丙交酯-共-乙交酯)。如果选择聚-(D、L-丙交酯-共-乙交酯)作为聚合物以形成本发明的药物制剂,则所述聚-(D,L-丙交酯-共-乙交酯)可以含有约55至约80摩尔%丙交酯单体和约45至约20摩尔%乙交酯。所述聚-(D、L-丙交酯-共-乙交酯)还可以含有约65至约75摩尔%丙交酯单体和约35至约25摩尔%乙交酯。所用的聚-(D、L-丙交酯-共-乙交酯)可以含有末端酸基团。
在另一个实施方式中,所述可生物降解聚合物为包含用烷基部分取代的丙交酯单元的聚乳酸聚合物或共聚物。所述可生物降解聚合物可以例如包含聚(己基-取代的丙交酯)或聚(二己基-取代的丙交酯)、基本上由聚(己基-取代的丙交酯)或聚(二己基-取代的丙交酯)组成、或由聚(己基-取代的丙交酯)或聚(二己基-取代的丙交酯)组成。
在药物制剂的另一个实施方式中,将所述可生物降解聚合物配制成包封所述缀合物的微粒或纳米粒子。在此类实施方式中,本发明的制剂可以基于由可生物降解聚合物和本发明的脂质运载蛋白突变蛋白或缀合物的组合形成的微粒或纳米粒子,以提供控制释放;所述可生物降解聚合物例如聚乳酸(PLA)、聚乙醇酸(PGA)、及其共聚物,所述缀合物例如脂质运载蛋白突变蛋白与亲水聚合物的缀合物,所述亲水聚合物例如聚乙二醇或聚丙二醇。
在另一个实施方式中,所述药物制剂可以是植入物或可植入装置的形式。所述植入物或可植入装置可以具有任何形状,并且包括但不限于可植入的棒、塞、盘、粒、膜或片。可以通过已知方法生产此类可植入装置,所述已知方法例如由合适聚合物模塑、挤出和膜制备。如果所述药物制剂为植入物或可植入装置,植入物材料可以是可生物降解的或不可生物降解的,例如可生物降解或不可生物降解聚合物。适合的可生物降解聚合物包括但不限于上文所述的那些。
不可生物降解聚合物可以在活生物体中保持较长的时间周期,例如数年,而无显著降解。此类物质包括聚乙烯醇(PVA)和乙烯醋酸乙烯酯(EVA)的聚合物等等。
所述药物制剂、特别是可植入装置还可以含有例如抗代谢物,以防止植入部位的纤维化。
在一个实施方式中,本发明采用可生物降解的微粒用于控制释放脂质运载蛋白突变蛋白或聚合物缀合的脂质运载蛋白突变蛋白。
本文使用的“微粒”是指直径优选小于1.0mm、通常为1.0至200.0微米且更优选1.0至100.0微米的颗粒。微粒可以具有任何适合的粒径,只要同样提供另人满意的目标脂质运载蛋白突变蛋白的持续释放即可。在示例性实施例中,微粒的平均粒径(直径)可以小于100微米,例如在约20至约80微米的范围内或平均粒径(直径)在约40至约50微米的范围内。术语“微粒”包括通常为固体球形微粒的微球。术语“微粒”还包括微胶囊,其通常为具有不同于围绕的外壳的组合物核心的球形微粒。为了本发明的目的,可互换地使用术语“微球”、“微粒”和“微胶囊”。
应该选择用于制备本发明的药物组合物/制剂的脂质运载蛋白突变蛋白的质量与聚合物的质量的比率,使得实现另人满意的控制持续释放。在示例性实施例中,脂质运载蛋白突变蛋白的质量占用于制备制剂(例如微粒或纳米粒子)的聚合物质量的15%或更少。在其它实施方式中,药物组合物中突变蛋白的质量占聚合物质量的10%或更少。
可以使用多种本领域中熟知的用于控制释放制剂的可生物降解聚合物来制备用于本申请的微粒。如上文所述,合适聚合物例如包括但不限于聚丙交酯、聚乙交酯、聚(丙交酯-共-乙交酯)、聚乳酸、聚乙醇酸、聚(乳酸-共-乙醇酸)、聚己内酯、聚碳酸酯、聚酯酰胺、聚酸酐、聚氨基酸、聚原酸酯、聚缩醛、聚氰基丙烯酸酯、聚醚酯、聚二噁烷酮、聚烷基亚烷基酯、聚乙二醇和聚丙交酯或聚(丙交酯-共-乙交酯)的共聚物、可生物降解的聚氨酯和某些类型的蛋白质和多糖聚合物、以及其共混物和共聚物。上文已公开了蛋白质和多糖聚合物的实例,且包括但不限于透明质酸、壳聚糖、明胶、胶原、淀粉、糊精和纤维素。
为了本申请的目的,术语“可生物降解的”是指在具体治疗情况下可接受的某时间段内体内溶解或降解的聚合物。这种溶解或降解的产物可以包括较小的化学物质。可以例如通过酶、化学和/或物理过程形成降解。生物降解通常在暴露于生理pH和温度的少于五年且通常少于一年发生,所述生理pH和温度例如在6至9范围内的pH和在22℃至38℃范围内的温度。
优选的聚合物包括聚(羟基酸),尤其是聚(乳酸-共-乙醇酸)(聚(D,L-丙交酯-共-乙交酯);“PLGA”)、聚乙交酯(PGA)和聚丙交酯(PLA)或其衍生物,它们在暴露于生理pH,例如身体水性环境后,通过水解降解。然后所述聚合物水解产生乳酸和乙醇酸单体,这些单体是细胞代谢的正常副产物。聚合物分解的速率可以从数周至超过一年的时间段变化,这取决于数个因素,包括聚合物分子量、聚合物链中丙交酯与乙交酯单体的比率和单体亚单元的立构规整度(L和D立体异构体的混合物破坏聚合物结晶度,从而促进聚合物降解)。在示例性实施例中,用于制备制剂(例如本发明的微粒)的聚-(D,L-丙交酯-共-乙交酯)可以含有约55至80摩尔%丙交酯单体和约45至20摩尔%乙交酯醇酸。或者,所述聚-(D、L-丙交酯-共-乙交酯)可以含有约65至75摩尔%丙交酯单体和约35至25摩尔%乙交酯。使用的聚-(D、L-丙交酯-共-乙交酯)可以含有末端酸基团。微球也可含有两种或更多种具有不同分子量和/或单体比率的可生物降解聚合物的共混物。
衍生的可生物降解聚合物也适用于本申请,包括连接至PLGA的亲水聚合物等。在具体实施方式中,所述亲水聚合物选自但不限于聚乙二醇、聚丙二醇、聚乙二醇和聚丙二醇的共聚物、和聚乙烯吡咯烷酮。
具体而言,可以使用本领域熟知的多种技术以形成微球。这些技术包括,例如单或双乳化步骤,然后去除溶剂。可以通过萃取、蒸发或喷雾干燥以及其它方法实现溶剂去除。
在溶剂萃取方法中,将所述聚合物溶解于有机溶剂中,所述有机溶剂可至少部分溶解于萃取溶剂例如水中。然后将可溶形式或分散成细小颗粒的脂质运载蛋白突变蛋白缀合物加入到聚合物溶液中,并将混合物分散到含有表面活性剂的水相中,所述表面活性剂例如聚乙烯醇。将所得的乳液加入到更大体积的水中,在水中,将有机溶剂从聚合物/生物活性剂中去除以形成硬化的微粒。
在溶剂蒸发方法中,将所述聚合物溶解于挥发性的有机溶剂中。然后将可溶形式或分散成细小颗粒的生物活性分子(即脂质运载蛋白突变蛋白缀合物)加入到聚合物溶液中,并且将混合物悬浮于含有表面活性剂的水相中,所述表面活性剂例如聚乙烯醇。搅拌所得的乳液直到大部分有机溶剂蒸发,从而留下固体微球。
在喷雾干燥方法中,将所述聚合物溶解于适合的溶剂中,例如二氯甲烷(例如0.04g/ml)。然后将已知量的脂质运载蛋白突变蛋白缀合物悬浮(如果不可溶)或共溶(如果可溶)于聚合物溶液中。然后喷雾干燥溶液或分散液。可以获得具有形态的直径范围为1至10微米的微球,所述形态取决于聚合物的选择。
本领域已知其它已知方法,例如相分离和凝聚以及上述方法的变形也可在本申请中采用。
在另一个实施方式中,本发明采用可生物降解的纳米粒子用于控制释放脂质运载蛋白突变蛋白和聚合物缀合的脂质运载蛋白突变蛋白。本文使用的术语“纳米粒子”是指直径优选为约20.0纳米至约2.0微米、通常约100纳米至1.0微米的颗粒。
可以如上文针对微粒所述实现纳米粒子的配制,不同之处在于使用高速混合或匀质化以将聚合物/生物活性剂乳液的尺寸降低至约2.0微米以下,优选地约1.0微米以下。例如,WO 97/04747中描述了用于制备纳米粒子的适合的技术,所述专利申请的全部内容通过引用并入本文。
可以通过注射、通过吸入、经鼻或口服而给予这些控制释放制剂。
通常,药剂包封于可生物降解聚合物微球和纳米球中可以比给予药物自身延长保持治疗药物水平。取决于制剂和包封的活性分子,可将持续释放延长长达数月。然而,由于治疗性蛋白质易于被将它们包封于聚合物载体中所需的程序造成损坏,一些蛋白质的极性性质可限制在聚合物药物载体中包封的程度,且可导致所包封的生物活性分子的部分在首次给予时迅速损失(“突释”),因此,有利的是,将亲水聚合物连接至这些治疗性蛋白质以在包封于药物载体中的条件下保护它们免于降解和变性。此外,相对于未经修饰的蛋白质,这会提高可以被包封的经修饰的蛋白质的量。并且,相对于载体中的未聚乙二醇化的蛋白质,降低了包封于可生物降解聚合物药物递送载体中的聚乙二醇化的治疗性蛋白质的免疫原性,特别是当通过皮下或肌内注射或吸入或粘膜递送(例如口服或经鼻递送)给予时。当可生物降解聚合物用于口服递送时,这种降低的免疫原性是特别有利的。并且,已报道了通过以可生物降解聚合物体系、特别纳米球给予,可以使得通常情况下不能从胃肠道吸收的聚乙二醇化的蛋白质、肽、寡糖和寡核苷酸可生物利用。在PCT公开文本WO 02/36169中更详细地描述了这些益处,所述专利申请的内容通过引用全文并入本文。
可以将本申请的药物组合物用于在数个方面改善脂质运载蛋白突变蛋白或其缀合物的体内递送,所述方面例如免疫原性降低、生物利用度提高、持续时间增加、稳定性提高、突释减少和/或脂质运载蛋白突变蛋白体内的控制持续释放。
本申请的药物组合物可以包含未缀合形式的脂质运载蛋白突变蛋白或上述脂质运载蛋白突变蛋白的缀合物。如果脂质运载蛋白突变蛋白不缀合至另一个部分,则所述脂质运载蛋白突变蛋白定义为上文公开的与本发明的缀合物相关的脂质运载蛋白突变蛋白。
在本发明制剂的一个实施方式中,所述脂质运载蛋白突变蛋白缀合至亲水聚合物,优选聚乙二醇部分。所述聚乙二醇可以是线性或分支的或者可以是活化的PEG衍生物,且可具有约5、7、10、12、15、20、25、30、35、40、45、50、55、60、65或70kDa的分子量,优选约5至约40kDa,更优选约15至25kDa,例如20kDa。在具体实施方式中,将该缀合的脂质运载蛋白突变蛋白与聚丙交酯、聚乙交酯或其共聚物例如聚(D,L-丙交酯-共-乙交酯,包括丙交酯和乙交酯的烷基化衍生物一起配制。所述聚(D,L-丙交酯-共-乙交酯)可以含有约55至80摩尔%丙交酯单体和约45至20摩尔%乙交酯。所述聚-(D,L-丙交酯-共-乙交酯)还可以含有约65至75摩尔%丙交酯单体和约35至25摩尔%乙交酯。优选地,这种制剂为微粒或纳米粒子的形式。在一个具体实施方式中,所述突变蛋白为人泪脂质运载蛋白或hNGAL突变蛋白。
在其它实施方式中,将未缀合形式的脂质运载蛋白突变蛋白与聚丙交酯、聚乙交酯或其共聚物,包括丙交酯和乙交酯的烷基化衍生物一起配制。这种制剂可以是微粒或纳米粒子的形式,且所述突变蛋白为人泪脂质运载蛋白或hNGAL突变蛋白。
在本发明制剂的一个实施方式中,约30至90%、优选地40至70%的脂质运载蛋白突变蛋白以活性形式释放,即保持它们结合给定配体的能力。取决于制剂的实际组成,所述释放可以经过一定时间段。所述时间段可以包括一天或多天,例如2至约70天,优选20至60天。
在另一个方面,本申请表征了用于将脂质运载蛋白突变蛋白全身性或局部控制递送至受试者的方法,所述方法包括给予所述受试者本发明的缀合物或药物制剂。
在此类方法中,通过给予至受试者的限定的身体腔室或器官,可以促进脂质运载蛋白突变蛋白的局部控制递送。已发现,将脂质运载蛋白突变蛋白制剂给予至受试者(例如哺乳动物,优选人)的限定的身体腔室或器官,可以促进持续释放效果,这是因为脂质运载蛋白突变蛋白在相应身体腔室或器官中的半衰期可以与全身半衰期不同。因此,通过局部给予本发明的脂质运载蛋白突变蛋白缀合物或制剂,可实现局部持续释放,同时避免全身性暴露的延长。这可以归因于下述事实:在不同的身体腔室或器官之间,脂质运载蛋白突变蛋白的代谢性降解或分泌不同。例如,相比血清半衰期,脂质运载蛋白突变蛋白在眼内腔中的半衰期延长约150倍。因此,释放的突变蛋白在相应的身体腔室或器官中的半衰期可以比血清中的半衰期长,从而离开限定的身体腔室或器官的脂质运载蛋白突变蛋白迅速被肾过滤降解或清除,而它们在身体腔室或器官内的半衰期足够长以达到期望的药理效果。
在本发明的一些实施方式中,所述限定的身体腔室为眼、关节、脑、脊椎、肿瘤,或身体腔、室。
在这些方法的一个实施方式中,口服、通过吸入、粘膜递送、皮内注射、皮下注射、肌内注射、静脉内注射、玻璃体内注射、关节内注射、颅骨内注射、脊椎内注射、肿瘤内注射或植入给予所述缀合物或制剂。
在这些方法中,所述受试者可以是哺乳动物,优选人。
在另一个方面,本申请还包括用于制备脂质运载蛋白突变蛋白-聚合物缀合物的方法,所述方法包括在至少一种有机溶剂和至少一种金属螯合剂存在下,在促进突变蛋白和聚合物的缀合物形成的条件下,使脂质运载蛋白突变蛋白与亲水聚合物接触;以及分离所述缀合物。可以使用多种标准技术例如柱色谱法分离所述缀合物。
在本发明的具体实施方式中,所述亲水聚合物选自聚乙二醇、聚乙二醇/聚丙二醇共聚物、聚氧乙烯化丙三醇、以及其线性、分支的和氨基-反应性衍生物。适合的氨基-反应性衍生物包括,例如醛、PEG-羧酸的N-羟基琥珀酰亚胺酯、PNP-碳酸酯和苯并三唑封端的亲水聚合物衍生物。其它优选的PEG的活化衍生物包括但不限于N-马来酰亚胺PEG衍生物。通常,所述亲水聚合物和脂质运载蛋白突变蛋白以约10:1-1:1、优选3:1至1.2:1、更优选1.7:1至1.5:1的摩尔比接触。
适合用于本发明的有机溶剂包括多种已知溶剂,包括但不限于水可混溶的有机溶剂,例如二氯甲烷、乙醇、甲醇、DMSO、二噁烷、DMF和NMP。通常,所述有机溶剂(优选二噁烷)以约0至25%、优选2-20%、更优选5-15%或0.1-10%的浓度(v/v)存在。
适合用于本发明的金属螯合剂也包括多种已知化合物,包括但不限于氨基多羧酸,例如乙二胺四乙酸(EDTA)、二乙烯三胺五乙酸(DTPA)、氨三乙酸(NTA)、N-2-乙酰氨基-2-亚氨基二乙酸(ADA)、双(氨基乙基)乙二醇醚-N,N,N',N'-四乙酸(EGTA)、反式-二氨基环己烷四乙酸(DCTA)、谷氨酸和天冬氨酸;和羟基氨基羧酸,例如N-羟基乙基亚氨基二乙酸(HIMDA)、N,N-双-羟基乙基甘氨酸(bicine)和N-(三羟基甲基甲基)甘氨酸(tricine);和N-取代的甘氨酸,例如双甘氨肽。其它适合的螯合剂包括2-(2-氨基-2-氧代乙基)氨基乙烷磺酸(BES)和去铁胺(DEF)。本申请方法中使用的适合的螯合剂包括,例如在溶液中结合金属离子的那些,从而使得金属离子无法与可用的氧反应,由此防止与蛋白质反应和降解蛋白质的自由-OH残基的产生,或者使所述自由-OH残基最少化。此类螯合剂可以降低或防止在无螯合试剂保护下配制的蛋白质的降解。
用于本发明的螯合试剂可以以其盐的形式存在,所述盐的形式例如前述螯合剂的羧基或其它酸性官能团。此类盐的实例包括与钠、钾、钙和其它弱结合的金属离子形成的盐。如本领域已知,盐的性质和要中和的电荷的数量将取决于存在的羧基的数量和提供稳定化螯合剂时的pH。同样如本领域已知,螯合试剂以不同的强度与具体靶标结合。通常,重金属离子比类似电荷的较低分子量的金属离子的结合力更强。
用于本申请方法的螯合剂也可选自EDTA、EGTA和本领域已知的其它多价阳离子螯合剂。
在一个实施方式中,所述螯合剂选自EDTA、去铁胺(DEF)、二乙烯三胺五乙酸(DTPA)和双(氨基乙基)乙二醇醚-N,N,N',N'-四乙酸(EGTA)。
通常,所述螯合剂(优选EDTA)以约0.1-10mM、优选1-5mM、更优选1-3mM的浓度存在。
在本发明的具体实施方式中,所述脂质运载蛋白突变蛋白和亲水聚合物(例如PEG)在蛋白质浓度为约0.1-5重量%、优选0.5-1.5%的水性溶液中接触(即反应或缀合)。在本发明的另一个实施方式中,所述脂质运载蛋白突变蛋白和亲水聚合物在pH为约5.0-7.5、优选pH 6.5至7.2、更优选约7.0的水性溶液中接触。可以通过包括缓冲盐、加入有机酸/碱、或加入常用的无机酸/碱来控制pH。在另一个具体实施方式中,所述亲水聚合物和脂质运载蛋白突变蛋白在约4℃至50℃、优选约15℃至25℃的温度下接触。
形成后,从不期望的反应副产物和未反应的脂质运载蛋白突变蛋白中分离所述蛋白质-聚合物缀合物。这可以使用多种已知技术、例如色谱法得以实现。在具体实施方式中,采用离子交换色谱(例如阳离子交换),并且可以收集、浓缩、脱盐和干燥期望的缀合物。
在另一个实施方式中,本申请的方法还包括在分离缀合产物之前淬灭所述脂质运载蛋白突变蛋白和亲水聚合物的反应(即缀合)的步骤。在具体实施方式中,这通过将反应的pH降低至约1-4、优选约2-3、更优选约2.4至2.6得以实现。
在一个方面,本申请还涉及根据前述用于制备脂质运载蛋白突变蛋白聚合物-缀合物的方法获得的缀合物。
通过本申请的方法生成的具体蛋白质-聚合物缀合物包括,例如脂质运载蛋白突变蛋白-聚合物缀合物,优选脂质运载蛋白突变蛋白-PEG缀合物(聚乙二醇化的脂质运载蛋白突变蛋白)。其可以包括任何上述脂质运载蛋白突变蛋白。在具体实施方式中,所述脂质运载蛋白突变蛋白特异地在脂质运载蛋白突变蛋白的N末端、C末端或氨基酸侧链反应(聚乙二醇化)。为了促进缀合,PEG部分可包含反应性基团。如上文所述,适合的氨基酸侧链可天然存在于各脂质运载蛋白的氨基酸序列中,或者可以通过突变引入。在经由突变引入适合的结合位点的情况下,一种可选方法是在适当的位置用半胱氨酸残基进行替代。在一个实施方式中,该突变包括下述的至少一种:针对人泪脂质运载蛋白(SWISS PROT数据库条目P31025)的线性多肽序列的Thr40→Cys、Glu 73→Cys、Arg 90→Cys、Asp 95→Cys或Glu 131→Cys替换,或者蛋白质的脂质运载蛋白家族其它成员的相应替换。可以在如本领域熟知的任何合适制剂中治疗性地给予所得的聚乙二醇化的脂质运载蛋白突变蛋白。在具体实施方式中,通过例如在给予前将缀合物包封于可生物降解聚合物中而给予在持续释放制剂中的缀合物。
此单步骤方法能够迅速且有效地制备脂质运载蛋白突变蛋白-聚合物缀合物。在PCT公开文本WO 2004/091494中针对胰岛素更详细地公开了所述制备方法,所述专利申请通过引用全文并入本文。
在另一方面,本申请还涉及制备控制释放组合物的方法,所述方法包括:合并包含脂质运载蛋白突变蛋白和聚合物的有机相与水相;以及回收所述组合物。在一个实施方式中,所述水相可以包含有机离子,其中存在所述有机离子从而降低可能的脂质运载蛋白突变蛋白的降解。
为了本申请的目的,术语“有机相”和“不连续相”可以互换,并且是指在本申请方法中产生的溶剂、聚合物和脂质运载蛋白突变蛋白的溶液,所述溶液之后通过乳化法与水相接触,以便产生本申请的控制释放组合物。
为了本申请的目的,术语“降解”是指对所述脂质运载蛋白突变蛋白的任何不期望的修饰,例如酰化;或对所述聚合物的任何不期望的修饰,例如分解。
为了本申请的目的,术语“水相”和“连续相”可互换,并且是指在本申请方法中产生的水和有机离子试剂的溶液,所述溶液之后通过乳化法与有机相接触,以便产生本申请的控制释放组合物。
为了本申请的目的,术语“合并”是指将两种或更多种物质放在一起的任何方法。此类方法包括但不限于,混合、共混、掺和、调制、匀质化、掺入、互混、融合、接合、弄混、搅拌、联合、并入、混入、接合、一体化等等。
为了本申请的目的,本文可以以从“约”或“大致”一个具体数值和/或至“约”或“大致”另一个具体数值来表示范围。当表示这样的范围时,另一个实施方式包括从一个具体数值和/或至另一个具体数值。类似地,当通过使用前缀“约”表示数值为近似值时,将被理解的是,该具体数值形成另一个实施方式。还将被理解的是,每个范围的端点的意义既与另一个端点相关也与另一个端点独立。通常,术语“约”包括给定数值的±10%。
为了本申请的目的,术语“有机离子”是指阳离子和阴离子物质。示例性有机离子包括扑酸盐、萘甲酸盐、胆酸盐等等。有机离子可以以其盐或酸形式存在。用于本申请的有机离子包括阴离子和阳离子物质。阴离子物质包括但不限于下述有机酸及其盐:十二烷基硫酸、胆酸、三氟甲基对苯甲酸、2-萘磺酸、2,3-萘二羧酸、1-羟基-2-萘甲酸、3-羟基-2-萘甲酸、2-萘甲酸和双水杨酸。此外,硫酸盐、磺酸盐、磷酸盐和膦酸盐的有机形式是适合的有机离子。阴离子物质的盐形式可以包括钠、铵、镁、钙等等。阳离子分子包括但不限于具有铵或胍基团或取代的铵基团的那些。和生物活性剂一起使用有机阴离子试剂,所述生物活性剂具有一个或多个带有或能够获得正电荷的官能团,例如铵或胍基团。可以与生物活性剂一起使用有机阳离子,所述生物活性剂具有一个或多个带有或能够获得负电荷的官能团,例如羧基、硫酸盐、磺酸盐、磷酸盐或膦酸盐基团。用于本申请的有机离子试剂可以溶解于水和有机相中达到提高包封效率和药物载量的程度。在具体实施方式中,经由生物活性剂的降解降低而实现包封效率和药物载量提高。在具体实施方式中,有机离子试剂在水相中的浓度范围为约0.5至100mM。在另一个具体实施方式中,所述有机离子范围为约5至40mM。
为了本申请的目的,“控制释放组合物”应指具有与天然生物活性剂不同的释放曲线(release profile)的任何制剂。通常释放曲线将包括生物活性剂经至少一周、至少一个月、至少45天或长于45天的时间段的生理可检测浓度。
在该方法的一个实施方式中,所述方法还包括在有机相中合并脂质运载蛋白突变蛋白和聚合物和/或在水相中合并有机离子的步骤。
在所述方法的另一个实施方式中,在乳化法中合并有机相和水相以产生控制释放组合物。乳液可包含分散于水相中的有机相的小滴。之后可以从乳液小滴中去除溶剂以形成硬化的微粒。在具体实施方式中,通过蒸发去除所述溶剂。在另一个具体实施方式,通过萃取到萃取液中去除溶剂;例如,所述萃取液可以是水。在另一个具体实施方式中,通过过滤去除溶剂。
然后可以从水相回收硬化的微粒并干燥。
在另一个实施方式中,通过搅拌有机相和水相产生乳液。在另一个实施方式中,通过使用混合器产生乳液。在具体实施方式中,所述混合器为静态混合器。在某些实施方式中,通过采用紊流混合产生乳液。在另一个实施方式中,不经紊流混合而产生乳液。
可以在组分的沸点至凝固点的任何温度实施所述乳化法。在一个实施方式中,温度范围为约0℃至约100℃,且通常为5℃至75℃。在具体实施方式中,在约15℃至约60℃实施所述乳化法。
在某些实施方式中,所述方法包括使包含聚合物和生物活性剂的有机相与包含有机离子的水相接触,其中有效量的有机离子离开水相并进入有机相。
在一个实施方式中,所述有机相包含溶剂,所述溶剂选自但不限于二氯甲烷、乙酸乙酯、苄醇、丙酮、乙酸和碳酸丙烯酯。所述有机相还可以包含可生物降解聚合物在其中可溶的其它溶剂。在优选的实施方式中,所述溶剂是乙酸乙酯或二氯甲烷。
在具体实施方式中,所述有机相还包括共溶剂。任选地将它们用于提高生物活性剂在有机相中的溶解性。
所述共溶剂可以选自但不限于二甲基亚砜、二甲基甲酰胺、N-甲基吡咯烷酮、PEG200、PEG400、甲醇、乙醇、异丙醇、苄醇和水。
在另一个具体实施方式中,所述共溶剂可以以有机相的溶剂的0至90%w/w存在。在另一个具体实施方式中,所述共溶剂可以以有机相的溶剂的0至50%w/w存在。
可以先将脂质运载蛋白突变蛋白溶于适当体积的共溶剂,然后将其加入到任选其中具有溶解的可生物降解聚合物的有机相的溶剂中,以形成有机相所有组分的溶液。普通技术人员可以调整加入的体积和顺序,以实现脂质运载蛋白突变蛋白和可生物降解聚合物的溶液。在具体实施方式中,所述可生物降解聚合物将以2-40%w/w的浓度存在于有机相中。在另一个具体实施方式中,所述可生物降解聚合物将以5-20%w/w的浓度存在于有机相中。
在另一个实施方式中,所述水相还包括乳化剂。所述乳化剂可以选自但不限于,聚(乙烯基醇)、白蛋白、卵磷脂、维生素E-D-α-生育酚聚乙二醇(TPGS)和聚山梨醇酯。在具体实施方式中,所述乳化剂可以以终浓度范围为约0.1至10%(w/w)、终浓度为约1.0%至约8%(w/w)、约1.0%至约6%(w/w)、约1.5%至约5%(w/w)、或0.5至5%(w/w)的浓度存在。
在制备本发明的控制释放组合物的乳化法的某些实施方式中,含有脂质运载蛋白突变蛋白的有机相包含选自二氯甲烷、乙酸乙酯、苄醇、丙酮、乙酸和碳酸丙烯酯的溶剂。
在其它实施方式中,所述有机溶剂包含作为溶剂的二氯甲烷以及作为共溶剂的甲醇、乙酸和H2O(参加实施例6)。在其它实施方式中,所述有机溶剂由二氯甲烷(仅)组成且包含作为共溶剂的甲醇、乙酸和H2O(参见实施例6)。
在有机溶剂包含作为溶剂的二氯甲烷以及作为共溶剂的甲醇、乙酸和H2O或者在有机溶剂由作为溶剂的二氯甲烷以及作为共溶剂的甲醇、乙酸和H2O组成的实施方式中,有机相包含占有机相总体积的约70%至约85%(v/v)的二氯甲烷、约10%至约15%(v/v)的甲醇、约5至约10%的乙酸(v/v)和约0.1%至约1.0%(v/v)的水。在其它此类实施方式中,有机相包含占有机相总体积的约75%至82%(v/v)的二氯甲烷、约11%至约14%(v/v)的甲醇、约6%至约8.5%的甲醇和约0.1%至约0.8%(v/v)的水。在此方面,应注意,在有机相仅含有作为溶剂/共溶剂的二氯甲烷、甲醇、乙酸和H2O的情况下,当然可以选择它们以上述范围给出的相应体积,使得总体积总共是100%(同样参见实施例6)。
在某个实施方式中,所述有机离子的终浓度范围为约0.1至1000mM。在具体实施方式中,它们以1至100mM的浓度溶解。可以调整每个具体有机离子试剂和生物活性剂的浓度,以实现期望的药物载量和包封效率。
在某个实施方式中,所述控制释放组合物选自但不限于微粒和纳米粒子。在具体实施方式中,所述微粒和纳米粒子是可生物降解的。
在另一个实施方式中,所述聚合物可以选自但不限于,聚丙交酯、聚乙交酯、聚(丙交酯-共-乙交酯)、聚(乳酸)、聚(乙醇酸)、聚(乳酸-共-乙醇酸)、聚己内酯、聚碳酸酯、聚酯酰胺、聚酸酐、聚(氨基酸)、聚原酸酯、聚缩醛、聚氰基丙烯酸酯、聚醚酯、聚二噁烷酮、聚亚烷基烷基酯、聚乙二醇和聚原酸酯的共聚物、可生物降解的聚氨酯、及其共混物和共聚物。
在某个实施方式中,所述乳化法选自水包油和油包水。
在具体实施方式中,可以用任何已知的乳化法实施本申请的方法。
在具体实施方式中,所述有机离子选自阴离子和阳离子物质。在一个实施方式中,所述有机离子是有机酸的盐。在具体实施方式中,所述有机离子选自三氟甲基对苯甲酸盐、胆酸盐、2-萘磺酸盐、2,3-萘二羧酸盐、1-羟基-2-萘甲酸盐、3-羟基-2-萘甲酸盐、2-萘甲酸盐和双水杨酸或硫酸盐、磺酸盐、磷酸盐和膦酸盐的有机衍生物。
在另一个实施方式中,降解包括酰化。在具体实施方式中,所述酰化反应涉及脂质运载蛋白突变蛋白的氨基(例如氨基酸侧链的N末端氨基或氨基)指向聚酯(例如聚(D,L-丙交酯-共-乙交酯))的羰基碳的亲核进攻。在所制备的组合物中,通过促进潜在亲核试剂(例如氨基)的质子化,由此使得亲核试剂较不易于参与和PLGA聚合物骨架或其片段的酰化反应,来防止或降低脂质运载蛋白突变蛋白的降解。
在另一个实施方式中,降解包括聚合物的分解。过度分解会导致聚合物分子量的损失和生物活性剂过早释放。
在另一个特定实施方式中,本申请提供控制释放组合物,所述控制释放组合物包括聚合物和与有机离子的复合物形式的脂质运载蛋白突变蛋白。当有机离子和脂质运载蛋白突变蛋白形成紧密的物理结合时,可以形成这样的复合物。
在另一个实施方式中,在不存在有机离子的情况下,可以相对于通过本申请方法制备的生物活性剂的含量,提高脂质运载蛋白突变蛋白的含量。
在另一方面,本申请涉及通过上述方法制备的控制释放组合物。
在又一方面,本申请包括根据本发明的缀合物、药物制剂或控制释放组合物用于控制递送脂质运载蛋白突变蛋白、延长脂质运载蛋白突变蛋白的体内半衰期、提高脂质运载蛋白突变蛋白的生物利用度、或降低脂质运载蛋白突变蛋白给予受试者后的免疫免疫原性的用途。
可以经由任何治疗上有效用于蛋白质类药物的肠胃外或非肠胃外(肠)途径给予根据本发明的脂质运载蛋白突变蛋白缀合物、药物制剂或控制释放组合物。肠胃外应用方法包括,例如皮内、皮下、肌内、气道内、鼻内、玻璃体内、关节内、颅骨内、脊椎内、肿瘤内或静脉内注射和输注技术,例如以注射液、输注液或酊剂的形式;以及气溶胶装置和吸入,例如以气溶胶混合物、喷雾或粉末的形式。例如J.S.Patton等人(The lungs as a portal of entry for systemic drug delivery.Proc.Amer.Thoracic Soc.2004Vol.1pages338-344)提供了关于肺部药物递送即经由吸入气溶胶(还可用于鼻内施用)或气道内滴注的综述。肺部是全身性药物递送的入口(例如Proc.Amer.Thoracic Soc.2004Vol.1pages 338-344)。非肠胃外递送模式例如口服,例如以丸剂、片剂、胶囊、溶液或悬浮液的形式,或者经直肠,例如以栓剂的形式。可以以含有常规非毒性的药学上可接受的赋形剂或载体、如期望的添加剂和溶媒的制剂全身性或局部给予本发明的脂质运载蛋白突变蛋白缀合物、药物制剂或控制释放组合物。
在本申请的一个实施方式中,所述脂质运载蛋白突变蛋白缀合物、药物制剂或控制释放组合物经肠胃外给予哺乳动物,特别是人。相应的给予方法包括但不限于,例如皮内、皮下、肌内、气道内或静脉内注射和输注技术,例如以注射液、输注液或酊剂的形式,以及气溶胶装置和吸入,例如以气溶胶混合物、喷雾或粉末的形式。在化合物的血清半衰期相对较短的情况下,静脉内和皮下输注和/或注射的组合可能是最方便的。药物组合物可以是水性溶液、水包油的乳液或油包水的乳液。
在此方面,应注意如Meidan VM和Michniak BB 2004Am.J.Ther.11(4):312-316中描述的经皮递送技术,例如离子渗透、超声渗透或微针促进递送,也可用于经皮递送本文描述的突变蛋白。非肠胃外递送模式是,例如口服,如以丸剂、片剂、胶囊、溶液或悬浮液的形式,或者直肠施用,例如以栓剂的形式。可以在含有多种常规的非毒性药学上可接受的赋形剂或载体、添加剂和溶媒的制剂中全身性或局部递送本发明的脂质运载蛋白突变蛋白缀合物、药物制剂或控制释放组合物。
给予的突变蛋白的剂量可在宽泛的限值内变化,以实现期望的预防作用或治疗反应。这将取决于例如化合物对于所选配体的亲和力以及突变蛋白和配体的复合物的体内半衰期。此外,理想的剂量将取决于突变蛋白或其融合蛋白或其缀合物的生物学分布、给予模式、要治疗疾病/障碍的严重性以及患者的健康状况。例如,当用于局部给予的膏剂时,可以使用高浓度的脂质运载蛋白突变蛋白。然而,如果需要,还可以在持续释放制剂中提供本发明的脂质运载蛋白突变蛋白缀合物,所述持续释放制剂例如脂质体分散体或水凝胶基聚合物微球,如PolyActiveTM或OctoDEXTM(参见Bos等人,Business Briefing:Pharmatech 2003:1-6)。可得的其它持续释放制剂例如PLA-PEG基水凝胶(Medincell)和PEA基聚合物(Medivas)。
因此,使用药学上可接受的成分以及已建立的制备方法,可以将本申请的突变蛋白配制成组合物(Gennaro,A.L.和Gennaro,A.R.(2000)Remington:The Science and Practice of Pharmacy,20th Ed.,Lippincott Williams&Wilkins,Philadelphia,PA)。为制备本发明的制剂或组合物,可以使用药学上惰性的无机或有机赋形剂。为制备例如丸剂、粉末、明胶胶囊或栓剂,可以使用例如乳糖、滑石、硬脂酸及其盐、脂肪、蜡、固体或液体多元醇、天然的和硬化油。用于制备溶液、悬浮液、乳液、气溶胶混合物或者在使用前重配成溶液或气溶胶混合物的粉末的适合的赋形剂包括水、醇、甘油、多元醇、及其适合的混合物以及植物油。
制剂或组合物还可含有添加剂,例如填料、粘合剂、润湿剂、助流剂、稳定剂、防腐剂、乳化剂以及其它溶剂或助溶剂。
可以通过多种方式对制剂进行灭菌,所述方式包括以细菌阻留滤器进行过滤或通过掺入无菌固体组合物形式的灭菌剂,所述无菌固体组合物可以在临使用之前溶解或分散于无菌水或其它无菌介质中。
本申请的另一个方面涉及治疗疾病或障碍的方法,所述方法给予有其需要的受试者上述本发明的脂质运载蛋白突变蛋白缀合物、药物制剂或控制释放组合物。
有此治疗需要的受试者可以是哺乳动物,示例性实例例如人、狗、小鼠、大鼠、猪、猿例如cymologous等等。
要根据本发明方法治疗的疾病和障碍的确切性质取决于所利用的突变蛋白预期要结合的配体。因此,本申请的突变蛋白可以用于治疗任何疾病,只要已知所述疾病或障碍发展中涉及的靶标分子可以显示为本申请的核酸文库的表达产物,或者显示为以其它方式获得的脂质运载蛋白的突变蛋白。
在一个具体实施方式中,用于本发明的缀合物、制剂和组合物的脂质运载蛋白突变蛋白是以高亲和力结合VEGF的人泪脂质运载蛋白突变蛋白。在另一个示例性实施方式中,所述脂质运载蛋白突变蛋白具有SEQ ID NO:22中所述的氨基酸序列。在一个实施方式中,该脂质运载蛋白突变蛋白缀合至亲水聚合物,例如聚乙二醇,优选大致20kDa的分子量,且任选与可生物降解聚合物一起配制,所述可生物降解的合物为聚(D,L-丙交酯-共-乙交酯)聚合物。在一个实施方式中,所述制剂为微粒或纳米粒子的形式。
该缀合物、制剂或组合物可用于疾病或障碍、例如VEGF相关的疾病或障碍的治疗方法中。在一个实施方式中,所述疾病或障碍,例如VEGF相关的疾病或障碍与血管化增加有关,例如癌症、哮喘、关节炎(包括骨关节炎和类风湿关节炎)、炎症、慢性阻塞性肺病(COPD)、肺动脉高血压、新生血管性湿性年龄相关性黄斑变性(neovascular wet age-related macular degeneration,AMD)、糖尿病性视网膜病变或黄斑水肿、早产儿视网膜病变或视网膜静脉阻塞。所述癌症可选自下述示例性实例:胃肠道癌、直肠癌、结肠癌、前列腺癌、卵巢癌、胰腺癌、乳癌、膀胱癌、肾癌、子宫内膜癌、和肺癌、白血病以及黑素瘤等。
附图简述
通过下述非限制性实施例和所附的附图进一步说明本发明,其中:
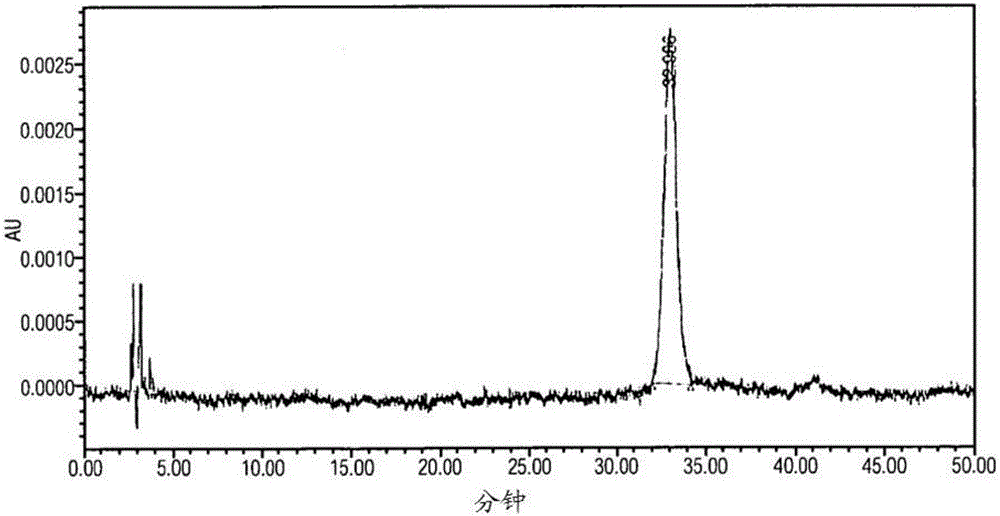
图1示出了对VEGF具有结合亲和力的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22)的溶液的RP-HPLC色谱图。
图2示出了对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG20)的RP-HPLC色谱图。
图3示出了对VEGF具有结合亲和力且缀合至分子量为40kDa的分支的PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG40)的RP-HPLC色谱图。
图4示出了对VEGF具有结合亲和力且缀合至分子量为5kDa的线性PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG05)的RP-HPLC色谱图。
图5示出了对VEGF具有结合亲和力且缀合至分子量为10kDa的线性PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG10)的RP-HPLC色谱图。
图6示出了对VEGF具有结合亲和力且缀合至分子量为5kDa的线性PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG5)的RP-HPLC色谱图,所述人泪脂质运载蛋白突变蛋白从微球颗粒中萃取。
图7示出了对VEGF具有结合亲和力且缀合至分子量为10kDa的线性PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG10)的RP-HPLC色谱图,所述人泪脂质运载蛋白突变蛋白从微球颗粒中萃取。
图8示出了对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG20)的RP-HPLC色谱图,所述人泪脂质运载蛋白突变蛋白从微球颗粒中提取。
图9示出了对VEGF具有结合亲和力且缀合至分子量为40kDa的分支的PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG5)的RP-HPLC色谱图,所述人泪脂质运载蛋白突变蛋白从微球颗粒中萃取。
图10示出了对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG20)的RP-HPLC色谱图,所述人泪脂质运载蛋白突变蛋白经24小时从微球颗粒中释放。
图11示出了对VEGF具有结合亲和力且缀合至分子量为40kDa的分支的PEG的人泪脂质运载蛋白突变蛋白(SEQ ID NO:22#2PEG40)的RP-HPLC色谱图,所述人泪脂质运载蛋白突变蛋白经24小时从微球粒子中释放。
图12示出了本发明的微球制剂体外经3周的时间段释放的脂质运载蛋白突变蛋白的绝对量。
图13示出了本发明的微球制剂体外经3周的时间段的脂质运载蛋白突变蛋白的每日释放。
图14示出了来自本发明的3个控制释放制剂(批次372-56、372-57和372-58)的活性VEGF结合的人泪脂质运载蛋白突变蛋白的经计算体外每日释放,所述VEGF结合的人泪脂质运载蛋白突变蛋白对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG(SEQ ID NO:22#2PEG20)。以深灰色条示出了从批次372-56的制剂释放的突变蛋白SEQ ID NO:22#2PEG20的活性脂质运载蛋白突变蛋白的量(以ng/天计),以中等灰色条示出了从批次372-57的制剂释放的活性脂质运载蛋白突变蛋白的量,以浅灰色条示出了从批次372-58的制剂释放的活性脂质运载蛋白突变蛋白的量。
图15示出了来自3个控制释放制剂(批次372-56、372-57和372-58)的活性VEGF结合的人泪脂质运载蛋白突变蛋白的累积体外释放,所述VEGF结合的人泪脂质运载蛋白突变蛋白对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG(SEQ ID NO:22#2PEG20)。以深灰色三角形示出了从批次372-56的制剂释放的突变蛋白SEQ ID NO:22#2PEG20的活性脂质运载蛋白突变蛋白的量(相比于突变蛋白的初始量),以中等灰色三角形示出了从批次372-57的制剂释放的活性脂质运载蛋白突变蛋白的量(以百分比计),以浅灰色三角形示出了从批次372-58的制剂释放的活性脂质运载蛋白突变蛋白的量(以百分比计)。
图16示出了来自3个控制释放制剂(批次372-56、372-57和372-58)的总人泪脂质运载蛋白突变蛋白的经计算体外每日释放,所述人泪脂质运载蛋白突变蛋白对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG(SEQ ID NO:22#2PEG20)。如图14所示,以深灰色条示出了从批次372-56的制剂释放的突变蛋白SEQ ID NO:22#2PEG20的活性脂质运载蛋白突变蛋白的量(以ng/天计),以中等灰色条示出了从批次372-57的制剂释放的活性脂质运载蛋白突变蛋白的量,以浅灰色条示出了从批次372-58的制剂释放的活性脂质运载蛋白突变蛋白的量。
图17示出了来自3个控制释放制剂(批次372-56、372-57和372-58)的总人泪脂质运载蛋白突变蛋白的累积体外释放,所述人泪脂质运载蛋白突变蛋白对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG(SEQ ID NO:22#2PEG20)。如图15所示,以深灰色三角形示出了从批次372-56的制剂释放的突变蛋白SEQ ID NO:22#2PEG20的活性脂质运载蛋白突变蛋白的量(相比于突变蛋白的初始量),以中等灰色三角形示出了从批次372-57的制剂释放的活性脂质运载蛋白突变蛋白的量(以百分比计),以浅灰色三角形示出了从批次372-58的制剂释放的活性脂质运载蛋白突变蛋白的量(以百分比计)。
图18示出了从控制释放制剂(批次372-56、372-57和372-58)释放的活性VEGF结合人泪脂质运载蛋白突变蛋白相比于经84天释放的总脂质运载蛋白突变蛋白的百分比,所述人泪脂质运载蛋白突变蛋白对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG(SEQ ID NO:22#2PEG20)。使用和图14和16相同的标记以示出从控制释放制剂(372-56、372-57和372-58)释放的脂质运载蛋白突变蛋白的量。
图19示出了从3个控制释放制剂(批次372-56、372-57和372-58)释放的总人泪脂质运载蛋白突变蛋白的血浆水平,所述人泪脂质运载蛋白突变蛋白对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG(SEQ ID NO:22#2PEG20)。使用和图14和16相同的标记以示出从控制释放制剂(372-56、372-57和372-58)释放的脂质运载蛋白突变蛋白的量。
实施例
包括下述实施例来说明本发明的具体实施方式。本领域技术人员应该理解,在下文实施例中公开的技术表示由本发明人发现的在实施本发明中良好应用的技术,并且因此可以被认为组成具体的方式以用于其实践。但是,见于本公开内容,本领域技术人员应该理解可以在公开的具体实施方式中作出很多变化,并且仍获得类似或相似的结果,为没有脱离本发明的精神和范围。
除非另有说明,使用已建立的重组基因技术方法,例如如Sambrook等人(上文)所述。
实施例1:人泪脂质运载蛋白的聚乙二醇化的突变蛋白的制备
将对人VEGF具有纳摩尔结合亲和力的SEQ ID NO:22的人泪脂质运载蛋白突变蛋白缀合至聚乙二醇聚合物(PEG)。
通过点突变引入未配对的半胱氨酸残基来代替SEQ ID NO:22突变蛋白的位置91的氨基酸Asp,以便提供用于偶合活化的PEG的反应性基团。然后如前所述在E.coli中生成携带自由Cys残基的重组突变蛋白(例如参见PCT公布WO 2008/015239)。
将三(2-羧乙基)盐酸膦(TCEP)用作还原剂,以获得SEQ ID NO:22的自由半胱氨酸91残基,以用于用PEG修饰。TCEP浓度是关键因素,因为TCEP过量可以导致PRS-055的分子间二硫键的还原。该经还原的SEQ ID NO:22突变蛋白的存在可以通过RP HPLC和通过离子交换HPLC进行检测。如果二硫键被打开,则半胱氨酸的所有巯基都可以被聚乙二醇化,从而导致期望的PEG-SEQ ID NO:22物质与误聚乙二醇化的和寡聚乙二醇化的SEQ ID NO:22突变蛋白的混合物。如果TCEP浓度过低,则SEQ ID NO:22突变蛋白的活化不完全,从而导致聚乙二醇化产量降低。通过混合含SEQ ID NO:22突变蛋白的溶液与TCEP溶液进行活化。在室温下通过轻柔混合约2至3h孵育活化批次。
通过混合活化批次与马来酰亚胺偶联的PEG(平均分子量2、5、10、20kDa,线性碳链;或平均分子量40kDa,分支的碳链(例如PEG40Sunbright GL2-400MA01;NOF,Singapore))而进行聚乙二醇化。在活化时间段最后结束时立即加入PEG。基于SEQ ID NO:22突变蛋白的量计算PEG的量。SEQ ID NO:22突变蛋白与PEG的靶标摩尔比为1:2。
聚乙二醇化之后,进行另一次离子交换色谱步骤(Macro cap)以改善聚乙二醇化的蛋白质的纯度,并且去除游离PEG和未反应的SEQ ID NO:22突变蛋白。
由此获得的产物称为SEQ ID NO:22#1(SEQ ID NO:22;未缀合的突变蛋白)、SEQ ID NO:22#002PEG05(缀合至分子量为5kDa的线性PEG的突变蛋白)、SEQ ID NO:22#002PEG 10(缀合至分子量为10kDa的线性PEG的突变蛋白)、SEQ ID NO:22#002PEG20(缀合至分子量为20kDa的线性PEG的突变蛋白)、和SEQ ID NO:22#002PEG40(缀合至分子量为40kDa的分支的PEG的突变蛋白)。
实施例2:SEQ ID NO:22突变蛋白及其PEG缀合物的冻干
在尝试将SEQ ID NO:22突变蛋白溶解于有机溶剂中之前,需要从蛋白质溶液中去除盐并且冻干该物质。选择透析作为去除盐的方法。在含有碳酸氢铵的挥发性缓冲液中透析蛋白质,以在冻干后产生相对不含盐的粉末。
用0.02%碳酸氢铵将已知体积的药物储液(300μL PRS-055,2mL SEQ ID NO:22#002PEG20,3mL SEQ ID NO:22#002PEG40)稀释为10mL的体积。将经稀释的溶液转移到透析管(SpectraPor 3500MWCO)中,然后在4℃下同时以105rpm搅拌针对3L体积的0.02%碳酸氢铵进行透析。排空0.02%碳酸氢铵体积,然后以3小时的间隔用新鲜缓冲液替换2次。在第3次替换后,使透析持续过夜。之后将溶液转移到50mL Falcon管中,然后置于–80℃冷藏室中。溶液冷冻后,将所述管冻干大致72小时。
在确认活性后,透析SEQ ID NO:22#002PEG20和SEQ ID NO:22#002PEG40的剩余药物溶液。用0.02%碳酸氢铵将大致5mL的SEQ ID NO:22#002PEG20和6.5mL的SEQ ID NO:22#002PEG40分别稀释成20mL和25mL的体积。将经稀释的溶液转移到透析管中,然后在4℃下同时以105rpm搅拌针对4L体积的0.02%碳酸氢铵进行透析。排空0.02%碳酸氢铵体积,然后以2小时的间隔用新鲜缓冲液替换2次。之后将溶液转移到50mL Falcon管中,然后置于–80℃冷藏室中。溶液冷冻后,将所述管冻干大致45小时。
用0.02%碳酸氢铵将大致25mL的SEQ ID NO:22#002PEG05和40mL的SEQ ID NO:22#002PEG10分别稀释成50mL和40mL的体积。将经稀释的溶液转移到透析管中,然后在4℃下同时以105rpm搅拌针对4L体积的0.02%碳酸氢铵进行透析。排空0.02%碳酸氢铵体积,然后以2小时的间隔用新鲜缓冲液替换2次。之后将溶液转移到50mL Falcon管中,然后置于–80℃冷藏室中。溶液冷冻后,将所述管冻干大致45小时。将SEQ ID NO:22#002PEG05和SEQ ID NO:22#002PEG10的一部分再次透析,将32.5mg SEQ ID NO:22-PEG05和31.6mg SEQ ID NO:22-PEG10分别溶解于大致37.5mL和35mL的0.02%碳酸氢铵中。将溶液和4L的0.02%碳酸氢铵放在透析管中,置于4℃下16小时,然后将碳酸氢铵替换为新鲜的。2小时后,在多个50mL falcon管中收集样品,冷冻,然后冻干大致48小时。
实施例3:SEQ ID NO:22突变蛋白及其PEG缀合物的溶解性
测定SEQ ID NO:22及其PEG缀合物在有机溶剂中的溶解性。精确称量少量SEQ ID NO:22或相应的缀合物,加入4或6mL玻璃管中。以小体积增量(微量移液器)加入所选的有机溶剂。旋转所述管,然后目测观察澄清度。加入其余体积的溶剂直到获得澄清的溶液。
实施例4:缀合物的RP-HPLC分析
在PBS中稀释三种不同化合物(SEQ ID NO:22、SEQ ID NO:22#002-PEG20、和SEQ ID NO:22#002-PEG40)的溶液,使得所述溶液的浓度为~0.1mg/mL。通过反相HPLC(RP-HPLC)分析这些溶液。在HP/Agilent系统上,使用Waters xBridge BEH300C183.5μm,(4.6x250mm)柱进行所述反相方法。使用梯度法,在30℃下以1ml/min(表1)开始。使用15μl注射体积。
表1:反相梯度法
表2具有得到的结果。RP-HPLC浓度基于作为标准物的突变蛋白SEQ ID NO:22,以测定脂质运载蛋白突变蛋白含量,分子量用于测定聚乙二醇化的类似物的浓度。每种化合物的注入溶液的色谱图示于图1、2和3。
表2:RP-HPLC测定的样品的浓度
在冻干和重悬于PBS后获得SEQID NO:22#002PEG05和SEQ ID NO:22#002PEG10的色谱图(图4和5)。
实施例5:SEQ ID NO:22及其PEG缀合物在有机溶剂中的溶解性
微球制剂的制备需要将药物和聚合物(PLGA)共溶于有机溶剂中。在二氯甲烷(DCM)中进行SEQ ID NO:22缀合物的初始测试。由于SEQ ID NO:22和缀合物在该溶剂中的溶解性很差,在二甲基亚砜(DMSO),DCM与苄醇(BnOH),DCM/乙酸(AcOH),DCM/甲醇(MeOH),乙酸乙酯(EtOAc),EtOAc/AcOH,和DCM、MeOH、AcOH、和H2O的溶剂体系中进行SEQ ID NO:22和缀合物的其它溶解性测试。
将PLGA聚合物加入到这些溶液中的一些中,以测定药物的溶解性在聚合物存在下是否会改变。加入聚合物使得聚乙二醇化的药物/聚合物的所得重量/重量比率是15/85。在一些情况下,聚合物的加入得到不透明溶液,因此加入更多溶剂直到混合物变得澄清。
SEQ ID NO:22#001在测试的所有溶剂中的溶解性都很差(<3mg/mL)(表3)。
表3:SEQ ID NO:22#001溶解性
SEQ ID NO:22#002PEG20和SEQ ID NO:22#002PEG40均在由DCM/MeOH/AcOH/H2O组成的溶剂体系中溶解性最高(表4和5)。
表4:SEQ ID NO:22#002PEG20溶解性
表5:SEQ ID NO:22#002PEG40溶解性
当将聚合物加入到SEQ ID NO:22#002PEG20溶液中时,所述溶解性保持大约相同。当将聚合物加入到SEQ ID NO:22#002PEG40溶液中时,所述溶液浑浊,不得不稀释几乎2倍以使药物变得可溶。所述缀合物在更简单的DCM/AcOH或DCM/MeOH的溶剂体系中的溶解性不比在DCM/MeOH/AcOH/H2O中好。
还可在含有DCM、MeOH、AcOH、和H2O的溶剂体系中测试SEQ ID NO:22#002PEG05和SEQ ID NO:22#002PEG10。在表6中比较了这些缀合物与更高分子量缀合物的溶解性。
表6:SEQ ID NO:22缀合物在79%DCM、12.8%MeOH、7.9%AcOH、0.3%diH2O和PLGA聚合物中的溶解性比较
SEQ ID NO:22#002-PEG05和SEQ ID NO:22#002PEG20缀合物在具有PLGA聚合物的该溶剂混合物中具有最高溶解性(基于蛋白质)。
实施例6:SEQ ID NO:22突变蛋白的缀合物与微球产生技术的相容性
为确定SEQ ID NO:22突变蛋白的缀合物是否与微球形成技术相容,通过微球方法制备样品。然后通过将缀合物从微球中萃取出来,或者通过将释放自微球的缀合物收集到PBS中,而制备用于活性分析的样品。
将每种SEQ ID NO:22突变蛋白的缀合物溶解于由79%DCM、12.8%MeOH、7.9%AcOH、0.3%H2O和聚合物组成的溶剂体系中。获得用于制备微球的澄清的匀质油相溶液。通过合并油相与由1%聚乙烯醇(PVA)和1%DCM组成的水相而制备乳液。在0.3%PVA中收集乳液,然后使DCM蒸发以便硬化微粒。然后通过过滤收集硬化的颗粒,用水洗涤然后冻干。
当颗粒干燥时,通过将微球样品溶解于乙腈(ACN)中然后用0.1%TFA(在水中)沉淀聚合物而测定药物含量。离心含有萃取的药物的溶液,然后通过RP-HPLC(图6-9)分析上清液。
SEQ ID NO:22#002PEG05和SEQ ID NO:22#002PEG10的色谱图具有宽偏移的保留时间,说明它们在某些方面已经改变。从微球中萃取的SEQ ID NO:22#002PEG20和SEQ ID NO:22#002PEG40的色谱图含有额外的峰,暂时将此归因于PEG结构的一个分支的部分损失(参见Guiotto、A.等人(2004)Bioorg.Med.Chem.12,5031-5037)。40kDa PEG缀合物的新峰的洗脱时间(31.8分钟)类似于完整的20kDa PEG SEQ ID NO:22的洗脱时间(例如32.8分钟),如果该额外的峰还具有连接的20kDa of PEG的话,这是所期待的。
不仅将从微球中萃取的缀合物样品暴露于用于生产微球的条件,还暴露于用于从微球中萃取蛋白质的严格条件。为获得已暴露于微球法的更具有代表性的样品,将微球样品在离心管中悬浮于1.5mL PBS中,并放在37℃孵育箱的架子上。24小时后,以3000rpm离心该离心管3分钟。通过RP-HPLC分析所得的含释放的缀合物的上清液(图10-11)。因为24小时时SEQ ID NO:22#002PEG05和SEQ ID NO:22#002PEG10不具有可检测水平的释放的药物,本文仅示出SEQ ID NO:22#002PEG20和SEQ ID NO:22#002PEG40的色谱图。
从微球中释放的聚乙二醇化的脂质运载蛋白突变蛋白的色谱图(图10和11)类似于萃取的蛋白质样品的色谱图(图8和9)。
测定了药物含量和药物从微球中的初始24小时释放。结果示于表7,为通过使用所有蛋白质相关峰的面积而进行的测定。
表7:药物含量和SEQ ID NO:22微球的初始释放
ND=未检测
实施例7:SEQ ID NO:22突变蛋白的缀合物在PLG微球中的包封
期望的产品情况是以1-5μg/天的范围持续释放SEQ ID NO:22至少3个月且通过不大于25号针递送、体积不超过100μL的可生物降解PLG微球悬浮液。
平均直径小于50μm的微球可通过25号针注射。
取决于注射介质的设计,微球悬浮于注射介质的显示可通过25号针注射的百分比范围为10%至30%。对于100μL注射使用1注射介质中20%微球的平均值,将递送20mg微球。
在微球中,包封是聚(丙交酯-共-乙交酯)聚合物的多达13.5重量%的SEQ ID NO:22-PEG20。假设注射20mg微球,则每次注射将施用2.7mg活性药物。将总质量除以3个月的天数(90天),可以持续30μg/天的最大递送速率。通过改变微球注射质量,可以降低该递送速率。结合当前参数,可以获得指定递送速率的持续时间。
实施例8:体外释放研究
施用300mg聚乙二醇化的SEQ ID NO:22(SEQ ID NO:22#002PEG20),制备1个批次的微球以进行体外释放研究。
评估了SEQ ID NO:22PEG20含量、粒径分析和残留溶剂(通过TGA)(表8)。
表8:所述批次的微球的总结数据
通过SEC和RP-HPLC二者评估含量。这些测量值在分析技术之间是一致的,并且测量值低于之前的观察。
通过检测活性药物在37℃下漏槽条件(sink condition)缓冲液中的量,进行SEQ ID NO:22-PEG20装载微球的体外释放溶解。简而言之,将大致10mg微球(记载准确质量)称量并加入14mL聚丙烯测试管中。将4mL体积磷酸盐缓冲盐水(PBS)(1x PBS,无菌过滤,HyClone)加入每个管中。代替离心,使用血清分离滤器。
将牢固封盖的体外释放组件水平放入37℃、120rpm运行的Fisher水填充摇床水浴中。在记录的间隔,使用16x 100mm血清分离滤器(Fisher,目录号02-681-52)从微球中分离上清液。使用转移移液器,去除接近4mL上清液,部分体积直接转移到HPLC玻璃管中。弃除过量上清液。离心HPLC管中收集的样品,直至48小时内通过HPLC进行分析。将4mL体积的新PBS直接加入到聚丙烯管中,然后将管再次牢固封盖。把管放回摇床水浴中。这些实验的结果示于图12和13。
初始突释显著大于之前的观察。7天后,该制剂持续释放7至14天,近似的质量释放速率为约5μg/天。释放百分比的范围为0.28%/天至0.42%/天。
释放从2至3周提高,其速率提高至10μg/天。随着聚合物降解而速率可能迅速提高的这一可能,能够由制剂方法解决,所述制剂方法例如包括酯端基聚合物与酸端基的混合物。
实施例9:37℃下在50%大鼠血浆中3个不同微粒制剂的体外释放
针对在设计的溶剂体系(描述于实施例6,有机相由79%DCM,12.8%MeOH,7.9%AcOH,0.3%H2O和聚合物组成,含水相即水相由1%聚乙烯醇(PVA)和1%DCM组成)中的高溶解性将蛋白质冻干循环进行优化后,使用各300mg聚乙二醇化的SEQ ID NO:22(SEQ ID NO:22#002PEG20),制备3个批次的微球,以进行体外和体内释放研究。
评估了SEQ ID NO:22PEG20含量、24小时突释、残余溶剂(通过TGA)、粒径和粒径分布(表9)。
表9:3个批次的微球的总结数据
来自3个不同颗粒批次(372-56、372-57和372-58)的微粒含有大致1.6mg的人泪脂质运载蛋白突变蛋白,所述人泪脂质运载蛋白突变蛋白对VEGF具有结合亲和力且缀合至分子量为20kDa的线性PEG(SEQ ID NO:22#2PEG20)(400μl,5%溶液、~8%载量),使用顶置式搅拌器,在40rpm下将所述微球在50%大鼠Li-肝素血浆中于37℃下孵育。通过定量ECL ELISA分析结合活性脂质运载蛋白突变蛋白经前84天释放到槽中的量。对于批次372-56和372-58,使用具有75摩尔%DL丙交酯单体和25mol%乙交酯且具有末端酸基团和0.45至0.55Dl/g固有粘度的聚(DL-丙交酯-乙交酯)聚合物。此外,对于批次#371-58,使用棕榈酸作为成孔剂。对于批次372-57,使用具有65摩尔%DL丙交酯单体和35mol%乙交酯且具有末端酸基团和0.4至0.55Dl/g固有粘度的聚(DL-丙交酯-乙交酯)聚合物(使用25型号Cannon-Fenske玻璃毛细管粘度计,在30℃下以CHCL3中0.5%w/v测量所述粘度)。
所有3个制剂都能观察到功能活性脂质运载蛋白突变蛋白的延长的释放。初始释放后为低平台期(28-56天),然后为向上的从63天开始的更高释放(图14和15)。扩散的控制初始释放相对较慢,并延长经过数天。邻苯二甲酸的加入确实将批次372-58的初始释放显著提高至~25%,推测是由于微粒的孔隙度提高。所有3个制剂的平台释放速率都相当类似。
为进行比较,还测定了总脂质运载蛋白突变蛋白的释放(图16和17)。比较活性脂质运载蛋白突变蛋白和总脂质运载蛋白突变蛋白的释放水平结果,经测定,40-65%的初始释放的脂质运载蛋白突变蛋白看起来在整个释放时间段中是有活性的(图17)。微球批次之一的活性和总脂质运载蛋白突变蛋白的比率最稳定(批次00372-58)。
实施例10:3个不同微粒制剂在单次皮下施用于雌性RNU大鼠的药代动力学
此实验的目的是,通过测量释放的脂质运载蛋白突变蛋白在4个月内的血浆水平而测定延长的体内释放曲线并且将其与体外释放相关联。
从经皮下注射给药150mg/kg微球溶液(5%,3ml/kg)的裸鼠获得2-78天血浆样品,通过定量ECL ELISA分析所述血浆样品,测量结合活性脂质运载蛋白突变蛋白。可以在血浆中检测到从皮下注射的微球释放的活性SEQ ID NO:22-PEG20,并且初始释放后为恒定的低平台期。直到43天的初始体内释放谱与体外观察到的谱都非常对应。
示于图19的此研究的结果显示,在43天后血浆水平远低于1ng/ml,这指示了体内释放可能以比体外释放更快的速率发生。在单独的研究中,以实验方法测定了SEQ ID NO:22-PEG20的皮下注射生物利用度为30%,清除率为18ml/h/kg。可以计算为消除速率的体内释放速率等于稳定状态的释放速率(输注)(Css=k0/Cl,k0为释放/输注的速率常数,Cl为清除率(Cl=18ml h-1kg-1),Css为稳定状态血浆浓度(在平台期,Css=0.015μg/ml))。因此,SEQ ID NO:22-PEG20微球缓释剂型(depot)的释放速率为,k0=Css*Cl=0.015μg/ml*18ml h-1kg-1=270ng h-1kg-1或k0=6.48μg/天*kg=1.3μg/天*大鼠。对于在200g大鼠施用3个月~2,4mg(150mg/kg,8%载量)缓释剂型,可以预测26μg/天的恒定释放速率,这是目前观察到的释放速率的10倍(还考虑到,脂质运载蛋白突变蛋白的总释放是活性浓度的~2倍)。
本文示例性描述的发明可以在不存在任何未在本文具体公开的元素、限制的情况下适当地进行实施。因此,例如术语“包含”、“包括”、“含有”等应该广泛理解而无限制。并且,本文采用的术语和表达方式用作描述的而非限制的术语,并且无意使用排除所示和所描述特征的任何等同方式或其部分的术语和表达方式,但是应理解,在本发明要求保护的范围内,多种修饰方式是可行的。因此,应该理解,接管本发明已经通过优选实施方式和任何特征予以具体公开,但是本领域技术人员可以获得本文在此公开的实施发明的修饰方式和改变,并且认为此类修饰方式和改变在本发明的范围内。本文已经广泛和通用地描述了本发明。落在通用公开内容之内的每个较窄种类和亚集也形成本发明的部分。这包括具有从该通类去除任何主题的负面限制的本发明的通用描述,无论该排除的物质是否在本文具体描述。并且,在本发明的特征或方面以马库什基团的方式进行描述的情况下,本领域技术人员将认识到,本发明还由此以任何单独的成员或马库什基团成员的子集进行了描述。从下述权利要求来看,本发明的其它实施方式将是显而易见的。
- 还没有人留言评论。精彩留言会获得点赞!