溶瘤痘苗病毒癌症治疗的制作方法
发明背景
1.发明领域
本发明涉及肿瘤学和病毒学领域。更具体地说,本发明涉及适于癌症治疗的痘病毒,尤其包含溶瘤痘苗病毒。
2.背景
正常组织的内环境稳定是一种细胞增殖和细胞死亡之间高度受控的过程。细胞增殖和细胞死亡之间失去平衡就会致癌(Solyanik等,1995;Stokke等,1997;Mumby和Walter,1991;Natoli等,1998;Magi-Galluzzi等,1998)。例如,宫颈癌、肾癌、肺癌、胰腺癌、结肠癌和脑癌只是可能发生的多种癌中的几种(Erlandsson,1998;Kolmel,1998;Mangray和King,1998;Mougin等,1998)。事实上,癌症的发生率相当高,仅在美国每年就有超过500,000的人死于癌症。
细胞增殖和细胞死亡的维持至少部分是由原癌基因和肿瘤抑制基因调节的。原癌基因或肿瘤抑制基因可编码诱导细胞增殖的蛋白质(如sis、erbB、src、ras和myc)、抑制细胞增殖的蛋白质(如Rb、p16、p19、p21、p53、NF1和WT1)或调节细胞调亡的蛋白质(如bcl-2)(Ochi等,1998;Johnson和Hamdy,1998;Liebermann等,1998)。但是,基因重排或这些原癌基因和肿瘤抑制基因的突变都会导致原癌基因转化为致癌的癌基因或使肿瘤抑制因子变成无活性的多肽。一般来说单一点突变就足以完成这种转化。例如,肿瘤抑制蛋白p53上的点突变就可以导致其完全失去野生型p53的功能(Vogelstein和Kinzler,1992)。
到目前为止,只有几种有效的方法用于治疗多种常见类型的癌。对于单个个体来说,选用何种治疗措施要根据诊断结果、疾病分期以及其他因素如年龄、性别及患者的健康状况。最常用的癌症治疗方法是手术、放疗和化疗。手术在肿瘤的诊断和治疗中扮演主要角色。通常活检和去除癌性生长物都需要通过手术方法。但是,如果肿瘤已经转移和扩散,则手术就不可能使患者痊愈,因此需要采用其他治疗措施。
放疗和化疗是手术治疗措施的最常见的替代方法(Mayer,1998;Ohara,1998;Ho等,1998)。放疗是指通过高能射线精确照射以摧毁癌细胞,与手术很相似,主要用于治疗未转移的局域化的癌细胞。放疗的副作用包括皮肤刺激、吞咽困难、口干、恶心、腹泻、脱发和无力(Curran,1998;Brizel,1998)。化疗是指利用抗癌药治疗癌症,是另一种癌症治疗模式,最常用的化疗方法包括多种抗癌药的联合化疗,这种方法被证实可以提高多种癌的反应率(美国专利5,824,348;美国专利5,633,016和美国专利5,798,339,本文已纳入作为参考)。然而,化疗药物的主要副作用是这些药物也可以影响正常组织细胞,最可能受影响的是那些在某些情况下快速分裂的细胞(如骨髓、胃肠道、生殖系统和毛囊)。化疗药物的其他毒性效应包括口疮、吞咽困难、口干、恶心、腹泻、呕吐、疲乏、出血、脱发和感染。
复制选择性的溶瘤病毒具有可用于癌症治疗的前景(Kirn等,2001)。这些病毒可通过直接的复制依赖性和/或病毒基因表达依赖性溶瘤效应引起肿瘤细胞的死亡(Kirn等,2001)。另外,该病毒还可以增强诱导宿主内细胞介导的抗肿瘤免疫反应(Todo等,2001;Sinkovics等,2000)。这些病毒经改造后还可以在肿瘤内表达治疗性目的基因以增强抗肿瘤效应(Hermiston,2000)。但是这种治疗方法还存在一些主要的限制。
因此,需要找到更多的癌症治疗措施。使用溶瘤病毒进行治疗是一个潜在的开发领域。
发明概述
本发明的实施方式涉及的方法包括给予胸腺嘧啶激酶缺陷的痘苗病毒。所述方法包括以升高的病毒浓度给予痘苗病毒。在某些方面,所述方法包括在具有肿瘤的对象中诱导溶瘤或肿瘤血管的崩解,包括给予所述对象至少1×108个足以诱导肿瘤中细胞溶瘤的TK缺陷型、表达GM-CSF、可复制的痘苗病毒载体的病毒颗粒。在本发明的另一方面,所述方法可排除用痘苗预处理对象,例如在进行本文所述疗法前1、2、3、4、5或更多天、周、月或年,对象无需接种疫苗。在一些方面,治疗性病毒能够感染非注射的肿瘤或癌,因此能够通过局部给予和全身散播对患者进行治疗。
在某些方面,向对象给予至少2×108、5×108、1×109 2×109、5×109、1×1010、5×1010、1×1011、5×1011、1×1012、5×1012或更多的病毒颗粒或噬斑形成单位(pfu),也包括其间的各种值和范围。给予的病毒剂量可以是0.1、1、2、3、4、5、6、7、8、9、10或更多毫升,包括其间的所有值和范围。在一个方面,所述剂量足以在患者血清中产生可检测的GM-CSF水平,如至少约、最多约或约5、10、40、50、100、200、500、1,000、5,000、10,000、15,000至20,000pg/mL,包括其间所有值和范围。考虑到,病毒的单剂量指在1、2、5、10、15、20或24小时期间对对象或肿瘤给予的量。该剂量可随时间散布于或通过多次注射散布。通常,对同一通用靶区域给予多个剂量,如在肿瘤附近,或当进行静脉内给药时,在对象血流或淋巴系统的特定进入点进行给药。在某些方面,病毒剂量由注射设备进行递送,该设备包括提供多端口的单个针头或与注射器连接的多个尖头或其组合。在另一方面,给予痘苗病毒载体2、3、4、5或更多次。在另一方面,在1、2、3、4、5、6、7或更多天或周中给予痘苗病毒。
在某些实施方式中所述对象是人。所述对象患有癌症和/或肿瘤。在某些实施方式中,所述肿瘤是治疗前不可切除的并在治疗后可切除的。在某些方面肿瘤位于肝脏上或肝脏中。在其它的方面,所述肿瘤可以是脑癌肿瘤、头颈癌肿瘤、食道癌肿瘤、皮肤癌肿瘤、肺癌肿瘤、胸腺癌肿瘤、胃癌肿瘤、结肠癌肿瘤、肝癌肿瘤、卵巢癌肿瘤、子宫癌肿瘤、膀胱癌肿瘤、睾丸癌肿瘤、直肠癌肿瘤、乳腺癌肿瘤或胰腺癌肿瘤。在其它实施方式中,所述肿瘤是膀胱肿瘤。在另一实施方式中,所述肿瘤是黑色素瘤。所述肿瘤可为复发的、原发的、转移的和/或多药耐药的肿瘤。在一些实施方式中,所述肿瘤是肝细胞肿瘤或源自其它组织或位置的转移性肿瘤。在一些实施例中,肿瘤位于肝脏中。
在一些方面,所述方法还包括给予对象第二种癌症疗法。第二种癌症疗法可为化疗、生物治疗、放疗、免疫治疗、激素治疗、抗血管治疗、冷冻治疗、毒素治疗和/或手术,包括其组合。在另一方面,化疗(药物)可为泰素(taxol)或索拉非尼(sorafenib)。在另一方面,手术包括经动脉栓塞化疗(TACE方案,参见Vogl等,European Radiology 16(6):1393,2005)。所述方法还可包括第二次给予痘苗病毒载体。本发明的方法还可包括在治疗之前、期间、之后或其组合评价肿瘤细胞活力。在某些实施方式中,通过血管内途径、瘤内途径或其组合给予病毒。在另一方面,向瘤块内注射给药。在另一实施方式中,向肿瘤血管或在肿瘤血管区域内注射给药。在另一实施方式中,对所述肿瘤局部的淋巴或血管内注射给药。在一些方面,所述方法包括在给药之前或期间进行肿瘤成像。在一些方面,用痘苗病毒疫苗预先免疫或不预先免疫患者。在另一方面,患者可以是天然或临床无免疫应答的。
在一些方面,给予病毒的量足以诱导所注射肿瘤内至少20%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%细胞的溶解。
在一些实施方式中,所述痘苗病毒包括一种或多种修饰的病毒基因。所述一种或多种修饰的病毒基因可包括以下一种或多种:(a)干扰素调节多肽;(b)补体控制多肽;(c)TNF或趋化因子调节多肽;(d)丝氨酸蛋白酶抑制剂;(e)IL-1β调节多肽;(f)非感染性EEV形式多肽;(g)抑制感染性病毒从细胞中释放的病毒多肽(抗感染性病毒形式多肽)或其组合。
本发明的实施方式靶向常见的关键癌症通路。靶向这些通路涉及调节不同的细胞机制(如细胞胸腺嘧啶激酶水平:E2F的响应;EGF-R通路的激活;免疫保护区(sanctuary)、抗病毒IFN反应(ras,p53));VEGF诱导的血管孔径大小:沉积IV),导致多种功效机制,如溶瘤性的:坏死、血管关闭、CTL攻击诱导、系统性的:IT、IV;肿瘤特异性CTL。
本发明的实施方式建立在I期临床试验的基础上,该试验展示了痘苗病毒用作癌症治疗的安全性和有效性。双周给药的剂量递增研究使用瘤内注射对7名预期生存期小于6个月的转移性黑色素瘤患者进行临床试验。该试验表明痘苗病毒安全、耐受性好,并且在5名患者中(71%)中引起了肿瘤反应,并且有两名患者无病长期生存。
I/II期试验的初步结果也表明JX-594的持续安全。在5-8天中观察到类似流感的症状。还观察到血小板(plt)、淋巴和绝对中性粒细胞计数(ANC)(通常为Gr1-2)的短时间降低。第8天有一例死亡,但检测得到与治疗无关。总体上,JX-591病毒血症耐受性好,给予后立即(15-30min)(反应):血液中最高3×108总基因组;以及复制峰(第5-8天):血液中最高1010总基因组。
本申请讨论了本发明的其它实施方式。任何关于本发明一个方面的实施方式也可应用于本发明的其它方面,反之亦然。实施例部分的实施方式可理解为适用于本发明所有方面的本发明的实施方式。
术语“抑制”、“减少”或“预防”或这些术语的其它变形,用于权利要求书和/或说明书中时,包括任何可检测的减少或完全抑制达到期望的结果。
在权利要求书和/或说明书中,术语“一个”与术语“包含”连用时意为“一个”(one),但也包含“一个或多个”、“至少一个”以及“一个或一个以上”的意思。
考虑到,本文讨论的任何实施方式可与本发明的任何方法或组合物一起实施,反之亦然。而且,本发明的组合物和试剂盒可用于实现本发明方法。
本申请中术语“约”表明包含测定某数值的设备或方法的标准差的数值。
权利要求书中使用的术语“或”用于表明“和/或”,除非明确表明仅仅指代候选物或候选物是互斥的,尽管本公开文本支持指唯一候选物和“和/或”的定义。
本说明书和权利要求书中所用术语“包含”、“具有”、“包括”或“含有”是包含性或开放式的,并不排除其它未叙述的元素或方法步骤。
本发明的其他目的、特征和优点通过下面的详细描述可以变得更加明晰。但是应当理解,阐述本发明具体实施方案的详述和具体实施例只是以说明的方式提供,因为本领域的技术人员根据这些详细描述可以很容易地想到包括在本发明的精神和范围内的各种变化和修改。
附图简述
附图构成本说明书的一部分,可用于进一步证明本发明的某些方面。通过参考这些附图中的一个或多个再结合具体实施方案的详细描述可以更好地理解本发明。
图1.瘤内注射JX-594治疗肝肿瘤的临床试验研究设计。
图2.瘤内注射JX-594治疗黑色素瘤的临床试验研究设计。
图3.具有多种新颖的癌症根除机制的靶向溶瘤病毒疗法。
图4.对转移性黑色素瘤进行JX-594I期临床试验后的长期无病生存者。患者1,上方,32岁女性:难治性:DITC,IL-2;注射肿瘤:CR;非注射转移灶:-皮肤:CR;乳腺:手术CR。存活,无病1.5+年。患者2,下方,75岁男性:多个转移位点(n=24);注射肿瘤:CR;非注射转移灶:-CR;存活,无病3+年。
图5.注射和非注射肿瘤中的JX-594临床I期试验反应。
图6. JX594IT-hep001-患者人口统计学和治疗状态-组1和组2。
图7. JX594IT-hep001-患者人口统计学和治疗状态-组3。
图8.在血流中静脉内散播JX-594:从肿瘤中早期流出量对应于剂量,至6小时大多被清除。
图9. 80%患者显示出JX-594复制的病毒血症。第1-7个周期血液中出现JX-594的第二个峰,(+)输入剂量清除后,(-)检测限以下的水平,方块=脱离研究的患者,(p)=未决数据。检测限=700个基因组/毫升。
图10. 80%患者显示JX-594复制的病毒血症。第1-7个周期,第3-22天血液中出现JX-594的第二个峰。
图11. JX-594复制相关性血管闭合的急性治疗诱导血管坏死(患者1,胃癌)。
图12. JX-594鳞状细胞癌对照(肺-组2)的长期稳定疾病。
图13.代谢(PET)反应:JX-594注射2个周期后注射JX-594的肿瘤黑色素瘤的反应(组3)。
图14.代谢(PET)反应:JX-594注射9个月后注射JX-594的肿瘤肝癌长期对照的反应(组2)。
图15.肿瘤标记物反应:血液标记物表明AFP快速肝癌破坏中减少99.9%。
图16. JX-594 10%的体重增加(6kg;14lb.)表明耐受性和疗效。
图17.全身性病毒血症和肿瘤反应:JX-594相关病毒血症,导致整体疗效(HCC-组2):AFP减少40%。
图18.将JX-594全身递送至肿瘤及其反应:肝脏met注射后,在远端非注射肿瘤中的疗效。2个周期后2个非注射肿瘤的PET代谢反应(患者304,群组3)。
图19.治疗、疗效和生存数据:CT和PET(检测的)长期存活者的肿瘤反应。
图20.试验图解。
图21A-21B.关键血液学检测和肝功能检测(误差棒:平均值的标准误差)。(A)ANC的增加与JX-594剂量以及hGM-CSF的表达增加相关。实心棒:ANC,空心棒:GM-CSF。X轴:患者识别号。(B)第1周期中,组3和组4患者的ALT水平。大多数患者ALT水平随时间未发生显著变化,也观察到轻微短暂的转氨酶升高(transaminitis)。
图22A-22C.血液学检测的变化。(A)剂量依赖的血小板减少,(B)血小板减少的程度与周期无关,(C)与后续周期相比,周期1中ANC、嗜酸性粒细胞和单核细胞的变化程度更为显著。白色棒:ANC,灰色棒:嗜酸性粒细胞,黑色棒:单核细胞。误差棒代表平均值的标准误差。
图23A-23F. JX-594的药代动力学、血源性传播以及对远端肿瘤的感染。(A)循环中急性基因组浓度。注射后15分钟即检测JX-594的基因组。组1-3中急性清除率一致。(B)显示了组1和3在周期1中的JX-594基因组浓度。JX-594基因组的浓度,包括第二病毒血症峰值的水平,与剂量相关。LOQ:定量限。误差棒代表平均值的标准误差。(C)周期1中代表性JX-594的基因组浓度。(D)黑色素瘤患者(组3)的JX-594回收和hGM-CSF表达。在循环和恶性体液中检测到高水平的JX-594基因组和GM-CSF(组3,黑色素瘤患者)。星号:未能检测到;PE:胸膜渗出液。(E)恶性渗出液中细胞表达lac-Z,表明感染性JX-594的存在。(F)非注射肝癌转移(颈部)的活检样品显示痘苗病毒B5R染色(箭头,棕色)。
图24A-24B.抗肿瘤效果。(A)非小细胞肺癌靶肿瘤的代表性CT扫描图和肿瘤测量值。圆圈:肿瘤。箭头:JX-594开始注射的时间。注意肿瘤截面积随时间的变化。(B)代表性的生理、CT和PET-CT扫描结果表明颈部转移瘤的目标肿瘤反应(4个周期后),在诱导高滴度JX-594中和抗体后注射。
发明详述
本发明涉及用溶瘤痘病毒治疗癌症。具体说是描述使用表达GM-CSF的痘苗病毒达到特定溶瘤程度。在另一实施方式中,可改造表达GM-CSF的痘病毒使其可以更有效地或更高效地杀伤癌细胞和/或对非癌细胞的毒性更低或损伤更小,具体是对基因产物进行突变或修饰,该改变可以使病毒更好地感染宿主、对宿主细胞毒性更小和/或更好感染癌细胞。一个具体的修饰是使病毒的胸腺嘧啶激酶(TK)功能缺陷。
I.痘病毒
对痘病毒已有数个世纪的了解,其特征是天花病毒(天花)产生的痘印,该病毒由此而得名。2000多年前天花可能首先出现在中国和远东。幸运的是,这种经常致命的病毒现在已经被根除,最后一次自然爆发发生在1977年的索马里。
痘病毒颗粒为卵圆形或砖块形状,长约200-400nm。外表面褶皱成平行排列,有时成螺旋状排列。这种颗粒相当复杂,含有超过100种的不同蛋白。胞外形式包含两层膜(EEV-胞外包膜病毒体),而胞内形式仅含有内膜(IMV-胞内成熟病毒体)。外表面由环绕核心的脂质和蛋白构成,核心由紧密压缩的核蛋白构成。作为抗原,痘病毒也很复杂,同时诱导特异性和交叉反应抗体。颗粒中至少出现10种酶,多数与核酸代谢/基因组复制有关。
痘病毒的基因组是130-300Kbp的线装双链DNA。基因组的末端具有含数个串联重复序列的末端发夹环。已测序数种痘病毒基因组,多数必需基因位于基因组中部,而非必需基因则位于末端。痘病毒基因组中有约250种基因。
复制发生于细胞质中,因为该病毒足够复杂已具有基因组复制的所有必需功能。有些细胞功能参与其中,但其参与的本质尚未可知。然而,尽管在除核细胞中可发生痘病毒基因表达和基因组复制,但其成熟被阻断,表明细胞具有某种作用。
痘病毒的受体一般未知,但可能有很多种类且因细胞类型而不同,对于牛痘,一种可能的受体是EGF受体(McFadden,2005)。其穿透也可能涉及不止一种机制。脱壳发生在两个阶段:(a)颗粒进入细胞和细胞质时去掉外膜,以及(b)颗粒进一步脱壳,核心进入细胞质。
一旦进入细胞质,与核心相关的病毒酶开始进行基因表达。表达分为两个时期:在基因组复制前表达的早期基因,占基因组约50%,以及在基因组复制后表达的晚期基因。活性依赖于DNA复制的晚期启动子进行表达的时间调控。基因组的复制被认为是自我启动的,引起高分子量串联子的形成,随后这些串联子被切割修复形成病毒基因组。病毒的组装发生于细胞骨架中,可能包括与细胞骨架蛋白(如肌动蛋白结合蛋白)的相互作用。包涵体产生于细胞质中,并最终成熟为病毒颗粒。细胞到细胞的传播为感染传播提供了另一机制。总体上,这种大的复杂病毒的复制很快,平均需要12小时。
至少有9种不同痘病毒能在人体中引起疾病,但对天花病毒和痘苗病毒最为了解。天花毒株分为重型天花(25-30%致死率)以及轻型天花(症状相同但死亡率小于1%)。两种病毒的自然感染都通过呼吸道进行,并且是全身性的,产生一系列的症状,但最值得注意的天花特征是皮肤的脓包和疤痕。
A.痘苗病毒
痘苗病毒是一种大的、包膜复杂的病毒,具有约190Kbp的线性双链DNA基因组,编码约250种基因。痘苗病毒被人熟知的角色是作为根除天花的疫苗。在根除天花后,科学家们寻求使用痘苗病毒作为将基因递送到生物学组织的工具(基因治疗和遗传学改造)。痘苗病毒仅在宿主细胞质中复制,这在DNA病毒中是独一无二的。因此,需要大的基因组编码病毒DNA复制所需的不同的酶和蛋白。复制期间,痘苗病毒产生外膜不同的数种感染形式:胞内成熟病毒体(IMV)、胞内包膜病毒体(IEV)、细胞相关包膜病毒体(CEV)以及胞外包膜病毒体(EEV)。IMV是最丰富的感染形式,并被认为负责宿主间传播。另一方面,CEV被认为参与细胞到细胞的传播,EEV则对于宿主有机体内长距离播散具有重要作用。
痘苗病毒编码数种使病毒具有干扰素抗性的蛋白。K3L是与eIF-2有同源性的蛋白质。K3L蛋白抑制干扰素激活剂PKR的作用。E3L是另一个抑制PKR激活的痘苗病毒蛋白,也能与双链RNA结合。
痘苗病毒与引起牛痘的病毒非常相近。痘苗病毒的准确来源未知,但最常见的观点是痘苗病毒、牛痘病毒和天花病毒(天花的病原体)均来源于共同的祖先病毒。也有推测认为痘苗病毒起先分离自马。痘苗病毒的感染是温和的,健康个体通常没有症状,但可引起轻微的皮疹和发热,死亡率极低。对痘苗病毒感染产生的免疫应答可以保护人体免遭致死性的天花感染。基于这个原因,使用痘苗病毒作为活病毒疫苗对抗天花。痘苗病毒疫苗不含天花病毒因此是安全的,但偶尔也会产生某些并发症和/或疫苗副作用,尤其当疫苗无免疫应答时。
如上文所讨论的,将痘苗病毒改造以表达数种外源蛋白。一种此类蛋白是粒细胞-巨噬细胞集落刺激因子,或GM-CSF。GM-CSF是一种由巨噬细胞分泌的蛋白,刺激干细胞产生粒细胞(中性粒细胞、嗜酸性粒细胞和嗜碱性粒细胞)和巨噬细胞。人GM-CSF在23位氨基酸残基(亮氨酸)、27位氨基酸残基(天冬氨酸)和39位氨基酸残基(谷氨酸)上糖基化(参见美国专利号5,073,627,通过引用纳入本文)。GM-CSF也称作莫拉司亭(molgramostim),或者当蛋白在酵母细胞中表达时,称作沙格司亭(sargramostim)(商品名),是一种化疗后用作刺激白细胞尤其是粒细胞和巨噬细胞产生的药物。曾经报道过表达GM-CSF的痘苗病毒。然而,它并非作为溶瘤剂递送,而仅仅是作为GM-CSF的递送载体。同样,其给予患者的剂量低于达到显著溶瘤效果的剂量。在本文的一些实施方式中,使用表达GM-CSF的痘苗病毒,其给药浓度大于1×108pfu或颗粒。
B.修饰的痘病毒
病毒经常被免疫调节分子如干扰素(-α、-β、-γ)和肿瘤坏死因子-α(TNF)灭活、抑制或清除(Moss,1996)。病毒感染后宿主组织和炎症/免疫细胞通常可分泌这些分子。
这些分子具有直接的抗病毒效应,和/或通过补充和/活化炎症细胞和淋巴细胞产生间接的效应。由于这些免疫清除机制的重要性,因此病毒经进化后可表达抑制这些细胞因子/趋化因子和干扰素诱导和/或功能的基因产物。例如,痘苗病毒(VV;以及其他一些痘病毒)可编码结合并抑制CC趋化因子(如RANTES,嗜酸细胞活化趋化因子,MIP-1-α)的分泌蛋白vCKBP(B29R)(Alcami等,1998)。某些VV病毒株还可以表达可结合并灭活TNF的分泌型病毒蛋白(如ListerA53R)(Alcami等,1999)。大多数痘病毒株具有编码结合并抑制干扰素α/β(如B18R)或干扰素(B8R)的分泌蛋白的基因。vC12L是一种IL-18结合蛋白,可抑制IL-18诱导IFN-γ分泌和NK细胞/细胞毒T细胞活化。
大多数痘病毒的毒力研究都是在小鼠体内进行的。这些蛋白中的许多,但不是全部(如B18R在小鼠体内是无活性的)在小鼠体内是有活性的。当这些蛋白具有抗鼠靶细胞因子的活性时,这些基因的缺失将导致其毒力减弱、安全性提高,产生出这些基因缺失或功能突变的VV突变株。另外,对这些突变株的炎症反应/免疫反应及对这些突变株的清除通常要比表达抑制蛋白的亲本病毒株高。例如,痘病毒分泌蛋白的T1/35kDa家族(趋化因子结合/抑制蛋白)的缺失可导致侵润到病毒感染组织内的白细胞显著增加(Graham等,1997)。VV内vC12L基因的缺失可导致病毒在接种到小鼠鼻内后的滴度/毒性下降;另外,NK细胞和细胞毒T淋巴细胞的活性随IFN-γ的诱导而升高(Smith等,2000)。
粘液瘤病毒T7基因(可结合IFN-γ和多种趋化因子)的缺失可导致病毒毒力下降,毒性模型内的组织炎症/侵润显著增高(Upton等,1992;Mossman等,1996)。粘液瘤病毒M-T2基因的缺失也可以导致病毒在兔模型内的毒力下降(Upton等,1991)。B18R抗干扰素α/-β基因产物的缺失也可以导致病毒对IFN介导的清除的敏感性升高,在正常组织内的滴度降低、毒力下降(Symons等,1995;Colamonici等,1995;Alcami等,2000)。总之,这些病毒基因产物具有降低抗病毒免疫反应和减少炎症细胞侵润到病毒感染组织内的功能。通过缺失/突变使蛋白功能丧失可导致病毒在宿主组织内的毒力降低和/或原炎症特性升高。
细胞因子和趋化因子具有很强的抗肿瘤活性效应(Vicari等,2002;Homey等,2002)。这些效应可直接作用于肿瘤细胞(如TNF),或者通过对非肿瘤细胞的作用间接发挥效应。后者的例子是TNF,它是通过对肿瘤相关血管的毒性而发挥抗肿瘤活性的;这样可导致肿瘤失去血液供应而发生坏死。另外,趋化因子可补充(在某些情况下可活化)免疫效应细胞,如中性粒细胞、嗜酸性粒细胞、巨噬细胞和/或淋巴细胞。这些免疫效应细胞可通过多种机制杀伤肿瘤。这些机制包括表达抗肿瘤的细胞因子(如TNF),表达fas配基,表达穿孔素和粒酶,补充自然杀伤细胞等。炎症反应最终可诱导出全身的肿瘤特异性免疫反应。最后,许多这些细胞因子(如TNF)或趋化因子可协同化疗或放疗措施来破坏肿瘤。
通过全身给予重组的免疫刺激蛋白产生临床疗效是不可行的,因为(1)全身给药会产生严重的毒性反应以及(2)刺激局部侵润和抗肿瘤效应需要在肿瘤组织内的局部表达。我们需要的是找到方法使这些分子在肿瘤块内达到局部高浓度,而循环中的浓度达到最低。病毒可以经改造后使其表达细胞因子或趋化因子基因以增强其效应。这些基因从复制选择性载体中的表达与从非复制选择性载体中的表达相比具有潜在的优势。从复制病毒中的表达可使其在肿瘤块内达到局部较高的浓度;另外,复制病毒有助于通过肿瘤细胞的破坏/溶解及在原炎症环境中释放肿瘤抗原来诱导抗肿瘤免疫反应。但是这种方法也有几个不足。如果病毒在局部高浓度表达,携带毒性基因的复制完整型病毒(即使是肿瘤特异性的)就有可能释放到环境中,从而引起人们对其安全性的严重关注。因而,基因组内表达有强原炎症基因的病毒可能会对患者和公众产生安全性风险。另外,病毒大小限制了利用病毒如腺病毒来表达多个基因和/或大基因;这些分子在联合使用时的效率肯定更高。最后,现在使用的许多溶瘤病毒表达抗炎症蛋白,因此这些病毒将会在其感染的肿瘤组织内诱导出炎症环境。结果会抑制抗肿瘤免疫反应、抗血管效应和化疗/放疗增敏性的诱导。
C修饰的痘苗病毒
1.干扰素调节多肽
干扰素α/-β可通过几种机制阻断病毒的复制。干扰素-γ直接的病毒抑制效应较弱,但是它可以通过几种机制诱导细胞介导的免疫反应。病毒经进化后可表达中和干扰素抗病毒效应的分泌型基因产物。例如,痘苗病毒(及其他痘病毒)编码可分别结合干扰素-γ和-α/-β的B8R和B18R(Smith等,1997;Symons等,1995;Alcami等,2000)。另外的一种可抑制干扰素产生的牛痘苗病毒基因产物是caspase-1抑制因子B13R,它可以抑制干扰素-γ-诱导因子IL-18的活化。干扰素调节蛋白包括而不限于B18R,在其他的病毒株如痘苗病毒的Copenhagen株中被称为B19R;B8R;B13R;vC12L;A53R;E3L以及其他具有相似活性或特性的病毒多肽。IFN调节多肽还可以非排他性地分为优先调节IFNα和/或β通路的一类(如B18R、B8R、B13R或vC12L)和调节IFNγ通路的一类(例如的B8R、B13R或vC12L)。
癌细胞通常对干扰素耐受。多种机制参与其中。这些机制包括ras转导通路的活化(如ras突变,上游生长因子受体过表达/突变等),这是癌细胞的一个常见特征,会导致PKR抑制。另外,肿瘤组织内的淋巴细胞通常通过各种机制被抑制,如肿瘤细胞产生IL-10和fas-L。由于淋巴细胞是干扰素-γ产生的主要来源,因此淋巴细胞的抑制会导致肿瘤内干扰素-γ的分泌减少。因而干扰素无法对肿瘤组织产生效应。另外,干扰素自身具有抗肿瘤效应。例如,IFN-γ可增强MHC I类分子相关抗原的提呈;这将使CTL可以更有效地杀伤肿瘤细胞。例如,IFN-α/β可阻止肿瘤组织内的血管形成,从而抑制肿瘤的生长。
2.补体控制多肽
病毒克隆被清除的主要机制是通过补体依赖的机制杀伤宿主内被感染的细胞或肌体内的病毒颗粒。被感染的细胞死亡后就不可能再产生有感染性的病毒了。另外,在调亡过程中细胞内可释放降解DNA的酶。这些酶可使病毒DNA降解,使病毒灭活。调亡可被各种机制诱导,其中包括活化的补体与补体膜攻击复合物的结合。痘病毒如痘苗病毒经进化后可表达基因产物抑制补体介导的病毒和/病毒感染细胞的清除。因此这些基因可阻止调亡的发生,抑制通过补体依赖的机制清除病毒,因此病毒感染得以进行,病毒的毒力升高。例如,痘苗病毒补体控制蛋白(VCP;如C21L)可阻止补体介导的细胞杀伤和/或病毒灭活(Isaacs等,1992)。
VCP还具有抗炎症效应,因为它的表达可使侵润到病毒感染组织内的白细胞减少。补体控制多肽包括而不限于VCP,也被称为C3L或C21L。
癌细胞通常过表达细胞抗补体蛋白;这将使癌细胞在受补体攻击时得以存活。因此,那些由于其自身对补体介导的杀伤具有耐受性的优先作用于肿瘤细胞的药物对各种人源肿瘤具有选择性和较高的活性(Durrant等,2001)。另外,癌细胞的标志之一就是失去了正常的调亡机制(Gross等,1999)。凋亡抗性促进癌发生以及对抗肿瘤剂的抗性,这些抗肿瘤剂包括免疫试剂、化疗剂和放疗剂(Eliopoulos等,1995)。促凋亡分子(如bax)功能的丧失、抗凋亡分子(如bcl-2)水平/功能的增加和最后补体敏感性的丧失,均可介导凋亡的抑制。
3. TNF调节多肽
在各种病毒颗粒清除机制中有一种机制就是通过诱导如上所述的调亡杀伤宿主内被感染的细胞。调亡可由各种机制所介导,其中包括TNF和淋巴毒素α(LTα)与细胞TNF受体的结合,这种结合可激发细胞内的信号级联反应。TNF受体被活化后可参与免疫反应和炎症反应的调节以及诱导细胞调亡(Wallach等,1999)。
痘病毒的各种病毒株,包括某些痘苗病毒株,经进化后可表达基因产物抑制TNF介导的病毒和/和病毒感染细胞的清除。这些基因所编码的蛋白质通过结合及隔绝细胞内的TNF来抑制炎症反应和TNF的调亡诱导活性,结果导致病毒的清除被抑制。由于病毒不能被清除,因此病毒的感染得以继续,病毒的毒力增强。许多痘病毒科成员都可以表达分泌型病毒TNF受体(vTNFR)。例如,有几种痘病毒可编码vTNFRs,如粘液瘤病毒(T2蛋白)、牛痘病毒株和痘苗病毒株,如Lister可编码CrmB、CrmC(A53R)、CrmD、CrmE、B28R蛋白和/或其等价物。
这些vTNFRs具有抑制TNF介导的细胞杀伤和/或病毒灭活的功能(Saraiva和Alcami,2001)。TNF调节蛋白包括而不限于A53R、B28R(这个蛋白是存在的,但是在痘苗病毒的Copenhagen株中可能是无活性的)以及具有相似活性或特性的其他多肽。
癌细胞的一个标志是异常的基因表达,这可导致其对多种生长调节分子机制的敏感性丧失,如对TNF抗肿瘤活性的敏感性。因此,对于病毒在肿瘤微环境内的传播来说病毒免疫调节机制是不需要的。
4.丝氨酸蛋白酶抑制剂
病毒颗粒被清除的一个主要机制是诱导宿主内被感染的细胞调亡。感染细胞死亡后就无法继续产生有感染性的病毒了。另外,在调亡过程中细胞可以释放出降解DNA的酶。这些酶可降解病毒DNA使病毒灭活。各种机制都可以诱导调亡,其中包括细胞因子(如肿瘤坏死因子)的结合、细胞毒T细胞或fas配基结合而导致的粒酶表达;级联活化是常见的调亡通路的关键部分。病毒经进化后可表达基因产物抑制细胞内由某些分子所诱导的信号级联反应,这些分子包括fas配基或肿瘤坏死因子(TNF)/TNF相关分子(如腺病毒的E3 10.4/14.5、14.7基因(Wold等,1994);腺病毒的E1B-19kD(Boyd等,1994);牛痘病毒的crmA;痘苗病毒的B13R)(Dobbelstein等,1996;Kettle等,1997))。这些基因产物可抑制调亡诱导分子所诱导的调亡,因此即使存在抗病毒调亡诱导细胞因子、fas、粒酶或其他调亡刺激因子,病毒的复制也可以进行。
VV SPI-2/B13R与牛痘病毒CrmA高度同源;SPI-1(VV)与CrmA的同源性较低(Dobbelstein等,1996)。这些蛋白是serpins(丝氨酸蛋白酶抑制剂),CrmA和SPI-2都具有阻止各种形式的调亡的功能。
例如对白细胞介素-1β-转化酶(ICE)和粒酶的抑制可阻止感染细胞的调亡。这些蛋白可抑制IL-18的活化进而降低IL-18所诱导的IFN-γ释放。因而IFN-γ对细胞介导的免疫反应的免疫刺激效应被抑制(Kettle等,1997)。SPIs包括而不限于B13R、B22R以及其他具有相似活性或特性的多肽。
癌细胞的标志之一就是失去了正常的调亡机制(Gross等,1999)。对调亡的耐受促使其形成肿瘤并且对抗肿瘤药物,如免疫治疗药物、化疗药物和放疗耐受(Eliopoulos等,1995)。调亡抑制可由前调亡分子(如bax)功能的丢失、抗调亡分子(如bcl-2)表达水平的增加/功能的增强所介导。
5. IL-1β-调节多肽
IL-1β是一种具有局部和全身生物学活性的因子。已有的研究结果表明IL-1β和IL-1α之间的功能差异只有几种。IL-1β的许多生物学活性是用描述IL-1的许多不同缩写来描述的。除了在猪细胞内无活性外IL-1无种属特异性。IL-1的某些生物学活性是通过诱导其他介质的合成来间接介导的,其中包括ACTH(促肾上腺皮质激素)、PGE2(前列腺素E2)、PF4(血小板因子-4)、CSF(集落刺激因子)、IL-6和IL-8。
IL-1的合成可被其他细胞因子所诱导,其中包括TNF-α、IFN-α、IFN-β和IFN-γ以及细菌内毒素、病毒、丝裂原和抗原。IL-1的主要生物学活性是刺激T辅助细胞,该细胞被诱导后可分泌IL-2并表达IL-2受体。病毒感染巨噬细胞后可产生大量的IL-1抑制因子,这些抑制因子支持T细胞成熟缺陷患者体内细胞的机会感染和转化。IL-1可直接作用于B细胞,促使其增殖并合成免疫球蛋白。IL-1也可以作为启动因子使B细胞对IL-5发生反应。IL-1可刺激NK细胞、成纤维细胞、胸腺细胞、胶质母细胞的增殖和活化。
病毒蛋白阻断IL-1β的合成是病毒抑制或消除IL-1激发的系统抗病毒反应的一种策略。研究发现可有效阻断IL-1功能的结合蛋白也可由牛痘病毒的基因编码,这些蛋白的活性与B15R相似。痘苗病毒还可以编码被称为B8R的另外一种蛋白,该蛋白的功能类似于细胞因子的受体(Alcami和Smith,1992;Spriggs等,1992)。IL-1调节多肽包括而不限于B13R、B15R以及其他具有相似活性或特性的多肽。
癌细胞的一个标志是异常的基因表达,这可导致其对多种生长调节分子机制的敏感性丧失,如对IL-1抗肿瘤活性的敏感性。因此,对于病毒在肿瘤微环境内的传播来说病毒免疫调节机制是不需要的。
6. EEV形式
病毒传播到转移瘤部位、甚至固体瘤内的效率一般不高(Heise等,1999)。静脉内给予的病毒通常会被抗体(如腺病毒)(Kay等,1997)和/或补体系统(如HSV)(Ikeda等,1999)清除或灭活。除了这些免疫反应介导的机制之外,这些病毒的生物分布也会导致大多数病毒沉淀在正常组织而不是肿瘤组织内。例如,静脉内给予腺病毒主要集中在肝脏和脾脏内;只有低于0.1%的病毒可以进入肿瘤内,即使是免疫缺陷小鼠(Heise等,1999)。因此,虽然在免疫缺陷小鼠肿瘤模型内用极高剂量的病毒可以达到适度的抗肿瘤效果,但是静脉内递送的效率极低,并且对功效有严重影响。
痘苗病毒可在固体瘤内复制并引起肿瘤的坏死。另外,胸苷激酶缺失突变可使病毒具有感染肿瘤块和卵巢组织的能力,并且在小鼠肿瘤模型系统内可很好地表达标记基因(Gnant等,1999)。
但是,由于这些研究一般都是基于5天后标记基因表达的情况来确定肿瘤的变化,因此还不清楚病毒是否可以在肿瘤/卵巢组织内良好地沉淀、表达基因和复制(Puhlmann等,2000)。无论通过何种机制,不含其他目的基因的病毒所得到的抗肿瘤效果都是无统计学意义的(Gnant等,1999)。相反,瘤内注射病毒可以得到显著的抗肿瘤效果(McCart等,2000)。因此,如果提高静脉内递送到肿瘤内的病毒量则可以改善静脉内给予的效果。
痘苗病毒在细胞内的复制可产生胞内病毒(IMV,胞内成熟病毒;IEV,胞内包装病毒)和胞外病毒(REV,胞外包装病毒;CEV,细胞相关的胞外病毒)(Smith等,1998)。野生型痘苗病毒株复制后所产生的病毒中有约99%的是IMV。该病毒形式在环境中相对稳定,因此是个体之间传播的主要形式;但是,该形式的病毒在感染宿主内无法有效传播,因为病毒无法从细胞内有效释放以及对补体和/或抗体中和反应的敏感性。与此相反的是,EEV可释放到细胞外基质中,这种形式的病毒一般只占病毒产量的约1%(Smith等,1998)。EEV负责病毒在感染宿主内的传播,在宿主外则容易被降解。更重要的是,EEV进化出了几种机制来避免其在血液内被中和。首先,EEV对补体相对耐受(Vanderplasschen等,1998);这一特性归功于宿主细胞将补体抑制剂插入到外膜包被内以及痘苗病毒可将补体控制蛋白(VCP)分泌到细胞外环境中。其次,EEV与IMV相比对中和抗体的耐受性相对要高(Smith等,1997)。EEV还可以在感染后比IMV更早(如4-6小时)释放(IMV只在细胞死亡过程中或死亡后释放),因此EEV形式的病毒传播更快(Blasco等,1993)。
但是,不幸的是野生型痘苗病毒只能产生极少量的EEV。另外,用痘苗病毒治疗(即病毒的输入剂量)到目前为止只限于细胞内病毒。常规的痘苗病毒(VV)制备和纯化方法都可以导致EEV被灭活(Smith等,1998),病毒的制备一般用非人源细胞系;非人源细胞来源的EEV不能耐受补体介导的清除作用(EEV所产生的补体抑制蛋白具有种属特异性)。因此,痘苗病毒的疗效受IMV形式的病毒对中和反应的相对敏感性及病毒在实体瘤组织内的传播效率低下的限制;这种传播一般是指从一个细胞到临近的细胞内。IMF通过血流或淋巴系统传播到远端肿瘤组织内的效率也不高。
因此,EEV形式的痘苗病毒天然具有优于目前所用形式(IMV)的痘苗病毒的特征;EEV适于在实体瘤的局部及其临近或远端肿瘤位点内快速而有效地传播。由于EEV对补体效应相对耐受,因此当EEV从同一物种的细胞内制备时,通过血管内给予后该病毒形式在血液中的稳定性要比标准方法制备的痘苗病毒(只含IMV)高、并且维持活性的时间要更长(Smith等,1998)。由于EEV对抗体介导的中和反应耐受,因此该病毒形式通过血管内给予后在血液中维持活性的时间要比标准方法制备的痘苗病毒(只含IMV)长(Vanderplasschen等,1998)。这个特性对于在中和抗体升高的情况下需要重复给予时特别重要;所有被认可的抗癌疗法都需要重复给予。因此,痘苗病毒及其他痘病毒的EEV形式能高效地将治疗性病毒及其携带的基因通过血流转移到肿瘤内,与标准的痘病毒制备物相比其系统效应会提高。最后,病毒在大众中传播的危险会显著降低,因为EEV在体外是很不稳定的。
参与病毒EEV形式调节的多肽包括而不限于A34R、B5R以及可影响痘病毒EEV形式产生的其他各种蛋白。A34R上的密码子151突变可使赖氨酸残基变成天冬氨酸残基(K151D),这样A34R蛋白就不能将EEV形式的病毒定位到细胞膜上。B5R是一种可结合补体的EEV膜结合多肽。A34R的完全缺失可导致EEV的释放增加,但是病毒的感染性会大大降低,而K151D突变可增加EEV的释放,同时也能保持释放病毒的感染活性。B5R的序列与VCP(抗补体)同源,但是还未发现它具有补体抑制活性。
一种鉴定增强型EEV形式的方法简述如下。EEV稀释到冷MEM中,与含活性血清或热灭活血清(56℃,30min,对照)的冷MEM混合(1:1)(血清的最终稀释度为1/10、1/20或1/30)。在7℃孵育75分钟后,样品在冰上冷却,将mAb5B4/2F2加到新鲜的EEV样品中中和所有的污染物(IMV和破裂的EEV)。然后使病毒颗粒与RK13细胞在冰上结合1小时,将补体和未结合的病毒颗粒洗去,2天后计数形成的噬菌斑。噬菌斑的数量越多说明对补体的耐受性约高(Vanderplasschen等,1998,通过引用纳入本文)。描述分离EEV形式的痘苗病毒的示例性方法参见Blasco等,1992(本文已纳入作为参考)。
7.其他多肽
其他病毒免疫调节多肽包括可结合免疫反应的其他介质的多肽和/或调节免疫反应相关分子通路的多肽。例如,趋化因子结合多肽如B29R(在痘苗病毒的Copenhagen株中虽然存在该蛋白,但是无活性)、C23L、vCKBP、A41L以及具有相似活性或特性的多肽。其他痘苗病毒蛋白,如痘苗病毒生长因子(如C11L)是一种病毒EGF样生长因子,在本发明的某些实施方案中也是修饰的靶位。可被称为病毒免疫调节因子的其他多肽包括而不限于B7R、N1L或具有提高痘病毒毒力活性或特征的其他多肽。
8.痘苗病毒诱导的细胞融合
在本发明的某些实施方案中,A56R或K2L编码核酸的修饰、缺失或突变可导致细胞与细胞的融合或VV感染所诱导的合胞体(syncitia)形成。痘苗病毒诱导的细胞融合常常会增强VV的抗肿瘤效应,因为VV可在肿瘤内传播。由于细胞融合而导致的肿瘤内病毒传播通常可使病毒逃过中和抗体和免疫反应的攻击。这些基因中的一个或两个都发生突变的VV可以更有效地杀伤和感染临近的未感染细胞(即“旁观者效应”),这样就可以提高病毒的局部抗肿瘤效应。
D.其他痘病毒
痘苗病毒是痘病毒科、脊椎动物痘病毒、正痘病毒属的一员。正痘病毒属与脊椎动物痘病毒亚科的其他属相比其成员具有更高的同源性,正痘病毒属包括11个不同但是又密切相关的种,其中有牛痘病毒、天花病毒(天花致病原)、牛痘病毒、水牛痘病毒、猴痘病毒、鼠痘病毒和马痘病毒以及其他病毒(见Moss,1996)。如本文所描述,本发明的某些实施方案可以延伸到正痘病毒属以及副痘病毒属、禽痘病毒属、羊痘病毒属、兔痘病毒属、猪痘病毒属软疣痘病毒属和亚塔痘病毒属的其他成员。痘病毒家族的一个属通常用血清学方式来定义,其中包括在试验动物体内的中和反应和交叉反应。正痘病毒属的各个成员及Chordovirinae亚科的其他成员利用免疫调节分子,如本文所描述的那些调节分子来抑制宿主的免疫反应。因此,本文所描述的发明并不限于痘苗病毒,而是适用于多种病毒。
E.病毒制备
痘苗病毒可用Earl和Moss描述的方法制备,参见Ausubel等,1994,通过引用纳入本文。
II.蛋白和核酸组合物
本发明涉及痘病毒,包括那些经改造后与野生型相比含有一个或多个突变的痘病毒,这样的病毒具有可用于治疗癌细胞的优良特性,并且对非癌细胞来说是低毒的或无毒的。下文以实施例的方式描述了实现本发明方法和组合物的各种方法,如利用重组DNA技术制备突变病毒的方法。
在某些实施方案中,本发明涉及制备缺乏一种或多种功能性多肽或蛋白的方法和/或制备能更好释放的特殊形式病毒,如具有感染性的EEV形式病毒的方法。在其他的实施方案中,本发明涉及痘病毒以及和作为药用制剂一部分的蛋白质性组合物联合使用。
本文所用的术语“蛋白质”或“多肽”是指至少含一个氨基酸残基的分子。在某些实施方案中使用的是野生型的蛋白质或多肽,而在本发明的多数实施方案中,病毒蛋白或多肽是缺失的或者是经过修饰的,这样就可以使病毒更适用于治疗癌细胞或癌症患者。上述术语在本文中可以交互使用。“修饰蛋白”或“修饰多肽”是指化学结构相对于野生型蛋白或多肽发生改变的蛋白或多肽。在某些实施方案中,修饰蛋白或多肽至少具有一个改变的活性或功能(假设蛋白或多肽有多种活性或功能)。相对于野生型蛋白或多肽的活性或功能来说,改变的活性或功能可以是以某些其他方式(如特异性)降低、消失、消除、增强、提高或改变的活性或功能。需要特别说明的是修饰蛋白或多肽的一种活性或功能发生改变但是却保留了野生型蛋白或多肽的其他活性或功能。另外,修饰蛋白可以是完全失去功能的,或者其编码核酸序列被改变从而使其根本不再表达该多肽、因移码突变而表达截短的多肽或者表达不同的氨基酸序列。
在某些实施方案中,突变蛋白或多肽的长度可以包含但不限于5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、275、300、325、350、375、400、425、450、475、500、525、550、575、600、625、650、675、700、725、750、775、800、825、850、875、900、925、950、975、1000、1100、1200、1300、1400、1500、1750、2000、2250、2500或更多个氨基酸残基,以及上述数值之间的任何数目的氨基酸残基。需要说明的是多肽可通过截短而发生突变,使其比相应的野生型多肽短。
本文所用的术语“氨基分子”是指任何氨基酸、氨基酸衍生物或氨基酸类似物,这是本领域技术人员所熟知的。在某些实施方案中,蛋白分子的残基是连续的,其间无任何非氨基分子将氨基分子残基的序列中断。在其他的实施方案中,序列可包含一个或多个非氨基分子部分。在一些特殊的实施方案中,蛋白分子的残基序列可被一个或多个非氨基分子部分中断。
相应的,术语“蛋白质性组合物”包含氨基分子序列,该序列内至少含有一个含20个天然合成蛋白质中常见氨基酸的序列,或者至少含有一个修饰的或不常见的氨基酸。
蛋白组合可用本领域技术人员所熟知的任何技术制备,其中包括通过标准的分子生物学技术表达蛋白质、多肽或肽,从天然材料中分离蛋白化合物,或者用蛋白材料化学合成。各种基因的核苷酸和蛋白质、多肽和肽序列已经被鉴定出来,可在本领域技术人员所熟知的电子数据库中找到。一个这样的数据库是生物技术信息基因库国家中心(National Center for Biotechnology Information's Genbank)和GenPept数据库(www.ncbi.nlm.nih.gov)。这些已知基因的编码区可用本文所描述的方法或本领域技术人员所熟知的其他方法扩增和/或表达。
A.功能方面
当本申请提及病毒蛋白或多肽的功能或活性时,除非特别说明,是指该病毒蛋白或多肽在生理条件下的活性或功能。例如,干扰素调节多肽是指可直接或间接影响至少一种干扰素及其活性的多肽。多肽可直接或间接诱导、增强、提高、增加、消除、减弱、降低、抑制或屏蔽干扰素的活性。在某些实施方案中,直接影响干扰素的例子包括可特异性结合干扰素的干扰素调节多肽。确定哪个分子具有这种活性可通过本领域技术人员熟知的试验方法实现。例如,将编码调节干扰素或其突变体的产物的基因转移到可诱导出干扰素活性的细胞内,然后与转移这种基因的细胞进行比较,通过确定其对干扰素反应水平的不同就可以确定哪些分子具有干扰素调节功能。
需要特别说明的是调节因子可以是影响蛋白质性组合物表达的分子,这些蛋白质性组合物参与靶分子通路,如通过结合干扰素编码的转录子。确定哪个分子是干扰素、IL-1β、TNF或其他具有疗效的分子的调节因子可通过本领域技术人员所熟知的方法实现,本文中已经描述了一些,例如利用天然的和/或重组的病毒蛋白。
B.病毒多肽的突变体
本发明的多肽氨基酸序列的改变可以是替代、插入或缺失突变体。与野生型相比,编码病毒多肽的基因发生突变可影响1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76、77、78、79、80、81、82、83、84、85、86、87、88、89、90、91、92、93、94、95、96、97、98、99、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、275、300、325、350、375、400、425、450、475、500或更多个多肽的非连续或连续氨基酸。痘苗病毒编码的各种多肽可通过参考Rosel等,1986,Goebel等,1990和GenBank登录号NC_001559来确定,本文都已纳入作为参考。
缺失突变体缺少天然或野生型蛋白的一个或多个残基。每个残基都可以缺失,一个区(如催化区或结合区)的全部或部分也可以缺失。终止密码子被引入(通过替代或插入)到核酸编码序列中时可产生出截短的蛋白。插入突变一般是指在多肽的非终止点上插入残基,包括插入一个免疫原性表位或者仅仅插入一个或多个残基。也可以制备末端添加产物,这被称为融合蛋白。
替代突变体一般是指在蛋白质的一个或多个位点上将一个氨基酸替换成另一个氨基酸,这样可以使多肽的一个或多个特性发生改变而不会丢失其他的功能或活性。替代可以是保守替代,即一个氨基酸用另外一个具有相似形状或电荷的氨基酸替代。保守替代是本领域所熟知的,例如其中包括如下交换:丙氨酸到丝氨酸;精氨酸到赖氨酸;天冬酰胺到谷氨酰胺或组氨酸;天冬氨酸到谷氨酸;半胱氨酸到丝氨酸;谷胺酰胺到天冬酰胺;谷氨酸到天冬氨酸;甘氨酸到脯氨酸;组氨酸到天冬酰胺或谷胺酰胺;异亮氨酸到亮氨酸或缬氨酸;亮氨酸到缬氨酸或异亮氨酸;赖氨酸到精氨酸;蛋氨酸到亮氨酸或异亮氨酸;苯丙氨酸到酪氨酸、亮氨酸或蛋氨酸;丝氨酸到苏氨酸;苏氨酸到丝氨酸;色氨酸到酪氨酸;酪氨酸到色氨酸或苯丙氨酸;以及缬氨酸到异亮氨酸或亮氨酸。另外,替代也可以是非保守的,这样多肽的功能或活性会受到影响。非保守的替代一般是指用一个化学结构不同的残基来替代另一个残基,如极性氨基酸或带电荷的氨基酸替代非极性氨基酸或不带电荷的氨基酸,反之亦然。
本文所用术语“功能等同的密码子”是指编码相同氨基酸的密码子(见下表1)。
表1
密码子列表
还应当理解的是氨基酸和核酸序列还可以包含附加的残基,如附加的N-或C-末端氨基酸或者5’或3’序列,这在本文所描述的上述序列中是必需的,只要该序列复合上述标准,包括保持所表达蛋白的生物学活性。添加末端序列特别适合于那些包含各种非编码序列的核酸序列,其中非编码序列位于编码区的5’或3’端,或者那些包含各种内部序列,即内含子的核酸序列,内含子是大家所熟知的包含在基因内的序列。
下面的讨论是基于蛋白质的氨基酸发生交换而产生等价的或者是改进的第二代分子。例如,蛋白质内的某些氨基酸被其他氨基酸所替代,但是蛋白质与某些结构,如抗体上的抗原结合区或底物分子上的结合位点的结合能力并没有显著下降。由于蛋白质的相互结合能力和性质决定了蛋白质的生物功能活性,因此某些氨基酸替代可在蛋白质序列内或者其DNA编码序列内发生,但是却可以产生出具有相似特性的蛋白质。因此发明者认为在基因的DNA序列上进行的各种改变可以使其生物功能或活性不丢失,如下面所讨论的。表1显示的是编码特定氨基酸的密码子。
在进行这些改变时,需要考虑氨基酸的水性指数。氨基酸水性指数对蛋白质生物功能的重要性在本领域中是被普遍认知的(Kyte和Doolittle,1982)。氨基酸的相对水性特征可影响蛋白质的二级结构,从而也就决定了蛋白质与其他分子,如酶、底物、受体、DNA、抗体、抗原等的相互作用。
还应当理解的是在本领域中相似氨基酸的替代可在亲水性指数的基础上有效进行。美国专利4,554,101阐明一个蛋白质的局部最大平均亲水性指数是由其相邻的氨基酸所决定的,并且与蛋白质的生物学特性相关,本文已纳入作为参考。如美国专利4,554,101所详细描述的,下列亲水性指数值被赋予了各个氨基酸残基:精氨酸(+3.0);赖氨酸(+3.0);天冬氨酸(+3.0±1);谷氨酸(+3.0±1);丝氨酸(+0.3);天冬酰胺(+0.2);谷胺酰胺(+0.2);甘氨酸(0);苏氨酸(-0.4);脯氨酸(-0.5±1);丙氨酸(-0.5);组氨酸*-0.5);半胱氨酸(-1.0);蛋氨酸(-1.3);缬氨酸(-1.5);亮氨酸(-1.8);异亮氨酸(-1.8);酪氨酸(-2.3);苯丙氨酸(-2.5);色氨酸(-3.4)。
应当理解的是一个氨基酸被另外一个具有相似亲水性指数的氨基酸替代后可以得到一个生物等价和免疫等价的蛋白质。在这种替代中,亲水性指数值差异在±2之内的氨基酸替代是优选的,差异在±1之内的氨基酸替代是更优选的,差异在±0.5之内的氨基酸替代是尤其优选的。
如上面所指出的,氨基酸替代一般都是根据氨基酸侧链取代基的相对相似性来进行,如其疏水性、亲水性、电荷、侧链长度等。考虑了上述各种特征的典型替代是本领域技术人员所熟知的,其中包括:精氨酸和赖氨酸;谷氨酸和天冬氨酸;丝氨酸和苏氨酸;谷胺酰胺和天冬酰胺;以及缬氨酸、亮氨酸和异亮氨酸。
III.核酸分子
A.编码天然蛋白或修饰蛋白的多核苷酸
本发明涉及可从细胞中分离的多核苷酸,该多核苷酸可表达全长的或部分的蛋白或多肽。在本发明的某些实施方案中,涉及被特异性突变的病毒基因组,这种突变可使病毒缺乏某些功能性病毒多肽。多核苷酸可编码含全部或部分病毒氨基酸序列的肽或多肽,这些多核苷酸也可以被改造使之不编码这种病毒多肽,或者编码至少有一个功能或活性降低、消除或缺失的病毒多肽。重组蛋白可从产生活性蛋白的表达细胞内纯化。基因组及痘苗病毒编码区的定义见于Rosel等,1986,Goebel等,1990和/或GenBank登录号NC_001559,本文都已纳入作为参考。
本文所用的术语“DNA节段”是指从特定物种的基因组DNA中分离出来的游离形式的DNA分子。因此,编码多肽的DNA节段是指含野生型、多态性或突变多肽编码序列的DNA节段,可以从哺乳动物或人的基因组总DNA内分离或纯化。多肽、比多肽短的DNA节段以及重组载体,如质粒、粘粒、噬菌体、病毒等也包含在术语“DNA节段”内。
本申请所使用的术语“痘病毒多核苷酸”是指编码痘病毒多肽的核酸分子,可从基因组总核酸中分离。
同样,“痘苗病毒多核苷酸”是指编码痘苗病毒多肽的核酸分子,可从基因组总核酸中分离。“痘病毒基因组”或“痘苗病毒基因组”是指在存在或不存在辅助病毒的情况下可使宿主细胞产出病毒颗粒的核酸分子。与野生型病毒相比,基因组可以是重组突变的,也可以不是。
术语“cDNA”是指以信使RNA(mRNA)为模板制备的DNA。与基因组DNA或基因组非或部分处理的RNA模板来源的DNA多聚物不同的是,利用cDNA的优点在于cDNA主要包含相应蛋白的编码序列。有时也需要使用全部或部分基因组序列,比如要获得最佳表达需要非编码区时或者非编码区如内含子是反义策略的靶位时。
还需要说明的是一个给定物种的特定多肽可由天然突变体代表,该突变体的核酸序列有些许不同,但是所编码的蛋白是相同的(见上述表1)。
同样,含分离的或纯化的野生型或突变多肽基因的多核苷酸是指包含野生型或突变多肽编码序列的DNA节段,在某些情况下也包含调节序列,这些调节序列是从天然基因或蛋白编码序列中分离出来的。在这种情况下,术语“基因”也可以简单地指功能蛋白、多肽或肽编码单位(包括正确转录、翻译后修饰或定位所需要的任何序列)。如本领域技术人员所理解的,这个功能术语包含可表达或适于表达蛋白质、多肽、结构域、肽、融合蛋白和突变体的基因组序列、cDNA序列和人工基因小片段。编码全长的或部分的天然或修饰多肽的核酸含有编码该多肽的全部或部分的连续核酸序列,其长度可以是:10、20、30、40、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、210、220、230、240、250、260、270、280、290、300、310、320、330、340、350、360、370、380、390、400、410、420、430、440、441、450、460、470、480、490、500、510、520、530、540、550、560、570、580、590、600、610、620、630、640、650、660、670、680、690、700、710、720、730、740、750、760、770、780、790、800、810、820、830、840、850、860、870、880、890、900、910、920、930、940、950、960、970、980、990、1000、1010、1020、1030、1040、1050、1060、1070、1080、1090、1095、1100、1500、2000、2500、3000、3500、4000、4500、5000、5500、6000、6500、7000、7500、8000、9000、10000或更多个核苷酸、核苷或碱基对。
本发明的某些特殊实施方案涉及分离的DNA节段以及含DNA插入序列的重组载体,其中DNA插入序列编码野生型或突变的痘病毒多肽或肽,这些多肽或肽的氨基酸序列内包含与天然多肽一致或基本一致的连续氨基酸序列。因此,分离的DNA节段或含DNA节段的载体可编码抑制或降低INF活性的INF调节因子或TNF调节多肽。术语“重组的”可与多肽联用或作为特异性多肽的名称,一般是指在体外经改造的核酸分子编码的多肽或这种分子的复制产物所编码多肽。
本发明的其他实施方案涉及分离的DNA节段以及含DNA插入序列的重组载体,其中DNA插入序列编码多肽或肽,这些多肽或肽的氨基酸序列内包含与多肽一致或基本一致的连续氨基酸序列。
本发明所用的核酸片段不论其编码序列的长度如何都可以和其他的核酸序列连接,如启动子、多聚腺苷酸信号、额外的限制性内切酶位点、多克隆位点、其他编码序列等,这样其全长的变化就会相当大。因此需要说明的是几乎任何长度的核酸片段都可以使用,但是优选的核酸片段长度受制备方法及其在重组DNA技术中的用途限制。
需要说明的是本发明的核酸构建体可编码任何物种的全长多肽,也可以编码截短的多肽,如截短的痘苗病毒多肽,这时编码区的转录子代表了截短的版本。截短的转录子被翻译成截短的蛋白。另外,核酸序列可编码带有其他异源编码序列的全长多肽序列,这样有利于多肽的纯化、运输、分泌、转录后修饰或多肽的疗效如靶向性和活性。如上面所讨论的,修饰多肽编码序列可添加上标签或其他异源多肽,其中“异源的”是指与修饰多肽不一样的多肽。
在一个非限定性例子中,一个或多个核酸构建体可制备成包含与特定基因,如B18R基因一致或互补的一段连续核苷酸的构建体。核酸构建体可以至少含有20、30、40、50、60、70、80、90、100、110、120、130、140、150、160、170、180、190、200、250、300、400、500、600、700、800、900、1,000、2,000、3,000、4,000、5,000、6,000、7,000、8,000、9,000、10,000、15,000、20,000、30,000、50,000、100,000、250,000、500,000、750,000到1,000,000核苷酸,或者更长的构建体,甚至可以达到和包含染色体的长度(包括所有的中间长度和中间范围),假设核酸构建体如酵母人工染色体是本领域技术人员所熟知。很容易理解的是本文所用的“中间长度”和“中间范围”是指包含上述数值或上述数值之间的任意长度或范围(即包含这些数值和这些数值之间的所有整数值)。
本发明所用的DNA节段包含生物功能等价的修饰多肽和肽,如修饰的白树素毒素。这种序列的产生可能是密码子简并性和功能等价的结果,已知在天然状态下核酸序列及其编码的蛋白序列都可能发生。另外,功能等价的蛋白或肽可利用重组DNA技术制备,其中可根据修饰氨基酸的特性人工改变蛋白的结构。通过定点突变技术可将人为设计的改变引入到蛋白序列内,如可以提高蛋白质的抗原性、降低蛋白质对受体的体内毒性效应、或提高含蛋白的治疗药物的疗效。
本发明的某些实施方案涉及分离的DNA节段和序列内包含本文所描述的序列(和/或参考文献所描述的序列)来源的连续核酸序列的重组载体。但是,这种序列可被突变以制备活性不同于野生型蛋白的蛋白产物。
还应当理解的是本发明并不局限于这些已知序列的特定核酸序列和氨基酸序列。重组载体和游离DNA节段可以包含不同序列,包括痘病毒编码区自身、在基础编码区上具有特定改变或修饰的编码区,或者可以编码包含痘病毒编码区在内的更长的多肽、或编码具有突变氨基酸序列的生物等价蛋白或肽。
本发明的DNA节段包含生物等价的痘病毒蛋白和肽。这种序列的产生可能是密码子简并性和功能等价的结果,已知在天然状态下核酸序列及其编码的蛋白序列都可能发生。另外,功能等价的蛋白或肽可利用重组DNA技术制备,其中可根据修饰氨基酸的特性人工改变蛋白的结构。通过定点突变技术可将人为设计的改变引入到蛋白序列内,如可以提高蛋白质的抗原性。
B.诱导痘病毒多核苷酸发生突变
在各种实施方案中,痘病毒多核苷酸都可以被改变或突变。
改变或突变包括插入、缺失、点突变、颠换等,结果可导致某些通路或分子机制的调节、活化和/或灭活,或者改变基因产物的功能、定位或表达,特别是使基因产物失去功能。编码全长或部分痘病毒的多核苷酸的突变可用各种标准的突变方法完成(Sambrook等,1989)。突变是生物的量或结构发生改变的过程。突变包括单个基因的核苷酸序列的修饰、基因或整个染色体的阻断。单个基因的改变可以是点突变的结果,其中包括DNA序列内单个核苷酸的去除、添加或替代,也可以是插入或缺失多个核苷酸的结果。
突变可在接触化学或物理突变剂后被诱导。这些突变诱导剂包括离子辐射、紫外线和化学物质的不同组合,如烷化剂和多环芳香烃,这些物质都可以直接或间接(一般发生在经代谢生物转化后)与核酸发生相互作用。当被影响的DNA复制或修饰时这种诱导剂所诱导的DNA损伤可导致碱基序列的改变,从而发生突变。也可以通过特殊的靶向性方法进行定点突变。
为了在痘病毒基因组内引入突变,天然和修饰多肽可由包含在载体内的核酸分子编码。本文所用的术语“载体”是指携带核酸分子的载体,其中载体内插入的外源核酸分子可通过载体被导入到可以复制载体的细胞内。核酸序列是“外源性的”是指对于载体导入的细胞来说该核酸是外源的,或者该序列虽然与细胞内的序列同源但是其在宿主细胞核酸内的位置通常不包含该核酸。载体包括质粒、粘粒、病毒(噬菌体、动物病毒和植物病毒)以及人工染色体(如YACs)。本领域的技术人员可以通过标准的重组技术构建这种载体,这些方法在Sambrook等,(1989)和Ausubel等,1994中有描述,本文已纳入作为参考。除了可以编码修饰多肽如白树素以外,载体还可以编码未修饰的多肽序列,如标记物或靶向性分子。可以编码这种融合蛋白的载体包括pIN载体(Inouye等,1985)、编码一组组氨酸的载体和用于制备谷胱甘肽S-转移酶(GST)可溶性融合蛋白的pGEX载体以利于以后的纯化和分离或切割。靶向性分子是一种可以使修饰多肽特异性地定位到特定器官、组织、细胞或受体其他位置的分子。
术语“表达载体”是指含有核酸序列的载体,其中的核酸序列至少编码部分能被转录的基因产物。在某些情况下,RNA分子随后被翻译成蛋白、多肽或肽。在另外一些情况下,这些序列不翻译,而是转录出反义分子或核酶。表达载体含有各种“调控序列”,调控序列是指操作连接的编码序列在特定宿主内转录和翻译所需要的核酸序列。除了控制转录和翻译的调控序列外,载体和表达载体还可以包含具有其他功能的核酸序列,这些序列将在下文描述。
1.启动子和增强子
“启动子”是一种调控序列,是核酸序列上一个控制转录起始和速率的区域。可以包含调节蛋白和分子结合的元件如RNA聚合酶及其他转录因子。术语“操作位置的”、“操作连接的”、“控制下的”和“转录控制下的”是指相对于核酸序列来说启动子位于正确的功能位置和/或方向,可以调控该序列的转录起始和/或表达。启动子可以连接有“增强子”,也可以不连接,增强子是指参与核酸序列转录活化的顺式调节序列。
启动子可以是与某个基因或序列天然相连的序列,如通过分离位于编码片段和/或外显子上游的5’非编码序列而得到的启动子。这种启动子被称为“内源性的”。同样,增强子也可以是与某个序列天然相连、位于该序列上游或下游的序列。另外,将编码核酸片段置于重组的或异源的启动子控制之下也有某些优势,重组的或异源的启动子是指在天然状态下该序列不是与核酸序列相联系的。重组的或异源的增强子也是指在天然状态下不是与核酸序列相联系的增强子。这种启动子或增强子包括其他基因的启动子或增强子、从其他原核细胞、病毒或真核细胞中分离出的启动子或增强子以及非“天然的”启动子或增强子,即含有不同转录调节区和/或改变表达的突变的不同元件。除了可以通过合成方法制备启动子和增强子的核酸序列外,还可以利用重组克隆和/或核酸扩增技术,其中包括PCRTM,结合本文所描述的组合物来制备序列(见美国专利4,683,202,5,928,906,本文都已纳入作为参考)。另外,需要说明的是可控制无核细胞器如线粒体、叶绿体内的序列转录和/或表达的调控序列也可以使用。
很显然,利用启动子和/或增强子来有效地控制DNA节段在细胞、细胞群和生物体内的表达是十分重要的。分子生物学领域的技术人员一般都熟悉如何使用启动子和增强子,知道如何选择细胞以表达蛋白,例如Sambrook等,(1989)所描述的,本文已纳入作为参考。所使用的启动子可以是组成性的、组织特异性的、可诱导的启动子和/或可在适当的条件下使被导入的DNA节段高水平表达的启动子,例如在大规模制备重组蛋白和/或肽时。启动子可以是异源的,也可以是内源性的。
组织特异性启动子或元件的种类及鉴定其活性的测定是本领域技术人员所熟知的。这种区域的例子包括人LIMK2基因(Nomoto等,1999)、生长抑素受体2基因(Kraus等,1998)、鼠附睾视黄酸结合基因(Lareyre等,1999)、人CD4(Zhao-Emonet等,1998),鼠α2(XI)胶原(Tsumaki,等,1998)、D1A多巴胺受体基因(Lee,等,1997)、胰岛素样生长因子II(Wu等,1997)、人血小板内皮细胞粘附分子-1(Almendro等,1996)、以及SM22α启动子。
2.起始信号和内部核糖体结合位点
编码序列的有效翻译还需要特异性的起始信号。这些信号包括ATG起始密码子或其临近序列。包含ATG起始密码子的外源翻译控制信号也需要提供。本领域的技术人员能够很容易地确定这一点并提供必须的信号。大家都知道起始密码子必须位于目的编码序列阅读框架内才能确保整个插入序列被翻译。外源性的翻译控制信号可以是天然的,也可以是合成的。插入合适的转录增强元件可以提高表达的效率。
在本发明的某些实施方案中,使用内部核糖体进入位点(IRES)可以使多个基因或多个顺反子信使RNA表达。IRES元件可以使核糖体绕过5’甲基化帽子依赖性翻译的扫描模式在内部位点开始翻译(Pelletier和Sonenberg,1988)。小核糖核酸病毒家族的两个成员(脊髓灰质炎病毒和脑心肌炎病毒)来源的IRES元件以及哺乳动物信使RNA来源的IRES(Macejak和Sarnow,1991)。
IRES元件可以被连接到异源开放阅读框上。多个开放阅读框架可以一起转录,其中每个阅读框架都被IRES隔开,转录出一条多顺反子信使RNA。
利用IRES元件每个开放阅读框架都可以有核糖体结合从而可以高效翻译。利用单个启动子/增强子转录出一条信使RNA可以使多个基因都得到有效表达(见美国专利5,925,565和5,935,819,本文已纳入作为参考)。
3.多克隆位点
载体可包含多克隆位点(MCS),MCS是指包含多个限制性酶切位点的核酸序列,通过标准的重组技术每个酶切位点都可以用于消化载体(见Carbonelli等,1999,Levenson等,1998,和Cocea,1997,本文已纳入作为参考)。“限制性内切酶消化”是指利用内切酶催化切割核酸分子的过程,其中内切酶只在核酸分子的特定位置上切割。很多这样的限制性内切酶都可以买到。这种内切酶的使用方法是本领域技术人员所熟知的。载体通常被在MCS内切割的限制性内切酶线性化或片段化从而可以使外源序列连接到载体上。“连接“是指两个核酸片段之间形成磷酸二酯键的过程,其中两个核酸片段可以是彼此不相临近的。涉及限制性内切酶和连接反应的技术是重组技术领域的技术人员所熟知的。
4.剪接位点
大多数被转录出来的真核RNA分子都会发生RNA剪接以去除初级转录子内的内含子。含基因组真核序列的载体需要供体和/或受体剪接位点以保证转录子被正确处理,然后表达蛋白(见Chandler等,1997,本文已纳入作为参考)。
5.终止信号
本发明的载体或构建体一般都包含至少一个终止信号。“终止信号”或“终止子”由参与RNA聚合酶所转录的转录子特异性终止的DNA序列组成。因此,在某些实施方案中,需要结束RNA转录子合成的终止信号。终止子是在体内获得理想的信使RNA表达水平所必须的。在真核系统中,终止子区域还可以包含允许新转录子发生定点切割以暴露多聚腺苷酸信号的特异性DNA序列。
这种序列可向特异的内源性聚合酶发出信号,使之将一段约200A的残基(聚A)添加到转录子的3’末端。经这种聚A尾巴修饰的RNA分子更稳定,其翻译的效率也更高。因此,在其他涉及真核生物的实施方案中,优选包含RNA切割信号的终止子,更优选的是该终止子信号可促进信使RNA的多聚腺苷酸化。终止子和/或多聚腺苷酸位点元件可提高信使RNA的表达水平和/或使核糖体阅读过表达盒进入其他序列的可能降到最低。
本发明所用的终止子包括本文所描述的或者本领域技术人员所熟知的转录子的任何已知的终止子,其中包括而不限于基因的终止序列,如牛生长激素终止子,或者病毒的终止序列,如SV40终止子。在某些实施方案中,可转录或可翻译的序列可能缺少终止信号,如由于序列截短。
6.多聚腺苷酸信号
基因表达,特别是真核基因表达一般都包含多聚腺苷酸信号以使转录子正确多聚腺苷酸化。据信多聚腺苷酸信号的性质不是成功实现本发明的关键,任何这样的序列都可以使用。优选的实施方案包括SV40多聚腺苷酸信号和/或牛生长激素多聚腺苷酸信号,已知这些多聚腺苷酸信号在各种靶细胞内都有功能,并且使用方便。多聚腺苷酸化可提高转录子的稳定性,或者可以促进胞浆转运。
7.复制起点
为了在宿主细胞内扩增载体,载体内需包含一个或多个复制起始位点(通常被称为“ori”),这是一种复制在此处开始的特异性核酸序列。另外,如果宿主细胞是酵母,可以使用自主复制序列(ARS)。
8.选择和筛选标记
在本发明的某些实施方案中,可以通过在表达载体内添加一个标记而便于含本发明核酸构建体的细胞在体外或体内被鉴定。这种标记可以使细胞具有可鉴定的表型从而便于鉴定含表达载体的细胞。一般来说,筛选标记是一种可提供筛选特性的序列。阳性筛选标记是指在标记物存在的情况下可被筛选的标记,而负性标记是指在标记物存在的情况下不被筛选的标记。阳性筛选标记的例子有耐药标记。
一般来说,包含药物筛选标记有助于转化子的克隆和鉴定,例如提供新霉素嘌罗霉素、潮霉素、DHFR、GPT、zeocin和组胺醇耐药性的基因都是有用的筛选标记。除了能提供一种表型以便于区分转化子的标记外,其他类型的标记,包括筛选标记,也包括在本发明的范围之内,如GFP,其筛选的基础是比色分析。另外,可筛选的酶如单纯疱疹病毒胸苷激酶(tk)或氯霉素乙酰转移酶(CAT)也可以使用。本领域的技术人员了解如何使用免疫标记,可能结合FACS分析。用何种标记被认为是不重要的,只要该标记可与编码基因产物的核酸同时表达就可以。选择和筛选标记的其他例子是本领域技术人员所熟知的。
D.宿主细胞
本文所用的术语“细胞”、“细胞系”和“细胞培养物”可以互用。所有这些术语都包括其子代,可以是任何代和所有代的细胞。应当理解的是并不是所有的子代细胞都是一致的,因为可能会发生定向的或随机的突变。在用于表达异源核酸序列时,“宿主细胞”是指原核细胞或真核细胞,包括任何可被转化的生物,只要它能够复制载体和/或表达载体所编码的异源基因。宿主细胞可以而且已经被用作载体或病毒(如果不能表达外源多肽则不能作为载体)的受体。宿主细胞可以是“被转染的”或“被转化的”是指外源核酸,如修饰蛋白编码序列,被转移或导入到宿主细胞内的过程。转化细胞包括原代受体细胞及其子代细胞。
根据所要得到的结果是复制载体或是表达部分或全部载体编码核酸序列,宿主细胞可以是原核细胞或真核细胞,其中包括酵母细胞、昆虫细胞和哺乳动物细胞。许多细胞系和细胞培养物都可用作宿主细胞,可从美国模式培养物保藏所(ATCC)获得,这是收集和保存活体培养物和遗传材料的组织(www.atcc.org)。本领域的技术人员根据载体的结构及所要得到的结果能够确定选择何种合适的宿主。例如,质粒和粘粒可被导入到原核宿主细胞内以复制大量的载体。作为宿主细胞用于载体复制和/或表达的细菌包括DH5α、JM109和KC8,以及许多商业化的细菌宿主,如感受态细胞和SOLOPACKTM Gold细胞(公司,加州拉霍亚)(La Jolla,Calif.)。另外,细菌细胞如大肠杆菌LE392也可以作为宿主细胞复制噬菌体。合适的酵母细胞包括酿酒酵母(Saccharomyces cerevisiae)、粟酒酵母(Saccharomyces pombe)和巴斯德毕赤酵母(Pichia pastoris)。
用于复制和/或表达载体的真核宿主细胞的例子包括HeLa、NIH3T3、Jurkat、293、Cos、CHO、Saos和PC12。各种类型的细胞和生物来源的宿主细胞都可以得到,这是本领域技术人员所熟知的。同样,病毒载体可用于真核宿主细胞,也可用于原核宿主细胞,特别是能复制或表达载体的宿主细胞。
某些载体可包含调控序列以使其既可以在原核细胞,又可以在真核细胞内复制和/或表达。本领域的技术人员还了解培养上述宿主细胞并使其中的载体复制的条件。还清楚和了解大规模制备载体以及载体编码的核酸及其同源多肽、蛋白或肽的技术和条件。
E.基因转移方法
适于核酸转移以表达本发明的组合物的方法被认为可以包括能将核酸(如DNA,包括病毒载体和非病毒载体)导入到细胞器、细胞、组织或生物体内的任何方法,如本文所描述的和本领域技术人员所熟知的方法。这些方法包括而不限于通过注射(美国专利5,994,624、5,981,274、5,945,100、5,780,448、5,736,524、5,702,932、5,656,610、5,589,466和5,580,859,本文已纳入作为参考)、包括显微注射(Harland和Weintraub,1985;美国专利5,789,215,本文已纳入作为参考);电穿孔(美国专利5,384,253,本文已纳入作为参考);钙磷酸盐沉淀(Graham和Van Der Eb,1973;Chen和Okayama,1987;Rippe等,1990);DEAE-葡聚糖、然后用聚乙二醇(Gopal,1985);直接的声波负载(Fechheimer等,1987);脂质体介导的转染(Nicolau和Sene,1982;Fraley等,1979;Nicolau等,1987;Wong等,1980;Kaneda等,1989;Kato等,1991);微发射炮击(PCT专利申请WO 94/09699和95/06128;美国专利5,610,042、5,322,783、5,563,055、5,550,318、5,538,877和5,538,880,本文已纳入作为参考);碳化硅纤维搅拌(Kaeppler等,1990;美国专利5,302,523和5,464,765,本文已纳入作为参考);土壤杆菌介导的转化(美国专利5,591,616和5,563,055,本文已纳入作为参考);或PEG-介导的原生质体转化(Omirulleh等,1993;美国专利4,684,611和4,952,500,本文已纳入作为参考);脱水/抑制介导的DNA摄取(Potrykus等,1985)直接转移基因。通过使用这些技术,细胞器、细胞、组织或生物体可被稳定转化或瞬时转化。
F.脂质组分和部分
在某些实施方案中,本发明涉及包含与核酸、氨基酸分子如肽或其他小分子化合物相连接的一个或多个脂质的组合物。在本文所讨论的所有实施方案中,分子既可以是痘病毒多肽或痘病毒多肽调节因子,如编码全长或部分痘病毒多肽的核酸,也可以是编码全长或部分痘病毒多肽调节因子的氨基酸分子。脂质是不溶于水、能用有机溶剂提取的物质。本文所特别指出的化合物以外的化合物可以被本领域的技术人员理解为脂质,也包括在本发明的组合物和方法之内。脂质成分和非脂质成分可通过共价键或非共价键彼此结合。
脂质可以是天然的,也可以是人工合成的(即人工设计并制备的)。但是,脂质通常是一种生物物质。生物脂质是本领域所熟知的,包括中性脂肪、磷脂、磷酸甘油酯、类固醇、萜烯、溶血脂质(lysolipids)、鞘糖脂、糖脂、硫苷脂、含有醚和酯连接脂肪酸的脂质、可聚合的脂质、以及上述脂质的组合物。
与脂质相关的核酸分子或氨基酸分子、如肽可被分散在含脂质溶液中、用脂质溶解、乳化、混合、与脂质连接、共价结合、以悬液的形式悬浮于脂质中、以及以其他方式与脂质相联系。脂质或脂质/本发明的痘病毒相关组合物并不局限于任何特定结构的分子。例如,可以简单地分散在溶液中,可能形成不同大小和形状的凝集物。在其他的例子中,可以双层的结构存在,如微胶粒或萎陷的结构。在其他的非限定性例子中,也可以考虑lipofectamine(吉布科公司(Gibco BRL))-痘病毒或Superfect(凯杰公司(Qiagen))-痘病毒复合物。
在某些实施方案中,脂质组合物可以包含约1%、约2%、约3%、约4%、约5%、约6%、约7%、约8%、约9%、约10%、约11%、约12%、约13%、约14%、约15%、约16%、约17%、约18%、约19%、约20%、约21%、约22%、约23%、约24%、约25%、约26%、约27%、约28%、约29%、约30%、约31%、约32%、约33%、约34%、约35%、约36%、约37%、约38%、约39%、约40%、约41%、约42%、约43%、约44%、约45%、约46%、约47%、约48%、约49%、约50%、约51%、约52%、约53%、约54%、约55%、约56%、约57%、约58%、约59%、约60%、约61%、约62%、约63%、约64%、约65%、约66%、约67%、约68%、约69%、约70%、约71%、约72%、约73%、约74%、约75%、约76%、约77%、约78%、约79%、约80%、约81%、约82%、约83%、约84%、约85%、约86%、约87%、约88%、约89%、约90%、约91%、约92%、约93%、约94%、约95%、约96%、约97%、约98%、约99%、约100%或上述数值之间任何范围的特定脂质、脂类或非脂质组分,如药物、蛋白、糖、核酸或本文所描述的以及本领域技术人员所熟知的其他物质。在一个非限定性例子中,脂质组合物包含约10%到20%中性脂质、约33%到34%的脑苷脂和约1%的胆固醇。在另一个非限定性例子中,脂质体包含约4%到12%的萜烯,其中约1%的微胶粒是番茄红素,另外约有3%到11%的脂质体包含其他萜烯;约10%到35%的磷脂酰胆碱以及约1%的药物。因此,需要说明的是本发明的组合物包含任何组合或任何百分比的脂质、脂类或其他组分。
IV.药物制剂、递送和治疗方案
在本发明的一个实施方案中,提供了一种通过给予经修饰的痘病毒,如痘苗病毒治疗过度增殖性疾病、如癌症的方法。可用该方法治疗的肿瘤包括肝癌、肺癌、头颈癌、乳腺癌、胰腺癌、前列腺癌、肾癌、骨癌、睾丸癌、宫颈癌、胃肠癌、淋巴瘤、肺内的癌前病变、结肠癌、黑色素瘤、膀胱癌以及可被治疗的其他癌或肿瘤。
本文中药物组合物的有效量是指足以诱导溶瘤、破坏或裂解癌细胞,减缓、抑制或减少肿瘤生长或大小,包括在某些情况下根治肿瘤的用量。有效量也包括导致治疗性病毒全身性间接播散到肿瘤如感染非注射肿瘤的用量。
患者最好具有适当的骨髓功能(定义为外周粒细胞绝对值>2,000/mm3,血小板绝对值为100,000/mm3)、适当的肝功能(胆红素<1.5mg/dl)和适当的肾功能(肌氨酸酐<1.5mg/dl)。
A.给药
利用本发明的方法和组合物诱导溶瘤时,使肿瘤与表达GM-CSF的痘病毒接触。给药途径可视损伤的部位及损伤的性质而不同,包括例如皮内、透皮、胃肠外、静脉内、肌肉内、鼻内、皮下、局部(如在肿瘤附近给药,尤其是利用肿瘤血管或临近肿瘤的血管)、经皮、气管内、腹膜内、动脉内、膀胱内、瘤内、吸入、灌注、灌洗或口服等给药途径和制剂。
对于不连续的易接近的实体瘤来说,采用瘤内注射或直接注射到肿瘤血管内的方式给药是特别合适的。局部、区域或系统给药也可以采用。对于>4cm的肿瘤来说,注射的量约为4-10ml(优选10ml),而对于<4cm的肿瘤来说,注射的量约为1-3ml(优选3ml)。多点注射给药时单次剂量包含约0.1-0.5ml的体积。对于病毒颗粒来说最好通过多点注射来给药,点与点之间的间隔约1cm。
在进行手术干预的情况下,本发明可作为术前措施使无法进行手术的肿瘤变成可被切除的肿瘤。
在合适的情况下可进行连续给药,例如在肿瘤中或肿瘤血管中植入导管。这种连续给药可持续约1-2小时到2-6小时、约6-12小时、约12-24小时、约1-2天、约1-2周或更长时间。一般来说,通过持续灌注方式所给予的治疗性组合物的剂量与单点注射或多点注射所给予的剂量等价,并且在灌注期间可以进行适当调整。还需要说明的是四肢灌注方式也可用于给予本发明的治疗性组合物,尤其是用于治疗黑色素瘤和肉瘤时。
治疗方案可以不同,要根据肿瘤的类型、肿瘤的部位、疾病的进展情况以及患者的健康状况和年龄而定。显然,某些类型的肿瘤需要更积极的治疗措施,而同时某些患者可能无法耐受较繁重的治疗过程。临床医生最适宜根据治疗性制剂的效力和毒性(如果有)来作决定。
在某些实施方案中,被处理的肿瘤可能是无法被切除的,或者至少最初是无法被切除的。通过治疗性病毒构建体的处理可能能够提高肿瘤的可切除性,可能由于肿瘤的边界发生皱缩,或者由于去除了肿瘤某些侵入的部分。经过处理后就可能能够切除了。切除后可进一步采用其他处理措施来消除肿瘤部位残存的微小肿瘤组织。
治疗方案包括不同的“单位剂量”。单位剂量定义为预先确定的治疗性组合物的量。药物使用剂量、给药途径和剂型的选择是临床医学领域的技术人员所熟知的。单位剂量并不一定只通过一次注射来完成,也可以在一定的期间内持续输注。本发明的单位剂量为了方便用病毒构建体的噬菌斑形成单位(pfu)来表示。单位剂量范围为103、104、105、106、107、108、109,1010、1011、1012、1013pfu以及更高。另外,根据病毒种类及其可得到的滴度不同,可给予肿瘤或肿瘤位点1-100、10-50、100-1000、或者高达或至少约1×104、1×105、1×106、1×107、1×108、1×109、1×1010、1×1011、1×1012、1×1013、1×1014、或1×1015或更多的有感染性的病毒颗粒(vp),包括其间所有值和范围。
B.可注射的组合物和制剂
本发明中将编码全长或部分痘病毒基因组的表达构建体转移到肿瘤或肿瘤细胞内的优选方法是通过瘤内注射。但是,本文所描述的药物组合物也可以通过其他方式给药,如通过胃肠外、静脉内、皮内、肌肉内、透皮、甚至腹腔内途径给药,如美国专利5,543,158、5,641,515和5,399,363所描述(本文都已完整纳入作为参考)。
核酸构建体可用注射器或其他任何可注射溶液的方法注射,只要表达构建体能够通过特定孔径的注射针头就可以。最近所描述的一种无针注射系统(美国专利5,846,233)含有一个安装在安瓿腔内的喷嘴用于吸取溶液,一个动力装置用于将液体推出喷嘴转移到递送部位。在基因治疗中也可以用注射器系统将预先确定的精确量的溶液以任意深度注射到多个部位(美国专利5,846,225)。
活性化合物的游离碱溶液或药用盐溶液可通过在水中与适宜的表面活性剂,如羟丙基纤维素混合而制备。分散剂可用甘油、液体聚乙烯甘油或其混合物、或油来制备。在普通储存和使用条件下,这些制剂都要加入防腐剂以阻止微生物的生长。适于注射的药用制剂包括无菌水溶液或分散剂,以及在使用前可临时制备成无菌注射溶液或分散剂的粉末(美国专利5,466,468,本文已完整纳入作为参考)。无论何种情况,制剂都必须是无菌的,并且其流动性要足以能够通过注射器。在制造和储存条件下必须是稳定的,并且要防止微生物,如细菌和真菌的污染。载体可以是含水、乙醇、多元醇(如甘油、丙二醇和液体聚乙烯甘油等)、上述物质的适当混合物和/或植物油的溶剂或分散介质。可以通过包被,如卵磷脂,使颗粒在分散剂中保持所需的直径以及通过使用表面活性剂使制剂维持合适的流动性。可通过加入各种抗细菌和抗真菌物质,如对羟基苯甲酸酯类(parabens)、氯代丁醇、酚、山梨酸、硫柳汞等来防止微生物的生长。在许多情况下,加入一些等渗物质,如糖或氯化钠是优选的。可通过在组合物中加入吸收延迟物质,如单硬脂酸铝和明胶使可注射组合物的吸收缓慢。
对于通过胃肠外途径给药的水溶液来说,如果需要溶液应是缓冲的,液体稀释剂应首先用足量的盐或糖调节成等渗的。这些水溶液特别适于静脉内、肌肉内、皮下、瘤内和腹腔内给药。本领域的技术人员根据本说明书的描述可以选择应用这种水溶液所需要的无菌水溶剂。例如,一个剂量的药物可溶解到1ml等渗的氯化钠溶液中,然后添加到1000ml皮下补液溶液中或注射到输注的适当部位(见《雷明顿药物科学》(Remington's Pharmaceutical Sciences),第15版,1035-1038页和1570-1580页)。要根据被治疗的个体的情况来调整剂量。不论在任何情况下都负责给药的人来确定不同个体所需要的合适剂量。而且对于给人给药来说,制剂必须符合FDA生物标准办公室所制定的无菌、热源、一般安全性和纯度标准。
无菌注射液可通过使溶于合适溶剂中的所需量的活性化合物与上述各种其他组分混合、然后通过过滤除菌来制备。一般来说,分散剂可通过将各种无菌活性成分加入到含基础分散介质和所需要的上述各种其他组分的无菌载体内而制备。如果是用于制备无菌注射液的无菌粉末,优选的制备方法是真空干燥和冷冻干燥技术,该技术可从上述过滤除菌的溶液中制备出活性成分和其他所需成分的粉末。
本文所描述的组合物可制备成中性制剂和盐制剂。药用盐包括酸加盐(与蛋白质的游离氨基形成的盐),可用无机酸制备,如盐酸和磷酸,也可以用有机酸制备,如乙酸、醋浆草酸、酒石酸、扁桃酸等。与游离羧基形成的盐也可以来源于无机碱,如氢氧化钠、氢氧化钾、氢氧化铵、氢氧化钙和氢氧化铁,或者有机碱如异丙胺、三甲胺、组胺、普如卡因等。制成制剂以后,溶液就可以以剂型相配的方式、以治疗有效量给予。各种剂型都可以很容易地给药,如可注射溶液、缓释胶囊等。
本文所用的术语“载体”包含各种溶剂、分散介质、载体、包被物、稀释剂、抗细菌和抗真菌药物、等渗和吸收延迟物质、缓冲液、载体溶液、悬液、胶体等。利用这种介质和试剂制备药用活性物质的方法是本领域所熟知的。除了那些与活性组分不相容的常规介质或试剂以外,其在治疗性组合物中的应用也是本发明所涉及的。补充的活性组分也可以添加到组合物中。
术语“药用的”是指给予人体后不会产生变态反应或类似不良反应的分子实体和组合物。含有蛋白性活性组分的水性组合物的制备方法是本领域所熟知的。一般来说,这种组合物应制备成可注射的溶液或悬液,或者在注射前可制备成溶液或悬液的固体形式。
C.联合治疗
本发明的化合物和方法可用于治疗过度增殖性疾病,其中包括癌症。为了提高本发明的组合物,如表达GM-CSF的痘苗病毒的治疗效率,这些组合物可与其他能有效治疗这些疾病的药物联合使用。例如,肿瘤可用本发明的治疗性化合物联合其他的肿瘤治疗措施来治疗,如抗癌药或手术。
各种联合治疗措施都可以使用;例如,痘病毒如痘苗病毒是“A”,第二种抗癌措施是“B”:
A/B/A B/A/B B/B/A A/A/B A/B/B B/A/A A/B/B/B B/A/B/B
B/B/B/A B/B/A/B A/A/B/B A/B/A/B A/B/B/A B/B/A/A
B/A/B/A B/A/A/B A/A/A/B B/A/A/A A/B/A/A A/A/B/A
将本发明的痘病毒/痘苗病毒载体给予患者可按照第二种治疗措施的通用方法进行,并且需要考虑痘病毒治疗措施的毒性。如果需要,可重复进行几个治疗周期。还需要说明的是,各种标准的治疗措施以及手术干预都可以和本文所描述的癌症或肿瘤治疗措施联合使用。
“抗癌”药是对生物体内的肿瘤能产生消极影响的药物,如杀伤癌细胞,诱导癌细胞的调亡、降低癌细胞的生长速度、抑制转移的发生或转移的数目、缩小肿瘤、抑制肿瘤的生长、减少肿瘤或癌细胞的血供、激发抗癌细胞或肿瘤的免疫反应、阻止或抑制肿瘤的发展、或者延长癌症患者的生存期。抗癌药包括生物药(生物治疗)、化疗药和放疗药。最常见的情况是这些组合物各以有效量联合使用以杀伤或抑制细胞的增殖。这个过程包括细胞与表达构建体和药物或多个因子同时接触。这可以通过使细胞与一种包含两种药物的组合物或药用制剂接触,或者使细胞与两种不同的组合物或制剂同时接触,其中一种组合物含表达构建体,另一种组合物含第二药物。
肿瘤细胞对化疗药和放疗药耐受是临床肿瘤学领域遇到的一个主要问题。目前癌症研究的一个目标就是通过结合基因治疗找到提高化疗和放疗疗效的方法。例如,单纯疱疹病毒-胸苷激酶(HS-tK)基因用逆转录病毒载体系统转移到脑肿瘤内后可成功地诱导出肿瘤对抗病毒药物更昔洛韦的敏感性(Culver等,1992)。在本发明中,除了其他一些前调亡调节因子或细胞周期调节因子之外,痘病毒治疗同样可联合化疗、放疗、免疫治疗以及其他生物干预措施。
另外,痘病毒治疗可在其他药物治疗前或治疗后数分钟到数周内进行。在其他药物和痘病毒分别给予细胞的实施方案中,两种治疗措施实施的时间间隔一般不能太长,这样才能保证药物和痘病毒对细胞产生协同效应。在这种情况下,两种治疗措施最好在约12-24小时内分别给予细胞,更优选的是在6到12小时内。但是在某些情况下,人们希望将治疗的间隔时间延长,比如不同的治疗措施之间间隔数天(2、3、4、5、6或7)到数周(1、2、3、4、5、6、7或8)。
1.化疗
癌症的治疗措施还包括与化疗和放疗联合的各种治疗措施。例如,联合化疗包括顺铂(CDDP)、卡铂、丙卡巴肼、氮芥、环磷酰胺、喜树碱、异环磷酰胺、美法仑、苯丁酸氮芥、白消安、亚硝脲、放线菌素、柔红霉素、阿霉素、博来霉素、普里克霉素(plicomycin)、丝裂霉素、依托泊甙(VP16)、他莫昔芬、雷洛昔芬、雌激素受体结合药、紫杉醇、吉西他滨、新霉酰胺、法尼基蛋白转移酶抑制剂、反铂、5-氟尿嘧啶、长春新碱、长春花碱、氨甲蝶呤、太麻米德(Temazolomide)(DTIC的液态形似)、或上述药物的类似物或衍生物。化疗药与生物治疗联合被称为生物化疗。
2.放疗
可引起DNA损伤并被广泛应用的其他因素包括大家所熟知的γ射线、X射线和/或直接给予肿瘤细胞放射性同位素。本发明涉及的其他形式的DNA损伤因子还包括微波和紫外线辐射。最可能的情况是所有这些因子都可以对DNA、DNA的前体、DNA的复制和修复、染色体的装配和维持造成广泛的损伤。X射线的使用剂量范围为每日50-200伦琴持续一段时间(3到4周)到单次剂量2000-6000伦琴。放射性同位素的剂量使用范围很宽,要根据放射性同位素的半衰期、所激发的辐射的强度和类型以及肿瘤细胞的摄取量而定。
术语“接触”和“暴露于”在本文中用于细胞时是指治疗性构建体和化疗或放疗药转移到靶细胞或者与靶细胞直接并排放置的过程。为了达到杀死细胞或抑制细胞生长的目的,两种治疗措施都要以有效量给予细胞以便于杀死细胞或阻止其分裂。
3.免疫治疗
一般来说,免疫治疗依赖于利用免疫效应细胞和分子靶向并摧毁癌细胞。例如,免疫效应因子可以是肿瘤细胞表面某些标记物特异性的抗体。抗体本身可以单独作为治疗效应因子或者可以激活其他细胞来影响细胞的杀伤。抗体也可以被连接到药物或毒素(化疗药物、放射性核苷酸、蓖麻毒蛋白A链、霍乱毒素、百日咳毒素等)上,只作为一种靶向性药物。另外,效应因子可以是携带能与肿瘤细胞靶位直接或间接相互作用的表面分子的淋巴细胞。效应细胞包括细胞毒T细胞和NK细胞。联合治疗,即直接的细胞毒活性和某种痘病毒多肽的抑制或降低活性可以为癌症的治疗提供有益的帮助。
免疫治疗还可以作为联合治疗的一部分。联合治疗的常用方法将在下面讨论。在免疫治疗的一个方面,肿瘤细胞必须携带可作为靶位的某种标记,即在其他大多数细胞上不存在的标记。肿瘤细胞上存在多种标记物,这些标记物都可以作为本发明的适宜靶位。常见的肿瘤标记物包括癌胚抗原、前列腺特异性抗原、泌尿系统肿瘤相关抗原、胎儿抗原、酪氨酸酶(p97)、gp68、TAG-72、HMFG、Sialyl Lewis抗原、MucA、MucB、PLAP、雌激素受体、层粘连蛋白受体、erb B和p155。免疫治疗的另一个方面涉及利用免疫刺激效应产生抗癌效应。免疫刺激分子包括:细胞因子如IL-2、IL-4、IL-12、GM-CSF、IFNγ,趋化因子如MIP-1、MCP-1、IL-8,以及生长因子如FLT3配基。联合使用免疫刺激分子,无论是以蛋白的形式,还是肿瘤抑制因子如mda-7的基因转移,都可以增强抗肿瘤效应(Ju等,2000)。
正如以前所讨论的,目前正在研究或正在应用的免疫治疗措施有免疫佐剂(如牛型结核菌、恶性疟原虫、二硝基氯苯和芳香族化合物)(美国专利5,801,005;美国专利5,739,169;Hui和Hashimoto,1998;Christodoulides等,1998)、细胞因子治疗(如干扰素α、β和γ;IL-1,GM-CSF和TNF)(Bukowski等,1998;Davidson等,1998;Hellstrand等,1998)、基因治疗(如TNF,IL-1,IL-2,p53)(Qin等,1998;Austin-Ward和Villaseca,1998;美国专利5,830,880和5,846,945)以及单克隆抗体(如抗神经节苷脂GM2,抗HER-2,抗-p185)(Pietras等,1998;Hanibuchi等,1998;美国专利5,824,311)。赫赛汀(Herceptin)(曲妥单抗)是一种嵌合的(鼠-人)单克隆抗体,能够阻断HER2-neu受体。它具有抗肿瘤活性,已被批准用于治疗恶性肿瘤(Dillman,1999)。赫赛汀和化疗药物联合治疗癌症已被证明比单独的治疗措施更有效。
因此,本发明涉及利用一种或多种抗癌治疗措施与本文所描述的痘病毒相关治疗方法联合使用以治疗肿瘤的方法。
被动免疫治疗
现在已经有许多治疗癌症的被动免疫疗法。大致可以分为以下几类:单独注射抗体;注射连接有毒素或化疗药的抗体;注射连接有放射性同位素的抗体;注射抗独特型抗体;以及净化骨髓中的肿瘤细胞。
被动免疫治疗优选人源单克隆抗体,因为它们在患者体内的毒副作用很低或没有。人源化和嵌合单克隆抗体也成功用于癌症治疗。用作癌症治疗剂的单克隆抗体包括依决洛单抗(edrecolomab)、利妥昔单抗(rituximab)、曲妥珠单抗(trastuzumab)、吉姆单抗(gemtuzumab)、阿仑珠单抗(alemtuzumab)、替伊莫单抗(ibritumomab)、托西莫单抗(tositumomab)、西妥昔单抗(cetuximab)、贝伐单抗(bevacizumab)、尼妥珠单抗(nimotuzumab)和帕尼单抗(panitumamab)。
较好的是给予一种以上的抗两种不同抗原的单克隆抗体,甚至多种抗原的抗体。治疗方法还包括给予淋巴因子(lympholine)或其他免疫增强剂,如Bajorin等,(1988)的描述。有关人源单克隆抗体的研究进展会在本说明书的其他部分作进一步的详细描述。
主动免疫治疗
主动免疫治疗给予的是抗原性肽、多肽或蛋白质,或者自身的或同种异体的肿瘤细胞组合物或“疫苗”,一般需要和不同的细菌佐剂联合使用(Ravindranath和Morton,1991;Morton等,1992;Mitchell等,1990;Mitchell等,1993)。对于黑色素瘤的免疫治疗来说,那些激发出高滴度IgM反应的患者通常比未激发出或激发的IgM抗体滴度比较低的患者生存率更高(Morton等,1992)。IgM抗体一般都是瞬时抗体,但是抗神经节苷脂和抗糖类的抗体好像是例外。
获得性免疫治疗
获得性免疫治疗是指在体外分离出患者血液中的淋巴细胞或肿瘤侵润淋巴细胞,在体外用淋巴因子如IL-2活化或者转导肿瘤坏死基因,然后再输注到患者体内(Rosenberg等,1988;1989)。为达到这个目的,需要给动物或患者给予免疫有效量的活化的淋巴细胞和本文所描述的含佐剂的抗原肽组合物。活化的淋巴细胞最好是患者自身的易于从血液或肿瘤标本中分离并在体外被活化(或“扩增”)的细胞。这种形式的免疫治疗已对几例黑色素瘤和肾癌产生了抑制作用,但是发生反应的患者比未发生反应的患者明显要少。
4.基因
在另外一个实施方案中,第二治疗措施是基因治疗,其中治疗性多核苷酸在给予减毒痘病毒之前、之后或同时给予。痘病毒与编码下列基因产物之一的载体一起递送可对靶组织产生联合的抗癌效应。另外,痘病毒可改造成含治疗性多核苷酸的病毒载体。本发明包含多种蛋白质,其中一些将在下文描述。表7列举了本发明所涉及的可作为基因治疗靶位的各种基因。
细胞增殖诱导因子
诱导细胞增殖的蛋白质根据功能的不同可分为不同的类型。这些蛋白质的一个共同点就是其调节细胞增殖的能力。例如,一种形式的PDGF,sis癌基因,就是一种分泌型的生长因子。癌基因很少来源于编码生长因子的基因,到目前为止,sis是唯一已知的天然癌基因生长因子。在本发明的一个实施方案中,一种特殊的细胞增殖诱导因子的反义mRNA被用于阻止该细胞增殖诱导因子的表达。
蛋白质FMS、ErbA、ErbB和neu是生长因子受体。这些受体的突变可导致其调节功能的丧失。例如,Neu受体蛋白跨膜区的一个点突变可导致neu癌基因的产生。ErbA癌基因来源于甲状腺激素的胞内受体。被修饰的癌基因ErbA受体被认为可与内源性的甲状腺激素受体竞争,引起增殖失控。
最大的一类癌基因包括信号转导蛋白(如Src、Abl和Ras)。Src蛋白是胞浆蛋白酪氨酸激酶,在某些情况下它可以从原癌基因转化成癌基因,这是其酪氨酸残基527突变的结果。相反,在一个例子中,GTP酶蛋白ras从原癌基因转化成癌基因是其序列上的12位氨基酸从缬氨酸转化成甘氨酸的结果,这种突变可导致ras的GTP酶活性下降。
蛋白Jun、Fos和Myc可作为转录因子对核功能直接产生影响。
细胞增殖的抑制因子
肿瘤抑制癌基因的功能是抑制过度的细胞增殖。这些基因的灭活可使其抑制活性丧失,结果导致增殖失控。下面将要描述肿瘤抑制因子p53、p16和C-CAM。
除了上面所描述的p53之外,另外一个细胞增殖抑制因子是p16。真核细胞周期的转换主要被细胞周期素依赖的激酶或CDK激发。一种CDK,细胞周期素依赖的激酶4(CDK4)可调节细胞通过G1期。这个酶的活性可在G1晚期使Rb磷酸化。CDK4的活性受活化亚单位-D型周期素和抑制亚单位-p16INK4的调节,这两个亚单位是能够特异性结合并抑制CDK的蛋白质,因此可以调节Rb的磷酸化(Serrano等,1993;Serrano等,1995)。由于p16INK4蛋白是CDK4的抑制因子(Serrano,1993),因此这个基因的缺失可使CDK的活性升高,导致Rb蛋白的过度磷酸化。已知p16还可以调节CDK6的功能。
p16INK4属于新发现的一类CDK-蛋白,该类CDK蛋白还包括p16B、p19、p21、WAF1和p27KIP1。p16INK4基因位于染色体上的9p21区,这是一个多种类型的肿瘤经常发生缺失的染色体区。p16INK4基因在人源肿瘤细胞系中常常发生纯合的缺失和突变。这些现象说明p16INK4基因是一种肿瘤抑制基因。但是这种解释受到了挑战,因为已经观察到p16INK4基因在原发的未经培养的肿瘤中发生突变的几率比培养的细胞要低的多(Caldas等,1994;Cheng等,1994;Hussussian等,1994;Kamb等,1994;Kamb等,1994;Mori等,1994;Okamoto等,1994;Nobori等,1994;Orlow等,1994;Arap等,1995)。通过转染质粒表达载体恢复野生型p16INK4的功能可降低某些人源癌细胞系的克隆形成能力(Okamoto,1994;Arap,1995)。
可用于本发明的其他基因包括Rb、APC、DCC、NF-1、NF-2、WT-1、MEN-I、MEN-II、zac1、p73、VHL、MMAC1/PTEN、DBCCR-1、FCC、rsk-3、p27、p27/p16融合蛋白、p21/p27融合蛋白、抗血栓基因(如COX-1、TFPI)、PGS、Dp、E2F、ras、myc、neu、raf、erb、fms、trk、ret、gsp、hst、abl、E1A、p300、参与血管形成的基因(如VEGF、FGF、血小板凝血酶敏感蛋白、BAI-1、GDAIF或其受体)和MCC。
细胞调亡调节因子
调亡或程序性细胞死亡是正常胚胎发育、维持成年组织内环境稳定、以及抑制肿瘤形成的一个基本过程(Kerr等,1972)。Bcl-2家族的蛋白和ICE样蛋白酶已被证明在其他系统中是重要的调节因子和调亡效应因子。Bcl-2蛋白是一种与滤泡淋巴瘤有关的蛋白,在受到不同调亡刺激信号后在控制调亡和增加细胞的存活中扮演主要角色(Bakhshi等,1985;Cleary和Sklar,1985;Cleary等,1986;Tsujimoto等,1985;Tsujimoto和Croce,1986)。现在已经认识到进化保守的Bcl-2蛋白是相关蛋白家族的一个成员,这个家族可被分类为死亡激动剂或死亡拮抗剂。
Bcl-2被发现以后被证明可抑制各种刺激信号所激发的细胞死亡。现在已经清楚Bcl-2细胞死亡调节蛋白家族具有相似的结构和序列同源性。这个家族的不同成员或者具有与Bcl-2相同的功能(如BclXL、BclW、BclS、Mcl-1、A1、Bfl-1),或者与Bcl-2的功能相反,可促进细胞的死亡(如Bax、Bak、Bik、Bim、Bid、Bad、Haraliri)。
手术
大约60%的癌症患者都做过不同类型的手术,其中包括为了预防、诊断或分期、治愈和缓解的目的而进行的手术。以治愈为目的的手术可结合其他治疗措施,如本发明的治疗措施、化疗、放疗、激素治疗、基因治疗、免疫治疗和/或其他治疗方法。
以治愈为目的的手术包括切除肿瘤,其中癌组织的全部或部分被物理去除、切掉和/或毁坏。肿瘤切除是指通过物理方法至少去除肿瘤的一部分。除了切除肿瘤以外,手术治疗措施还包括激光手术、冷冻手术、电手术和显微手术(Mohs手术)。本发明还可用于配合浅表癌、癌前病变或少量正常组织的去除。
全部或部分癌细胞、癌组织或肿瘤被切除以后,在体内可能会形成空腔。本发明的治疗措施可通过灌注、直接注射或局部使用来实施,并配合以其他的抗癌治疗措施。这种治疗措施可重复进行,如每1、2、3、4、5、6或7天、每1、2、3、4和5周、每1、2、3、4、5、6、7、8、9、10、11或12月进行一次。这种治疗措施还可以选择不同剂量。
其他药剂
其他药剂也可与本发明联合使用以改善治疗的效果。这些其他药剂包括免疫调节剂、影响细胞表面受体和GAP连接表达上调的药剂、细胞增殖抑制和分化剂、细胞粘附抑制剂、增加过度增殖细胞对调亡诱导因子敏感性的药剂以及其他生物剂。
免疫调节剂包括肿瘤坏死因子;干扰素α、β和γ;IL-2及其他细胞因子;F42K及其他细胞因子类似物;或MIP-1、MIP-1β、MCP-1、RANTES及其他趋化因子。细胞表面受体或其配基如Fas/Fas配基、DR4或DR5/TRAIL(Apo-2配基)的表达上调可通过其对过度增殖细胞的自分泌或旁分泌效应提高本发明的调亡诱导能力。提高增加GAP连接的数目增强细胞内的信号水平可提高对临近过度增殖细胞群的抗过度增殖效应。在其他的实施方案中,细胞增殖抑制和分化因子可与本发明联合使用以改善本发明的抗过度增殖效应。细胞粘附抑制因子也可以提高本发明的疗效。细胞粘附抑制因子的例子有病灶粘附激酶(FAK)抑制因子和洛伐他汀。本发明还包括能提高过度增殖细胞对调亡的敏感性的其他因子,如抗体c225,这些因子也可与本发明联合以改善治疗的效果。
Apo2配基(Apo2L,也叫TRAIL)是肿瘤坏死因子(TNF)细胞因子家族的一个成员。TRAIL可激活多种类型的癌细胞发生快速调亡,但是对正常细胞没有毒性。各种组织中都有TRAIL mRNA。大多数正常细胞对TRAIL的细胞毒活性耐受,说明存在保护细胞不发生TRAIL诱导的调亡的机制。TRAIL的第一个被发现的受体被称为死亡受体4(DR4),含有胞浆“死亡域”;DR4可转导TRAIL携带的调亡信号。其他可结合TRAIL的受体也被鉴定出来。其中一个受体被称为DR5,含有一个与DR4非常相似的死亡域和调亡信号。
DR4和DR5mRNA在多种正常组织和肿瘤细胞系内都有表达。最近,诱骗受体如DcR1和DcR2被证明可通过DR4和DR5阻止TRAIL诱导的调亡。因此,这些诱骗受体代表了一种新型的对前调亡细胞因子在细胞表面的直接调节敏感性的机制。这些抑制性受体在正常组织内优先表达说明TRAIL可作为抑制抗癌因子诱导癌细胞的调亡,而对正常细胞起保护作用(Marsters等,1999)。
利用细胞毒性化疗药物进行肿瘤的治疗取得了很多成功,但是,化疗的后果之一就是药物耐药表型的出现/获得以及多药耐药现象的出现。耐药现象的出现依然是治疗这类肿瘤所遇到的主要问题,因此需要采用其他的方法,如基因治疗。
可与化疗、放疗或生物治疗联合应用的其他治疗措施包括温热疗法,该方法是将患者的组织置于高温下(可达106°F)。外部或内部发热装置可用于局部、区域或全身温热治疗。局部温热治疗是指只对一个小的区域加热,如肿瘤部位。外部加热通常是通过体外的装置发射高频微波靶向到肿瘤。内部加热是利用一个无菌的探针,如一个细的热金属丝或充满热水的中空细管、植入的微波天线或射频电极。
患者的器官或四肢可被加热以进行区域治疗,该疗法利用能产生高能量的装置进行,如磁体。另外,可以抽出患者的一部分血液,加热后再输注到能内部加热的区域。全身温热疗法在癌扩散到全身的情况下也可以使用。热水套、热蜡、感应线圈和热房可用于此目的。
激素疗法也可与本发明或上述其他癌症治疗措施联合使用。激素可被用于降低某些激素的表达水平或阻断某些激素的效应以治疗某些类型的肿瘤,如乳腺癌、前列腺癌、卵巢癌或宫颈癌。这种疗法通常与至少一种其他治疗措施联合使用,作为辅助治疗措施或降低转移的风险。表-US-00007表6癌基因来源人类疾病功能生长因子HST/KS转染FGF家族成员INT-2MMTV启动子FGF家族插入成员INTI/WNTI MMTV启动子因子样插入SIS猿肉瘤PDGF B病毒受体酪氨酸激酶ERBB/HER禽类扩增的,EGF/TGF-α/红-删除的鳞癌双调蛋白/囊胚(blastosis)细胞癌;β细胞素病毒(Hetacellulin virus);ALV胶质瘤受体启动子插入;扩增的人肿瘤ERBB-2/NEU/转染扩增乳腺,受到来自大鼠卵巢HER-2的调控,胃NDF/成胶质细胞瘤癌症调蛋白和EGF-相关因子FMS SM猫科CSF-1受体肉瘤病毒KIT HZ猫科MGF/Steel肉瘤病毒受体造血TRK转染NGF(来自人生长因子的神经)结肠癌受体MET转染分散因子/来自人HGF受体骨肉瘤RET转位零星甲状腺孤儿受体和点癌症;家族性Tyr突变骨髓甲状腺激酶癌症;多发性内分泌肿瘤2A和2B ROS URII禽类孤儿受体肉瘤病毒Tyr激酶PDGF受体转位慢性TEL(ETS样骨髓单核细胞转录白血病因子)/PDGF受体基因融合。
V.实施例
以下实施例用于展示本发明的多种实施方式,并无意以任何方式限制本发明。本领域的熟练技术人员应该知道,可改造本发明以实现目的并获得提及的结果和优势,以及本文所述的目的、结果和优势。实施例以及本文所述方法代表当前的优选实施方式,是示例性的,无意于限制本发明的范围。本领域熟练技术人员可对它们及其用途在权利要求书范围所限定的本发明构思范围内进行更改。
实施例1
肝癌的治疗
A.目的
(1)测定JX-594瘤内(IT)注射给药的最大耐受剂量(MTD)和/或最大可行剂量(MFD),(2)评价JX-594I.T.注射给药的安全性,(3)评价JX-594I.T.注射给药的复制/药代动力学,(4)评价I.T.注射给药后,对JX-594和肿瘤相关抗原的免疫反应(注射和非注射部位炎症浸润增加;中和抗体的形成;细胞因子反应;以及肿瘤和病毒特异性T淋巴细胞的诱导),(5)评价JX-594I.T.给药在注射和非注射部位的抗肿瘤效果。
B.研究设计
这是一个I期开放式剂量递增试验,对象是具有影像引导下可注射的浅表肿瘤结节的肝癌患者。难治性肿瘤患者接受下列剂量递增设计中的一种进行治疗:组1:1x 108pfu,组2:3x 108pfu,组3:1x 109pfu,组4:3x 109pfu。
该试验的目标治疗期是15个月。编入的患者每周期接受1次治疗。如果患者接受治疗时没有剂量限制性毒性(DLT),并且目标肿瘤没有进展,该患者将进入另一周期直至总数为4个周期。如果患者的目标肿瘤进展,或者因为DLT或其它原因退出,该患者将进行研究结束访问(End of Study Visit),并进入随访期。三周为一个周期。仅在第一个周期中观察DLT。
一次给药剂量可分布在三处病灶。注射病灶最大直径的总和必须小于10cm。每一剂量水平将对三个患者进行治疗直至观察到DLT。3名编入患者(enrollment)中若没有发生DLT,将进入到下一剂量水平;若3名患者中有1人发生DLT,则需编入另1名患者,直至观察到第二例DLT发生(在此定义为毒性剂量)或直至总共6名患者均接受治疗。如果组中未出现第二例DLT,患者进入下一剂量水平。
若2名患者在某一剂量JX-594治疗后发生DLT,则此剂量紧接着的前一剂量确定为MTD。当未确定MTD时,最高剂量水平确定为MFD。当MTD/MFD确定时,对另外6名患者进行治疗以获得更多在此剂量水平下关于安全性和毒性的数据。如果组4中未发生MTD,而此前组的剂量中发生多于2/3的PR效果,则用此剂量进行另外6名病人的临床试验。
DLT定义为JX-594造成的以下任一情况:1.任一时期的4级毒性,2.3级毒性(不包括流感类似症状:疲劳、恶心、肌肉酸痛、发烧)持续>5天。使用美国国立癌症研究所常见毒性标准指定本研究中发生毒性的严重程度。
1.对照肿瘤(非注射肿瘤)(周期1)和JX-594注射(周期2+)的决定
研究者在周期1中决定对照肿瘤位点。对照肿瘤应该是清晰的肿瘤结节,位于目标肿瘤所在肝叶之外的肝叶并且应该在目标肿瘤的淋巴回流之外。因此,对照肿瘤应该独立位于肝脏左叶或右叶。然而,如果肿瘤结节位于肝脏一侧的限制内,对照肿瘤可为JX-594的非注射肿瘤结节;然而当肿瘤具有大范围的单一结节时,不可确立对照肿瘤。以与JX-594治疗肿瘤相同的方式评价对照肿瘤。这样可对肿瘤生长和局部毒性/活性的对照效应进行评价。
如果病人继续进行周期2,以周期1中目标肿瘤相同的JX-594剂量水平注射对照肿瘤。如上所述,应当基于肿瘤大小使剂量成比例分布于肿瘤中。
2.非目标(非注射)肿瘤响应者
研究中非注射肿瘤可发生响应;在之前此类患者的JX-594临床I期试验中已报道该现象。了解该效应的机理是必需的;可能性包括病毒从注射肿瘤中扩散和/或肿瘤特异性细胞毒的诱导(T淋巴细胞(CTL)的肿瘤浸润,以及后续的细胞毒性T淋巴细胞介导的肿瘤破坏)。为了更好的理解这种效应的机理,进行下列研究。如果非注射肿瘤发生临床响应,与注射肿瘤在同一采集时间点进行核心活检或细针穿刺(参见附录A;非目标肿瘤活检总共不超过两个位点)。利用与获自注射肿瘤材料所用相同的方法对非注射肿瘤样品进行分析。
C.病人选择
1.入选标准
通常,患者应当符合下列所有标准:(1)年龄大于18岁,(2)临床或组织学确认(原发或转移)的肝癌患者,具有影像引导下可注射的浅表肿瘤(最长直径≤10cm),进行标准疗法仍发生进展(即对标准疗法是难治性),(3)进行标准治疗如手术切除、动脉化疗栓塞术、化疗和放疗,肿瘤仍然进展,(4)卡氏行为状态(Karnofsky Performance Status)(KPS)≥70的患者,(5)预期生存期至少16周的患者,(6)如果是性活跃患者,患者愿意在JX-594治疗后3个月内使用避孕方法,(7)患者具有了解并愿意签署知情同意书的能力,(8)患者具有配合研究步骤以及随访检验的能力,(9)患者具有合适的骨髓功能:WBC>3,000细胞/mm3,ANC>1,500细胞/mm3,血色素>10g/dL,血小板计数>75,000细胞/mm3,(10)患者具有合适的肾功能:血清肌酸酐<1.5mg/dL,(11)病人具有合适的肝功能:血清AST(≤2.5ULN),ALT(≤2.5ULN),总胆红素(≤2.0mg/dL);对于原发性肺癌患者,其Child-Pugh分级应当为A或B。
2.排除标准
患者禁止出现任一下列排除标准:(1)怀孕或哺乳婴儿,(2)HIV患者,(3)Child-Pugh分级为C的患者;分级为A或B中患者总胆红素>2mg/dL(当为原发性肝癌时),(4)患者具有临床显著活动的感染或被认为对新实验性药物治疗具有高风险的未控制的医学状况(如呼吸、神经、心血管、胃肠道、泌尿生殖系统),(5)患者显著免疫缺陷,或家族成员具有潜在疾病和/或服药引起的症状,(6)患者具有需要全身治疗的湿疹的病史,(7)患者具有不稳定的心脏疾病,包括在编入本研究中之前6个月诊断患有需要药物治疗的不稳定心脏疾病,包括MI、不稳定心绞痛、充血性心力衰竭、心肌炎、心率失常,以及其它任何具有临床显著状况的心脏状态,(8)患者在本研究药物治疗前4周内接受全身性皮质类固醇或其它任何免疫抑制药物治疗,(9)患者在编入本研究中之前4周内接受任何其它研究性药物研究、放疗、化疗或手术,(10)患者能够但不愿出具书面知情同意书,(11)患者对本研究药物组分过敏。
D.研究访问步骤
观察和测试计划中展示了本研究步骤的汇总表。通常允许自计划日+1/-1天的窗口,周末和假期不计入。
1.筛选访问(第–14至0天)
这是一项使用病毒的临床研究,该研究在与患者讨论后进行。任何意于参加的患者必须提供书面知情同意书。在签署知情同意书后,每位患者在研究开始前14天内将进行下列评估:
临床评估包括(1)完整的用药和手术史,包括抗癌治疗,(2)体重和生命指征(体温、脉搏速率和血压),(3)体检(全身系统),(4)卡氏行为评分(Karnofsky Performance Score),(5)胸部x-射线(前后及双侧),(6)12-导联ECG(加入本研究前3个月内进行的均可接受),(7)伴随药物评估(加入本研究前14天内所有使用的药物)。
实验室评估包括(1)常规血液测试(包括血小板计数和分类计数),(2)血清化学;钠、钾、BUN、肌酸酐、ALT、AST、碱性磷酸酶、总胆红素、LDH、钙、磷、镁、随机血糖、总蛋白、清蛋白和尿酸,(3)凝血测试:凝血酶原时间(PT)、部分促凝血酶原激酶时间(PTT)和国际标准化比值(INR);纤维蛋白原,(4)HIV、HBV和甲胎蛋白测试,(5)中和抗体滴度,(6)病毒基因组(Q-PCR),(7)常规尿分析(包括显微镜检),(8)怀孕测试(对于有生育可能的妇女),(9)在筛选测试时根据肿瘤类型进行合适肿瘤标记物测试(CA125、CEA、AFP、PSA、CA19-9等),当增加时,在每一周期的第22天进行测试。
基于影像的肿瘤评估和测量包括腹部CT扫描测量肿瘤结节(最长直径测量);可换为第1天时CT扫描(治疗前)。(病人加入本研究前2周内完成均可接受)。
第1天(周期1-4)-应当注意的是在JX-594给药前或给药后进行哪些评估。
第1天;治疗前—临床评估:体检(全身系统),体重和生命指征(体温,脉搏速率和血压),卡氏行为评分,同时治疗的鉴定,目标肿瘤的测试和评估,目标肿瘤的测量(n=1-3);其它非注射肿瘤的测量,(n=1-3),目标肿瘤的活检。
实验室评估:血液--1.常规血液测试(包括血小板计数和分类计数,2.血清化学;钠、钾、BUN、肌酸酐、ALT、AST、碱性磷酸酶、总胆红素、LDH、钙、磷、镁、随机血糖、总蛋白、清蛋白和尿酸,3凝血测试:凝血酶原时间(PT)、部分促凝血酶原激酶时间(PTT)和国际标准化比值(INR)、纤维蛋白原,4.细胞因子(包括GM-CSF),5.中和抗体滴度,6.病毒基因组(Q-PCR)
实验室评估:其它--1.pfu尿测试,2.pfu咽拭子
研究药物给药-1.如第8章所述进行JX-594给药
第1天:治疗后--1.体检。6小时内每小时检测两次生命指征(30分钟和60分钟时),随后常规测量,2.在如下时间点抽血进行细胞因子分析:治疗后1小时和3小时,3.在如下时间点抽血测量循环JX-594基因组:开始给药后10-15分钟,25-35分钟和4-6小时,4.治疗后3-4小时测定尿液和咽拭子样本中的病毒脱落,5.记录副反应和并发症。
第3天(周期1-4)--实验室评估:血液--1.常规血液测试(包括血小板计数和分类计数,2.血清化学、钠、钾、BUN、肌酸酐、ALT、AST、碱性磷酸酶、总胆红素、LDH、钙、磷、镁、随机血糖、总蛋白、清蛋白和尿酸,3凝血测试:凝血酶原时间(PT)、部分促凝血酶原激酶时间(PTT)和国际标准化比值(INR)、纤维蛋白原,4.细胞因子(包括GM-CSF),5.中和抗体滴度,6.病毒基因组(Q-PCR)
实验室评估:其它--1.pfu尿测试,2.pfu咽拭子
临床评估-记录副反应和并发症。
基于影像的评估:临床评估怀疑副反应时进行腹部CT扫描。
第5天(周期1-4)--临床评估记录副反应和并发症。
实验室评估:血液--1.常规血液测试(包括血小板计数和分类计数,2.血清化学、钠、钾、BUN、肌酸酐、ALT、AST、碱性磷酸酶、总胆红素、LDH、钙、磷、镁、随机血糖、总蛋白、清蛋白和尿酸,3凝血测试:凝血酶原时间(PT)、部分促凝血酶原激酶时间(PTT)和国际标准化比值(INR)、纤维蛋白原,4.细胞因子(包括GM-CSF),5.中和抗体滴度,6.病毒基因组(Q-PCR)
第8天(周期1-4)--临床评估-体检,CT扫描;目标肿瘤活检(也对至多1-2个表现出显著变化,包括炎症、坏死或缩小等的非注射肿瘤进行活检)。仅在周期1和2由PI主观衡量病人状况进行活检。记录副反应和并发症。
实验室评估:血液--1.常规血液测试(包括血小板计数和分类计数,2.血清化学、钠、钾、BUN、肌酸酐、ALT、AST、碱性磷酸酶、总胆红素、LDH、钙、磷、镁、随机血糖、总蛋白、清蛋白和尿酸,3凝血测试:凝血酶原时间(PT)、部分促凝血酶原激酶时间(PTT)和国际标准化比值(INR)、纤维蛋白原,4.细胞因子(包括GM-CSF),5.中和抗体滴度,6.病毒基因组(Q-PCR)
实验室评估:其它--1.pfu尿测试,2.pfu咽拭子,3.当发生坏死时细针穿刺坏死(仅在周期1和2时进行)。
第15天(周期1–4)--临床评估-体检。
实验室评估:血液--1.常规血液测试(包括血小板计数和分类计数,2.血清化学、钠、钾、BUN、肌酸酐、ALT、AST、碱性磷酸酶、总胆红素、LDH、钙、磷、镁、随机血糖、总蛋白、清蛋白和尿酸,3凝血测试:凝血酶原时间(PT)、部分促凝血酶原激酶时间(PTT)和国际标准化比值(INR)、纤维蛋白原,4.细胞因子(包括GM-CSF),5.中和抗体滴度,6.病毒基因组(Q-PCR)
实验室评估:其它--1.pfu尿测试,2.pfu咽拭子
第22天(周期1-4)--临床评估1.体检,2.基于影像的评估:腹部CT扫描(仅在周期2和4时进行),3.目标肿瘤的测量(n=1-3);测量其它非注射肿瘤,4.目标肿瘤活检(也对至多1-2个表现出显著变化,包括炎症、坏死或缩小等的非注射肿瘤进行活检),5.记录副反应和并发症,6.第22天当作在后续周期前的第1天。在第22天和后续周期的第1天间可有至多1周的间隔。
实验室评估:血液-1.常规血液测试(包括血小板计数和分类计数,2.血清化学、钠、钾、BUN、肌酸酐、ALT、AST、碱性磷酸酶、总胆红素、LDH、钙、磷、镁、随机血糖、总蛋白、清蛋白和尿酸,3凝血测试:凝血酶原时间(PT)、部分促凝血酶原激酶时间(PTT)和国际标准化比值(INR)、纤维蛋白原,4.中和抗体滴度,5.病毒基因组(Q-PCR),6.在筛选测试时根据肿瘤类型进行合适肿瘤标记物测试(CA125、CEA、AFP、PSA、CA19-9等),当增加时,在每一周期的第22天进行测试。
实验室评估:其它--1.pfu尿测试,2.pfu咽拭子
第28天或研究结束访问-临床评估-体检,记录副作用和并发症。
实验室评估:血液--1.常规血液测试(包括血小板计数和分类计数,2.血清化学测试:钠、钾、BUN、肌酸酐、ALT、AST、碱性磷酸酶、总胆红素、LDH、钙、磷、镁、随机血糖、总蛋白、清蛋白和尿酸,3.凝血测试:凝血酶原时间(PT)、部分促凝血酶原激酶时间(PTT)和国际标准化比值(INR)、纤维蛋白原,4.病毒基因组(Q-PCR)
周期3-4--1.在周期2第22天注射位点肿瘤最长直径未显示>25%增加的病人将进入周期3-4,2.在周期2第22天注射位点肿瘤最长直径显示25-50%增加的病人可进入周期3-4,3.在周期2第22天注射位点肿瘤最长直径显示>50%增加的病人将退出试验。
病人的随访和回顾—在研究结束访问后,以常规跟踪肝癌病人的方式对完成该临床研究的病人进行为期1年的随访跟踪。不论哪种临床研究,每三个月病人返回医院并进行检测,对存活的病人进行常规测试如肝癌血清测试和基于影像的评估。临床研究结束后,若确定有明显的临床受益,在签订另外的书面知情同意书后,再进行总数多至4次的给药。这时所有研究步骤与本研究最初4次给药方式相同。到周期4研究结束后,直到PI判断有显著的临床受益(不止是病情稳定),另外给予本研究药物共计4次。这种情况下,PI应事先与试验发起人讨论并取得发起人的同意。所有的研究计划与临床研究进行方式相同。
E.病毒复制、传播和特殊测试
1.血浆和尿液的Q-PCR和噬斑形成单位实验(药代动力学测试)
用定量聚合酶链反应(Q-PCR)测试评估传播到血流中的病毒。为了检测病毒是否在尿液和咽拭子中出现,在治疗后收集样品。
2.肿瘤活检和细针穿刺(免疫应答测试)
为了检查肿瘤位点病毒的复制,在治疗前后进行核心活检和细针穿刺(如果认为是安全且简便的)。分析这些活检样本,找到病毒复制、炎症和免疫细胞浸润、坏死和凋亡的证据。
为了获得组织,在影像引导下,使用核心活检或者使用细针穿刺活检。但是,这些活检有时会使病人处于紧急或危险的状况。因此当进行活检获取组织时,首先应关注病人的安全。当病人情况很有可能处于危险时(肝囊肿瘤等),应该通过安全的途径获取组织。
如果PI判断组织活检(细针穿刺)有可能引起病人的危险,则不进行活检(细针穿刺。此外,若病人安全需要,根据PI的判断,在进行组织活检(细针穿刺)和/或JX-594瘤内注射前后多至5天,病人可入院观察。
3.细胞因子分析(免疫应答测试)
用ELISA方法测定血清中GM-CSF、IL-1、IL-4、IL-6、IL-10、IFN-δ和TNF-α的浓度。
4.中和抗体实验(药代动力学测试)
用噬斑试验测定病人连续稀释血清中JX-594中和抗体滴度的出现。
5.药代动力学采血
药代动力学使用小黄头真空取血管采集3ml血液。
F.研究用药的给药
1.剂量、给药和治疗计划
剂量。剂量通常如下:组1:1x 108pfu、组2:3x 108pfu、组3:1x 109pfu、组4:3x 109pfu。
给药。JX-594可通过瘤内注射进行给药。由专家医生通过所述方式进行瘤内注射给药。使用21号或更小的针头,将相当于约25%肿瘤总体积(1-3个肿瘤)的含病毒的溶液直接注入瘤内。通常,注射在影像(如CT)的引导下进行。可注射1-3个肿瘤。每一肿瘤注射相等量的溶液。如果注射2-3个肿瘤,注入瘤内病毒溶液的体积与该肿瘤相对其它肿瘤的体积成正比(即如果一个肿瘤是另一肿瘤的2倍,则大肿瘤将注射2/3体积的病毒溶液)。
尽管周期1中所选目标肿瘤可能停止生长,但应当在所有周期中继续注射。然而,如果必要,在周期3中,研究者还可选择在周期1、2到3中未注射的非目标肿瘤,包括周期1中的目标肿瘤。注射肿瘤最大直径的总和必须≤10cm。瘤内注射病毒溶液的剂量与肿瘤体积成正比。
JX-594制备。JX-594以冷冻(-60℃或更低)的、含150μl病毒制剂(递送0.1mL)的一次性玻璃瓶的形式供应。100μl体积含有1.9x 108pfu病毒。在室温下竖直解冻小瓶。不要将JX-594放置于热水浴中。用移液管重悬。当稀释并运送至病人时,将病毒存储于4℃。解冻JX-5944小时后不可注射。
高级药师和其它指定药师应小心将JX-594竖直储存于生物安全柜中(2级)(使用手套、安全眼镜和防护衣)。所有稀释的起始步骤:使用注射器,取出需要量的无菌生理盐水溶液并转移至标准福尔肯(falcon)管中。病毒加上稀释液的终体积应当约等于目标肿瘤体积的25%。
组1:将一(1)小瓶JX-594用于组1中的病人。用微量移液器/注射器移取处方规定体积的转移至无菌生理盐水溶液的JX-594。
组2:将二(2)小瓶JX-594用于组2中的病人。混合后将第一个小瓶的内容物转至第二个小瓶中。用微量移液器/注射器移取处方规定体积的转移至无菌生理盐水溶液的JX-594。
组3和4:将四(4)或十一(11)小瓶JX-594分别用于组3或4中病人的给药。将所有的内容物转移到混和的小聚丙烯管中。用微量移液器/注射器移取处方规定体积的转移至无菌生理盐水溶液的JX-594。
所有稀释的最终步骤:于室温下将小管用铝箔包裹或将其置于避光袋中。给药前剧烈振荡10秒。在室温下暴露30分钟或解冻后4小时后不可进行注射。
治疗计划。通常编入病人每个周期接受1次治疗或1个剂量的JX-594。在周期结束时,JX-594注射目标肿瘤体积未进展的病人将接受后续周期的治疗(直到总共4个周期)。目标肿瘤进展的病人终止随访。一个周期定义为3周。可在1-3个病灶中平分剂量。注射病灶最大直径的总和必须≤10cm。
剂量递增。在临床研究的剂量递增期,每一组有2-6名病人。如果最初3名病人未经历DLT,研究进入下一组。如果一个组中3名病人有一名发生DLT,继续进行研究直到总共编入6名病人,或者包含第一名病人的两名病人经历DLT。
如果组1中6名病人中少于2名在首次注射后直至两周经历DLT,研究进入下一组。如果2名患者经历DLT,则此剂量紧接着的前一剂量确定为MTD。
在周期1中第1名病人首次注射后1周,编入第2名病人;编入下一名病人时应用该原则。如果一个组中发生DLT,在对所有原来编入病人完成周期1中第1次注射后2周,开始后续编入病人的治疗。在原来组的最后一名病人完成周期1中第1次注射后至少2周,病人进入下一剂量水平的组。
如果组1中超过2名病人经历DLT,终止临床研究。
G.安全性
治疗后可发生全身性副作用。发热、寒冷、肌肉痛、疲劳/无力、恶心和呕吐。注射肿瘤位点发生副作用如疼痛、坏死、溃疡和炎症。根据临床前研究和GM-CSF临床研究,可发生淋巴细胞、单核细胞和白细胞的短时间增加并伴随噬中性粒细胞的增加。在注射肿瘤位点可发生如下情况:疼痛、坏死、溃疡和炎症。
尽管在之前JX-594的I期试验中未描述散发性疫苗相关皮疹或脑炎,并且是可能性极小的,但仍理论上可能发生;这些并发症分别在约10,000名疫苗接种者中有1名和1,000,000名疫苗接种者中有1名。
剂量限制性毒性(DLT)
DLT定义为JX-594引起的任一3级或更严重的毒性,不包括流感类似症状(如疲劳、恶心或肌肉痛),持续时间超过5天或4级毒性持续任意时间。
确保病人安全远离操作风险(Security of Safety for Patients from Risk of Procedure)。活检可引起并发症,如腹膜内出血和/或肿瘤破裂引起的休克。尽管报道并发症的发生率<0.1%并可通过经导管动脉栓塞治疗术治愈,但首先应当考虑病人的安全。因此,如果治疗医师判断活检有可能引起病人的危险,可不进行活检。此外,若病人安全需要,根据PI的判断,在进行组织活检(细针穿刺)和/或JX-594瘤内注射前后至多5天,病人可入院观察。
H.效果
本研究的主要目的是临床I期安全性研究,并非为了临床受益。不过仍希望由于直接的病毒效应(即溶瘤效应)引起注射和/或非注射肿瘤缩小和/或发生治疗诱导的免疫介导的肿瘤破坏。
效果评估的标准是目标病灶的变化。如果非目标病灶发生任何变化,根据下表基于目标病灶的反应对其进行评估。
目标病灶的评价。完全反应(CR):所有目标病灶消失。部分反应(PR):目标病灶的总LD比作为对照的基线总LD减少至少30%。疾病进展(PD):目标病灶的总LD比作为对照的自开始治疗以来记录的最小总LD增加至少20%。疾病稳定(SD):与作为对照的自开始治疗以来记录的最小总LD相比,既没有足够的缩小符合PR,也没有足够增加符合PD。
下表列出了对于总体反应的评价标准。最好总体反应指从治疗开始点到疾病进展/复发所记录的最好的反应。
表2最好总体反应的评价
注意:对于健康状况整体恶化需要停止治疗但当时没有疾病进展的客观证据的病人应当归为“症状恶化”。应当尽全力检测客观疾病进展,即便是在治疗停止后。
在一些情况下,很难区分残余病灶和正常组织。当基于这些判断进行完全反应的评价时,推荐在确认完全反应状态前对残留病灶进行研究(细针穿刺/活检)。
可测量病灶的评价原则。所有的测量在周期2的最后一天(第22天)或周期4的最后一天(第22天)通过CT或MRI进行,使用尺或卡尺以度量符号记录。所有的基线测量应在尽可能接近治疗开始时进行,不可超过治疗开始前4周。
注意:之前照射过的病灶不可作为测量病灶。如果根据研究者的判断认为这些病灶可作为测量病灶,试验方案应描述考虑这些病灶的条件。还应当注意位于之前照射过区域的肿瘤病灶也被认为是不适合作为测量病灶的。如果研究者认为该病灶适合测量,则试验方案中必须明确考虑这类病灶的条件。
在基线(测定)和跟踪过程中,对于每一鉴定的和报道的病灶应使用相同的评估方法和技术进行表征。当使用两种方法评估治疗的抗肿瘤效果时,临床检查的评价优选基于影像的评价。
使用相邻切片进行常规CT,切片厚度应当为10mm或更低。操作螺旋CT时使用5mm相邻重建算法。如果可用的话,在筛选随访中使用PET-CT,在本文中,在周期2的第22天使用PET-CT。需要的话在周期4的第22天重复PET-CT。
测量确认/反应持续时间。确认:为了指定PR或CR状态,必须通过重复评价确认肿瘤测量值的变化,重复评价在首次复合反应标准的8周后进行。当为SD时,在进入研究后,最小16周间隔的随访测量必须至少有一次满足SD标准。
反应持续时间:总体反应持续时间定义为从首次记录CR或PR的日期(以先记录者为准)到最早客观确认疾病复发或进展日期(作为疾病进展参考的是自治疗开始后最小的测量记录)的时间。整体完全反应的持续时间定义为从首次记录CR的日期到最早客观确认复发的日期的时间。
疾病稳定的持续时间:SD定义为从治疗后首次记录SD的日期到最早客观确认PD的日期的时间(作为疾病进展参考的是自治疗开始后最小的测量记录)。
肿瘤反应的重新评估。需要的话本研究可由独立放射科医生进行肿瘤反应的评估。然而,这些评估结果仅用作研究目的而不影响临床结论。
I.统计方法和数据分析
1.样本量
估计样本量为18个病人,可能的范围是2-30个病人。本研究的主要目的是瘤内注射JX-594的安全性、MTD或MFD。本研究代表JX-594的第二次人体临床试验。因为之前没有基于有意义的统计学计算的临床试验,本研究样本量的选择基于临床安全性的考虑。本研究的结果可用于为将来的临床研究提供决定样本量变化的估计。
每一组中的病人有一次在到达真实MTD前停止研究的机会以及一次超过真实MTD继续进行的机会。下表显示了基于真实DLT发生率的每种后果的统计学可能性。下表代表了组中首先3个病人的每种后果的概率(每一病人群体给定不同真实发生率)。
表3
下表显示了6名病人的组中每种后果的概率。在最初3名病人中观察到1例DLT并在组中加入另外3名病人之后,代表在给定病人群体中各种真实发生率。
表4
*一名病人来自最初3名病人,一名来自第二次的3名病人。
2.统计方法/数据分析
概述群体为意向治疗(ITT)群体,定义为所有病人接受至少一次JX-594治疗。此外,可评估病人也评价为ITT群体的子集。可评估病人指那些接受了至少1个周期的治疗,在治疗前后合适时间阶段进行合适肿瘤测量的病人。
本研究分4个治疗组进行,根据组不同,每一组有2-6名病人。用合适的描述统计学、频率表格、图和数据表概述每一组的数据。将来自治疗组的数据合并用于挑选的数据显示。如下文所述产生特定数据显示。
对象年龄、体重和身高以描述统计学概述(平均值、中位数、标准差、最小值和最大值),性别和种族以频率表格概述。治疗组的数据以每一病人单独方式以及组合方式进行概述。为此产生单个病人列表。体检(Physical medical)历史数据以每一治疗组独立分开,并组合以频率表格进行概述。以描述统计学方式概述治疗给药(平均值、中位数、标准差、最小值和最大值)。任一接受研究药物的病人均纳入安全性分析。在每一治疗组终止时分别概述包括不良事件、实验室结果、毒性、生命指征和退出时间的安全性数据。用COSTART身体系统分类方案对AE进行编码并列表。产生AE对象的数量和比例以治疗组和治疗目的进行列表;此外,根据AE严重性和研究者指明的与JX-594的关系对数据进行分级(stratify)。
在终止时用显示治疗前后发生变化病人数目的移动表格对实验室结果进行概述。所选变量的实验室结果以图像显示。
除了整体肿瘤反应率,还报告目标和非目标位点的肿瘤反应率。报告目标和非目标位点的时间-肿瘤进展,还报告总体存活情况。因为本研究为自由非随机研究,每一组中病人数目很少,因此不单独对于来自本研究的测试数据做出假设。为了评价治疗组间的差异,可使用合适的参数或非参数方法对每一组进行比较。
实施例2
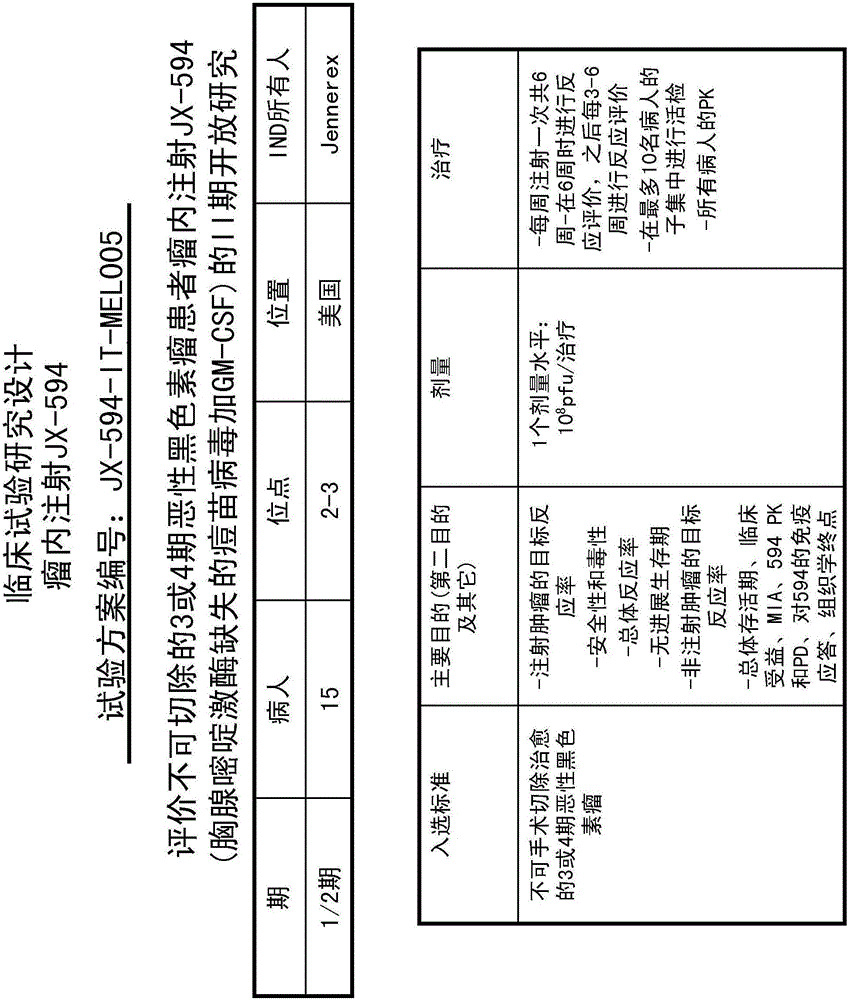
不可切除的恶性黑色素瘤的治疗
A.剂量和计划
1.剂量和计划的原理
每一治疗给予总剂量1x 108pfu。该剂量低于最高每周剂量1.6x 108pfu,这一剂量在首次JX-594治疗手术无法治愈的皮下黑色素瘤的I期临床试验中给药是安全的(Mastrangelo等,1998)。此外,1x 108pfu比正在进行的JX-594I期瘤内(IT)试验至今所知的安全给药的最高剂量(n=2)低10倍,比至今所知的最高剂量低3倍。在那次试验中,每3周进行一次给药,对1-3个肝脏肿瘤进行IT注射。该研究的的初步结果表明在JX-594治疗4天后(即第5天)流感类似症状和血液学参数恢复到基线水平。
因为在前述正在进行的肝脏IT研究中所有组的病人在第5天均从轻度到中度治疗相关毒性中恢复,所以选用每周1次的给药方案。此外,来自Mastrangelo等,1998的数据表明每周两次每次治疗高至8x 107pfu是安全有效的。
最初I/II期黑色素瘤研究(Mastrangelo等,1998)的证据表明,发现病人在再次接种后14-21天内,对痘苗病毒产生显著的体液免疫应答。发现尽管持续治疗但抗体滴度在暴露4-6周达到平台期。因此,在产生高滴度抗体和T细胞之前,本方案研究每周一次IT给药6周以产生最大的可能递送和JX-594的抗肿瘤效果。
2.研究原理
因为注射可到达疾病的比率相对高,且可见黑色素瘤中对于IL-2免疫疗法的阳性反应,而对于转移性黑色素瘤患者尚缺少有效可耐受的治疗方法,因此黑色素瘤是JX-594免疫疗法的理想靶点。此外,预计JX-594复制靶向黑色素细胞中高表达的EGFR通路。
最初I/II研究结果表明,瘤内注射JX-594能够安全有效治疗手术无法治愈转移性黑色素瘤病人的注射的和远端的疾病。显示了注射肿瘤(7名病人中的5名)和至少1个非注射肿瘤(7名病人中的4名)的反应,包括两名病人对JX-594治疗达到部分反应(6+个月)和一名病人完全反应(4+个月)。尤其值得注意的是,尽管所有病人都在治疗前接种过疫苗(因此预先存在抗牛痘免疫性),但仍产生疗效和基因的表达。
选择本研究设计为了扩展上述最初的I/II期研究,并在多至15名可评价的3期或4期不可切除的转移性黑色素瘤病人上评价注射肿瘤反应。此外,评价JX-594的安全性、药代动力学、药效学和对JX-594的免疫应答以及血液和肿瘤组织中GM-CSF转基因的表达。研究者也评价JX-594是否能够静脉内扩散并感染非注射局部和远端疾病,表明它能够产生与那些经历直接瘤内注射位点类似的抗肿瘤效果。这项发现除了对IT JX-594给药的整体临床经验做出补充外,也强烈支持IV JX-594用于治疗晚期/转移性疾病,尤其是用于晚期恶性黑色素瘤的治疗。
B.研究用产品描述
JX-594是靶向癌症,选择性复制的痘苗病毒,来源于常用的惠氏疫苗株(惠氏实验室)(Wyeth laboratories)。该病毒来源于一种胸腺嘧啶激酶(TK)失活的疫苗株。JX-594包含一种参与免疫应答的有效细胞因子hGM-CSF的基因和启动子。进一步改造JX-594插入lacZ基因以追踪组织中的病毒。
C.目的
目的包括评价(a)注射肿瘤的目标反应率,(b)JX-594IT注射给药的安全性和毒性,(c)JX-594IT注射给药后整体疾病负荷的目标反应率(RECIST标准),(d)无进展生存(PFS)时间,以及(e)非注射肿瘤反应率。
D.研究设计
1.研究概述
这是一项在不可切除的3或4期恶性黑色素瘤病人中开展的临床I/II期开放试验。病人在6周内接受总共六(6)次JX-594瘤内给予。每次治疗给予总剂量为1x 108噬斑形成单位,并均分在至多5个肿瘤中。如果病人在完成6次治疗后经历对于JX-594IT治疗的部分给予肿瘤反应,则可另外给予3次每周1次的治疗。
2.研究终点
临床研究的主要终点是注射肿瘤的反应率,包括完全反应率、部分反应率和反应持续时间。此类研究的第二个终点包括安全性,由治疗相关不良事件、严重不良事件(SAE)和与常规实验室参数中基线相比临床显著变化决定,常规实验室参数包括完全反应率、部分反应率、反应持续时间、无进展生存(PFS)、非注射肿瘤反应率,包括完全反应率、部分反应率和反应持续时间。其它终点可包括总体生存、临床受益(包括体重增加和行为状态的改善)、JX-594评价(如血浆和/或全血中病毒基因组(Q-PCR);血浆和/或全血中病毒感染性病毒,任选(噬斑实验)),免疫学评价(血清中JX-594中和抗体;血清中GM-CSF测量(ELISA实验)),组织学评价(组织中病毒基因表达;GM-CSF表达;lac-Z表达;炎症细胞浸润;坏死;凋亡;胞质中病毒复制工厂;EGFR通路状态;以及肿瘤胸腺嘧啶激酶状态)。
3.剂量
通常,如本文所述在无菌生理盐水中稀释病毒。每次治疗给予总剂量为1x108噬斑形成单位,并均分在至多5个肿瘤中。
4.总体研究持续时间和随访
研究期通常包括用于筛选的病人访问、研究治疗和治疗后随访评价。
筛选。在JX-594首次治疗前14天内确定符合研究条件的病人。
治疗。在6周内给予符合条件病人总共6次治疗,每周进行一次瘤内注射(第1、8、15、22、29和36天),给药剂量1x 108pfu。再次治疗前病人必须继续符合所有的合格标准。因为任何原因遗漏一次治疗,只要符合合格条件就在下一周给予遗漏的治疗,相应的调整访问计划,借此跟踪病人使病人总共接受6次治疗。注射延迟累计最大为4周。延迟治疗的病人仍需完成6次治疗并在第6次治疗后1周对反应进行评价。在最后一次给药后一周(即第43天)启动反应评价.如果病人在完成6次治疗后出现对JX-594IT治疗的部分注射肿瘤反应,则可另外给予3次每周1次的治疗。
治疗后随访。所有病人在JX-594最后一次治疗后28天(即第64天)返回进行跟踪随访。治疗结束后6个月或直到病人在注射位点发生疾病进展,开始新的癌症治疗或者死亡。最后一次注射后每3周病人返回门诊进行体检测量肿瘤(可能的话)并评价反应。每6周,通过PE和/或CT/MRI评价反应。在6个月随访后,病人每3个月返回门诊进行肿瘤测量和反应评价(包括CT/MRI)直到注射位点疾病进展、死亡或直到开始新的癌症治疗。
基因治疗产物的长期随访。在注射位点疾病进展或开始新癌症治疗后,根据当前FDA指导原则,继续监测病人存活和基因治疗的潜在长期效应。如果病人不再返回门诊治疗或治疗后随访,可通过邮件或电话收集数据。
E.研究群体
1.入选标准
通常病人满足下列所有条件:组织学确认的3期或4期恶性黑色素瘤;至少有一个瘤块能被CT/MRI和/或体检测量,在超声引导下能够直接目测进行注射;预计生存期至少16周;癌症无法手术切除治愈;KPS评分≥70;年龄≥18岁;有生育能力的男性和女性在用JX-594治疗期间和最后一次治疗后三月内愿意进行公认的生育控制方法;理解并愿意签署伦理审查委员会(IRB)/独立伦理委员会(IEC)(Institutional Review Board(IRB)/Independent Ethics Committee(IEC))批准的书面知情同意书表格;能够配合研究步骤和随访检查;肝功能尚可(总胆红素≤2.0ULN;AST、ALT≤2.0ULN);骨髓功能尚可(WBC>3,500细胞/mm3并<50,000细胞/mm3;ANC>1,500细胞/mm3,血红蛋白>10g/dL;血小板计数>125,000血小板/mm3);可接受的凝血状态(INR<(ULN+10%));以及可接受的肾功能(血清肌氨酸酐<2.0mg/dL).
2.排除标准
通常,病人不应该符合下列任一排除标准:目标肿瘤粘附于和/或侵袭主要血管结构(如颈动脉);怀孕或哺乳期;已知HIV感染;JX-594首次给药前4周内全身使用过皮质类固醇或其它免疫抑制药物;被认为对于研究用新药治疗具有高风险的临床显著活动的感染或不可控的医学状况(如肺部、神经、心血管、胃肠道、泌尿生殖系统);潜在疾病和/或药物(如全身性使用皮质类固醇)引起的显著性免疫缺陷;一些阶段需要全身治疗的湿疹病史;临床显著的和/或快速蓄积的腹水、心包和/或胸膜渗出液(如需要引流进行症状控制);严重或不稳定的心脏疾病,包括但不限于,筛选前6个月内出现下列任一情况:心肌梗死、不稳定心绞痛、充血性心力衰竭、心肌炎、确诊并需要药物的心率失常,或其它心脏状态临床显著性变化;筛选前4周内目标肿瘤经过放疗、化疗、手术或研究性药物治疗(若为丝裂霉素C和亚硝基脲则为6周内);之前对天花疫苗有严重反应或副作用;不能或不愿意给出知情同意书或配合本方案所需步骤;其家庭联系人为怀孕或哺乳、儿童<5岁、一些阶段需要全身治疗的湿疹病史、或潜在疾病(如HIV)和/或药物(如全身性使用皮质类固醇)引起的显著性免疫缺陷的病人也被排除,除非在病人活动给药期间和最后一次研究给药3周后能够另有生活安排。
3.其它符合标准的考虑
合格标准的偏差。如果预计偏差不会影响病人的安全、研究的进行或对研究结果的解释,也可编入与上述入选/排除标准有微小偏差(如实验室结果在预先明确的值之外)的病人。需要研究发起者或发起者代表对编入有微小偏差的病人的书面同意。
4.病人编入步骤
一旦研究者进行筛选评估并确认病人合格,发起者检查筛选和合格信息并向研究者提供所编入的每一位病人的确认书。一旦确认编入,使用预定病人编号方案对病人指定一个识别号。病人识别号由研究编号、位置编号、病人编号和病人姓名首字母组成。
F.研究产品
JX-594由杰尼瑞斯生物治疗公司(Jennerex Biotherapeutics)提供。通常,JX-594制剂为液体且冻存于供一次使用的玻璃小瓶中。每一小瓶含有0.15mL。病毒溶液为无色到淡黄色的澄清到轻微乳白色溶液。JX-594浓度为1.9x 109pfu/mL。
JX-594被认为是生物安全2级(BSL-2)的感染性物质。BSL-2名称和相关指导原则应用于对人体或环境有中度潜在危害的试剂。其它BSL-2试剂的例子包括麻疹病毒、沙门菌和乙肝病毒。参考机构感染控制政策。
通常JX-594储存于受监控的、进入受限的安全冰箱中。JX-594应当在-60℃或以下储存于第二层包装内标示清楚的小瓶中,放置于门上具有生物危害标志(表明试剂的性质)的冰箱或房间内。冰箱在-65℃设置报警限制使其在温度升至-60℃之前反应。长期处于>-60℃时需要将受影响的材料隔离直到其滴度得以再次确认。
在研究地点提供工作表以及补充研究信息以保证正确处理和准备JX-594。参考并执行机构感染控制政策[生物安全2级(BSL-2)]进行准备、运输和处理病毒载体。所有时间必须佩戴手套、长袍和护目镜。在特派药学家/科学家的指导下,根据BSL-2处理指导原则,于药房/实验室的垂直生物安全柜(2级)中进行关于JX-594的所有工作。在使用前后用70%酒精擦拭安全柜。
解冻。将小瓶直立在室温下进行解冻。不应将JX-594置于热水浴中。一旦解冻,将小瓶置于15mL聚丙烯锥形离心管中(如康宁(Corning)或福尔肯(Falcon)),盖上管子,100x g离心2分钟。用镊子或类似工具将JX-594小瓶从试管中取出。在稀释和递送给病人前,病毒制剂必须储存于冰上或冷藏(2-8℃)。在解冻后4小时内进行注射。
制备。将小瓶离心后,用微量移液器温和重悬(建议使用调到100μL的200μL微量移液器)。注意不要在制剂内吹入气泡。通常准备约2.75mL病毒溶液(JX-594+盐水),分装于5只注射器中,每只0.5mL。使用微量移液器将2.64mL无菌生理盐水转移到合适大小的聚丙烯试管中(如5mL福尔肯试管)。从一(1)小管JX-594中吸取116μL JX-594并将其转移到含盐水的福尔肯试管中。重新盖上福尔肯试管的盖子,使试管避光(用铝箔或置于避光容器中),并将盖好的试管马上放置于2-8℃(冷藏或防于湿冰上)。
在给药前30分钟内,剧烈涡旋振荡10秒。涡旋后,吸取0.5mL病毒溶液(JX-594+盐水)到5只注射器的每一只中。盖上注射器并送交研究者注射。在病毒制剂解冻后4小时内开始注射。在稀释和递送给病人前,病毒制剂必须储存于冰上或冷藏(2-8℃)。
1. JX-594给药
在6周内以每周一次共六(6)次用JX-594进行瘤内注射给药。在第1、8、15、22和36天进行给药。每次治疗病人接受剂量为1x 108pfu,分别给予≤5个病灶。只有能通过经皮注射(如皮肤可触及的结节或淋巴节转移)或超声波(US)引导注射可到达的病灶才符合治疗要求。
研究者决定每一次治疗注射的病灶(肿瘤)。根据大小注射肿瘤;最大的病灶应在每次治疗时都进行注射。根据研究者的判断,治疗一个肿瘤可使用一只或多只注射器。
在进针位点无菌备皮后进行局部麻醉。注射使用18–22号针头。如上所述使注射针头进入肿瘤。由主要研究者或副研究者进行注射。
将完整注射器体积(0.5mL)注入每个肿瘤中由中央进针位点辐射出的4个等距针道中。例如,如下进行病毒注射操作:(1)将针头(18-22号)插入肿瘤中央部位,(2)将针头向肿瘤边缘扩展(到肿瘤边缘1-3mm),(3)注射约25%注射器体积(约0.125mL)后向中央进针位点抽回,(4)无需将针头完全从肿瘤中撤回,以90°间隔重复上述步骤,总共产生4个针道。
预计毒性。治疗后预计产生如下全身毒性:发热、寒冷、厌食、肌肉痛、疲劳/衰弱和/或头痛。预计中性粒细胞、淋巴细胞、血小板和血细胞比容会短时下降。通常血液学参数在第5天回到基线(通常经历2-3天)。仅在周期1中,首次注射后的前4天内可能出现白细胞增多。在组3的两名病人给药后5-8天报道总白细胞计数为24,000/μL和118,000/μL。预计嗜酸性粒细胞在治疗后也会升高,通常在8天内保持升高。预计在注射位点可能出现下列毒性:疼痛、坏死、溃疡和炎症。其它病毒复制位点(如远端肿瘤)可能发生疼痛、坏死、溃疡和炎症。
尽管在任何治疗或接触JX-594后不大可能或未观察到散发性疫苗相关皮疹或脑炎,但仍可能发生这些状况;这些并发症分别在约10,000名疫苗接种者中有1名和1,000,000名疫苗接种者中有1名。此外,在最近的使用NYCBOH痘苗病毒株接种的项目中,证实了心肌炎的风险有统计学显著升高(每10,000接种者中有1-2人)(Arness等2004)。
G.统计学
1.后果定义
关于统计学分析的结果定义如下。本方案其它部分讨论了毒性代码,以及疾病进展、完全反应、部分反应、总体反应持续时间、可评价病人和治疗相关的定义。
无进展生存期。从首次JX-594治疗到研究者诊断为进展的诊断日期,或到无进展死亡日期的时间。对于最后一次了解到存活且无进展的病人在其最后一次评价进展时进行删除。在统计分析中对在记载为疾病进展前接受非本方案治疗的病人进行删除。
总体生存期。从首次JX-594治疗到死亡日期或最后一次知道其存活的日期的时间;在统计分析中对最后一次知道其存活的病人进行截尾。
2.分析集合或群体
对所有接受JX-594的病人进行分析,筛选时分析其人口学特征,而后则分析安全性、有效性、药代动力学和药效学。概述群体为意向治疗(ITT)群体,定义为接受至少一次JX-594治疗的所有病人。此外还评估可评价病人群体(ITT群体的子集)。如果一个病人接受了至少一次JX-594治疗,并在基线和治疗后的第一合适时间点具有合适的肿瘤测量值,则认为该病人是可评价的。
3.分析方法
使用描述性统计概述连续变量(n、平均值、标准差、中位数、最小值和最大值)。对于分类变量则通过显示每一分类内病人的数目和百分率(n,%)进行概述。基于可评价病人以及意向治疗群体进行分析。进行总体分析;此外,安全性和有效性分析与疾病阶段相关。
安全性:分析方法。将接受任何研究药物治疗的病人纳入安全性分析。包括不良事件、实验室结果、毒性、生命指征和撤销信息(withdrawal information)的安全性数据将随时间进行概述。用描述性统计概述病人年龄、体重和身高,用频率表概述年龄和种族。用频率表概述诊疗史。
用MedDRA分类方案对不良事件进行编码和列表。对治疗紧急AE的发生率进行列表;此外,根据不良事件严重性(等级)和研究者指明的与JX-594的关系对数据进行分级(stratify)。安全性分析关注3或4级非血液学不良事件和4级血液学不良事件。产生SAE的列表。
用描述性统计概述血液学和血清化学中的连续变量。此外,进行所选血液学参数的最低点分析并概述。在移动表格中随时间概述实验室结果,移动表格显示了相对参考范围、给药后由基线发生变化的病人的数目。所选变量的实验室结果以图片显示。
用描述性统计概述KPS行为评分中的分类变量。同时概述KPS行为评分中与筛选和/或基线相比的最大偏移。用描述性统计概述其余安全性变量。
药代动力学/药效学:分析方法。用下列血液中基因组的浓度评价随时间病毒复制和脱落进入血液。在所有的病人中测量JX-594的血液浓度和GM-CSF的水平,估测药代动力学参数。
要分析的药代动力学参数包括JX-594和GM-CSF对外周血计数、MIA和肿瘤活检组织的影响。评估并概述IT注射后对JX-594的免疫应答,包括白细胞亚组(绝对嗜酸性粒细胞计数、ANC、淋巴细胞)、细胞因子和JX-594中和抗体的形成与基线值相比的变化。
评价并概述组织学终点与基线值(肿瘤组织和正常组织对照)相比的变化,包括炎症细胞浸润、病毒基因表达、GM-CSF表达、lac-Z表达和肿瘤坏死。还评价凋亡、细胞质内病毒复制工厂、EGFR通路状态和肿瘤胸腺嘧啶激酶状态。
效果:分析方法。评价如下基于RECIST标准的治疗反应率:总体反应率、注射肿瘤反应率和非注射肿瘤反应率。概述完全反应率、部分反应率、疾病稳定率和疾病进展率。还报告无进展生存时间、至进展时间、反应持续时间和总体生存率。评价与疾病分期的相关性。
用卡普兰-梅尔(Kaplan-Meier)法估测无进展生存期和反应持续时间。展现中位数,中值、最小、最大持续时间的(双侧)95%置信区间,以及删除患者的数目。进行无进展生存期的描述性统计并描绘曲线。通过对JX-594治疗后病人体重增加和行为状态的改善的评估来评价病人的临床受益。评价黑色素瘤抑制活性蛋白(MIA)随时间的变化。还可将MIA与治疗反应进行比较。
反应评估的独立审阅。可要求中心向独立放射科审查员提供所选病人的所有放射学数据拷贝(优选数字形式或CD-ROM)。对于有皮肤损伤的病人,也将照片发送给IRR做独立审阅。来自中心和IRR的结果均进行报告。不对阅读者间的矛盾之处进行评价。
实施例3
难治性肝脏肿瘤的治疗
在JX-594的I期初步试验中,7名黑色素瘤病人接受注射入浅表皮肤转移瘤的递增剂量(Mastrangelo等,1999)。未报告最大耐受剂量(MTD);报告了肿瘤反应。当前试验的目的是为了明确下列内容:没有预先免疫(如在初步研究中所做的)、特别是在实体器官内治疗后,显著更高剂量(100倍)的安全性和MTD;药代动力学,包括3周的复制依赖性脱落进入血液;对抗广谱癌症类型的有效性。因此在该I期试验中,发明者通过瘤内注射治疗肝脏肿瘤(原发的或转移的)病人。发明者第一次报告了MTD,以及高水平JX-594复制和全身性GM-CSF表达,良好耐受剂量下的有效性和远端肿瘤靶向性。本文中该结果支持将来JX-594及来自该类型的产品的i.t.和i.v.试验。
A.材料和方法
1.研究设计
主要目的是为了确定JX-594的安全性和MTD。第二目的包括药代动力学、复制和脱落(尿液、咽拭子)、免疫应答(中和抗体、细胞因子)和肿瘤反应。在一个按组顺序剂量递增的设计中(每一剂量水平2-6名病人),病人接受4个剂量水平(108、3x 108、109、3x 109噬斑形成单位,pfu)中的一种剂量。若2名或多名患者在某一剂量治疗后观察到DLT,则此剂量紧接着的前一剂量水平确定为MTD。DLT定义为任何4级毒性,或3级毒性持续>5天。独立数据安全性监控委员会(Data Safety Monitoring Board(DSMB))审阅所有剂量递增的决定和主要的安全性评价。
2.病人选择
根据药物临床试验质量管理规范(GCP)指导原则,病人签署知情同意书。入选条件包括尽管经过标准疗法治疗但仍然进展的肝脏内不可切除的、可注射的实体瘤,正常的造血功能(白细胞计数>3,000mm3,血色素>10g/dL,血小板计数>75,000/mm3)以及脏器功能(包括肌氨酸酐<1.5mg/dL,AST/ALT≤2·5ULN,Child-Pugh A或B级),预计生存期>16周,且卡氏行为状态(Karnofsky Performance Status)(KPS)≥70。排除条件包括增高的接种并发症风险(如免疫抑制、湿疹),4周内接受过免疫抑制剂或癌症治疗药物的治疗、怀孕或哺乳。
3. JX-594的生产和准备
JX-594由惠氏牛痘毒株改造而来,在其TK基因区域插入人GM-CSF和lacZ基因使两种基因分别在合成早期启动子和p7·5启动子的控制之下。按照GMP指导原则在Vero细胞中产生并通过蔗糖梯度离心纯化得到临床试验材料。基因组-pfu比例约为70:1。在含10%甘油、138mM氯化钠的pH7.4的磷酸缓冲盐水中配制JX-594。最终产物的QC释放测试包括无菌、内毒素和功效的测定。用0.9%生理盐水稀释JX-594,其体积相当于目标肿瘤估计总体积的25%。
4.治疗步骤
使用21-号PEIT(经皮乙醇注射,多孔;日本东京汉考医疗公司)(HAKKOMedicals)针头通过影像引导的瘤内注射给予JX-594。每3周进行一次注射(n=1-3),从肿瘤中拔出针头的过程中,沿两个针头轨迹进行注射。最初的疗程为2个周期;如果肿瘤发生反应则可至多再进行6个周期。
5.病人监控
如表5所示进行病人监控。住院治疗后至少对病人监控48小时,门诊病人监控4周。
表5研究步骤
6.中和抗体(NAb)滴度
通过细胞病变效应抑制实验测定NAb滴度。用培养基以半对数稀释度连续稀释热灭活的血清。将50μL样品与1,000pfu JX-594一起孵育2小时,然后接种到A2780细胞上。3天后,用细胞计数试剂盒-8(日本熊本的岛京实验室)(Donjindo Laboratories,Kumamoto,Japan)检测细胞活力。NAb滴度定义为引起≥50%细胞存活的最高血清稀释度的倒数。
7. JX-594的定量PCR
用定量PCR(Q-PCR)连续测定血液中JX-594基因组,因为该方法能够检测产物并具有重现性而与抗体和/或补体中和无关。从QIAmp DNA血液微型试剂盒(德国希尔敦的凯杰公司)(Qiagen GmbH,Hilden,Germany)制备的样品中纯化JX-594DNA。如前所述进行Q-PCR(Kulesh等,2004)。JX-594检测和定量的下限分别是666和3,333拷贝/毫升血浆。
8. JX-594脱落检测
使用噬斑形成实验检测任何感染性JX-594脱落进入环境;感染性单位脱落有可能对公众健康有影响。尿液和唾液样本离心后重悬于10mM Tris(pH 9·0)中,用噬斑试验对A2780细胞进行滴定。检测限是20pfu/ml样品。
9.细胞因子实验
根据供应商指导用ELISA试剂盒(美国加州卡尔斯巴德市生物资源国际公司)(BioSource International;Carlsbad,CA,USA)检测GM-CSF。根据厂家指导用LINCOplex试剂盒(密苏里州查尔斯市林可公司)(LINCO;St.Charles,MO)检测血清中IL-1β、IL-6、IL-10、TNF-α和干扰素γ的水平。
10.血液和肿瘤样品中痘苗病毒蛋白和lacZ的组织病理学染色
用苏木精和伊红对福尔马林固定石蜡包埋的活检样品进行染色。对于免疫组织化学,使用B5R的小鼠单克隆抗体(Vac-14,α-B5R,46μg/mL;宾夕法尼亚大学Gary Cohen博士;1:50或1:100稀释),然后与达科(DAKO)EnVision+TM抗小鼠HRP标记的聚合物(加州卡平特里亚的达科公司(DAKO,Carpinteria,CA))孵育,最后用DAB显色(马里兰州盖瑟斯堡市基尔克和派瑞实验室)(Kirkegaard&Perry Laboratories;Gaithersburg,MD)。对于LacZ染色,将细胞900rpm离心1分钟,在玻片上用0.5%戊二醛固定。然后冲洗细胞并用X-gal溶液染色4小时至过夜。
11.肿瘤反应评价
每2个周期后评价肿瘤反应。标准方法是对比度增强CT扫描(除非禁忌)。获取最大肿瘤直径和杭斯费尔德单位(Hounsfield unit)(HU;密度估计值)。使用RECIST和Choi反应标准(Choi等,2007)。对基线处升高的肿瘤标记物进行跟踪。
12.统计学问题
安全性问题决定研究样本大小。使用意向治疗群体(≥1剂量)和标准剂量递增设计。考虑到治疗群体中真实DLT率不同,根据I期剂量递增试验的常规计算剂量递增的可能性。
B.结果
1.病人特征
共编入14位病人(表6列出特征;图20显示试验概况)。3名病人在组1-2中进行治疗,6名在第三组中,2名在最高剂量组中。因为肿瘤进展引起非相关病人死亡,根据DSMB的要求6名病人在组3中进行治疗。2名病人(组1、3)由于无关不良事件在1个周期后暂停治疗,最高剂量组的病人由于DLT接受了1个周期的治疗(见下)。
表6.病人统计
2.治疗相关毒性
a.不良事件(AE)
JX-594在MTD(109pfu)之内耐受良好。研究中未发生治疗相关的死亡。所有病人均经历1-2级流感样症状(治疗后4-16小时)。剂量相关低血压(2级,无器官功能紊乱)在4-12小时内发生。表7中列出了可能与JX-594相关的最常见AE。仅有1例严重AE病例(厌食和腹痛)被认为是治疗相关。报告了10例严重的无关(根据PI)AE归因于肿瘤进展相关并发症。在AE报告期间,4名病人因为肿瘤进展死亡。
组4中2名病人经历了DLT。两者均经历了肿瘤肿胀和阻塞肝内胆管引起的3级直接胆红素升高,以及III级厌食和腹痛。
b.实验室数据
在最初3天内观察到治疗相关的淋巴细胞、血小板和血细胞比容的瞬时减少。在最初4天内,9名病人的绝对中性粒细胞计数(ANC)有显著增加(7例增加>100%;图2A)。ANC的增加是剂量相关的并常与血液中检测到GM-CSF相关。组3和4中70%的病人ANC显著升高(≥5,000/μL)(相对组1和2中的17%;图21A);观察到单核细胞和嗜酸性粒细胞的升高。血小板减少是剂量依赖的(图22A),但是不依赖周期(图22B)。在周期1中ANC升高最多(图22C)。2名病人发生淋巴细胞减少和白细胞减少(表7)。MTD时未发生显著的转氨酶升高(图21B)。
3.药代动力学和药效学终点
a.血清GM-CSF
13名病人在基线时血清GM-CSF呈阴性。MTD时注射JX-594后3名病人检测到GM-CSF的时间>48小时(46-16,000pg/mL)(图21A),浓度高于那些皮下注射GM-CSF蛋白的病人(Cebon等,1992)。GM-CSF浓度与WBC诱导相关(图21A)。
b.中和抗体(NAb)
79%病人在基线时抗JX-594抗体(NAb)水平很低(<10)或检测不到。所有病人在22天内产生NAb。45%病人在首次给药后NAb滴度达到峰值,而55%则继续增加。
未发现基线值或治疗后NAb滴度与任一临床或实验室终点,包括JX-594药代动力学、复制、GM-CSF表达或效果的相关性。3名有目标RECIST肿瘤反应的病人具有可检测的基线NAb滴度以及治疗后高滴度(32,000、32,000和10,000)。此外,在高水平NAb诱导后对两名病人新产生的颈部转移进行治疗,两者的肿瘤都产生目标反应(如下;表8和图24B)。
表8.目标肿瘤反应和生存时间
1第一个数字反映剂量水平(如103在剂量水平1中)
2RECIST标准:部分反应(PR)是最大直径减少≥30%;疾病进展(PD)是增加≥20%;疾病稳定则是直径变化在PR和PD范围之间。
3Choi标准:最大直径减少≥10%或密度减少≥15%;+表明反应
4肿瘤标记物反应的定义:减少≥50%:PR;增加≥25%:PD;减少<50%或增加25%:SD;在病人201、301、402中标记物为甲胎蛋白(AFP);在病人401中为PIVKA2;在病人302、305、306中为癌胚抗原(CEA)。
5生存期:+表明没有癌症相关死亡;m:月;d:日
6仍然生存
7HU:杭斯菲尔德单位(Hounsfield unit)
8第3周进行CT扫描显示肿瘤进展
c.细胞因子
白介素6、IL-10和TNF-α在3小时达到峰值。也观察到较晚的峰值(3–22天)。周期2-8中的细胞因子诱导强于周期1。白介素-6诱导与血清中GM-CSF相关。未观察到IL-1β和IL-4诱导。
d. JX-594药代动力学
所有病人在注射后(50个周期中的49个)立即检测到JX-594基因组。浓度与剂量相关(图23A和23B),在15分钟内减少~50%,在4-6小时内减少~90%。最初的清除速率不依赖剂量,也不依赖抗体滴度。在血液中注射的JX-594最初释放和清除之后,经常检测到循环JX-594的延迟再度出现,这与复制相一致。15名病人中有12名(80%)在第3-22天检测到基因组(血液或血浆)。第二峰值浓度常与剂量相关,药代动力学相似(图23B)。在周期2-7中重复给药后检测到较低的第二浓度峰值(11名病人中的4名)。图23C显示了代表性的药代动力学。
e.非注射远端肿瘤位点中JX-594的散播、复制
在非注射肿瘤组织中检测到JX-594,表明远端肿瘤选择性的感染和复制(图23D-F)。例如,一名病人的恶性腹水和胸膜渗出液比同时间点的血液中具有更高的基因组浓度(分别高17-倍和12-倍)和GM-CSF浓度(分别高24-倍和13-倍)(图3D)。胸膜渗出液中的LacZ(+)细胞确认了JX-594的感染(图23E)。对另一病人进行远端颈部肿瘤活检,组织学检验证明存在复制的JX-594(图23F)。
f. JX-594脱落
在任一咽喉和尿液样中未检测到感染性JX-594。
g. JX-594的抗肿瘤效果
评价10名病人的目标肿瘤反应;未评价的病人有禁忌症用以对比(2)或者未进行治疗后扫描(2)。9名(90%)有符合CT RECIST标准的目标反应(30%)或疾病稳定(60%)。8名病人(80%)有符合Choi标准的目标反应(表8)。同时具有符合RECIST和Choi标准的反应的病人患有非小细胞肺癌(图24A)、HCC和黑色素瘤(表8)。目标反应是持续的;反应肿瘤位点未发生再次生长(随访4–18个月)。在肝脏中进行4个预先周期后,对两名病人原来未注射的颈部肿瘤进行直接注射,尽管产生高水平的JX-594中和抗体,但仍然得到了Choi和/或RECIST反应(表8,图24B);因此再次治疗的效果是可行的。
同样评价了远端非注射肿瘤的反应。在7名有远端非注射肿瘤的病人中,6名病人出现符合RECIST标准的远端疾病稳定;这些远端肿瘤的至进展时间从6+到30+周。这些病人中的3名出现符合Choi标准的反应(n=2)或符合PET-CT标准的反应(n=1;减少25-100%;表9)。
表9.具有目标肿瘤对照的病人的远端肿瘤反应(RECIST PR或SD)
1第一个数字反映剂量水平(如103在剂量水平1中)
2通过CT RECIST;+表明没有癌症相关的死亡
HU:杭斯菲尔德单位;LN:淋巴结;SC:锁骨上;PA:耳前;CR:完全反应;PR:部分反应;SD:疾病稳定;PD:疾病进展。
至今,8名病人(57%)已经存活至少8个月,4名超过1年并且1名超过20+个月。中位生存期是9个月。
*********
根据本公开,本说明书和权利要求书所描述的所有组合物和/或方法无需特别的试验就可以制备和实施。虽然本发明的组合物和方法是利用优选实施方案来描述的,但是,本领域的技术人员在不背离本发明的概念、精神和范围的情况下,可以对本发明的组合物和/或方法以及本文所描述的方法的步骤或步骤的顺序作出改动。更具体地说,本文所描述的因子可用某些化学和物理相关的因子代替,但是可以得到相同或相似的结果。对于本领域技术人员来说很明显的是,所有这种相似的替代和修改都被认为包含在权利要求所定义的本发明的精神、范围和概念之内。
参考文献
下列参考文献,在本文列举的范围内提供示例性步骤或其它补充本文所列举的细节,将其纳入本文作参考。
美国专利4,554,101
美国专利4,683,202
美国专利4,684,611
美国专利4,952,500
美国专利5,073,627
美国专利5,302,523
美国专利5,322,783
美国专利5,384,253
美国专利5,399,363
美国专利5,464,765
美国专利5,466,468
美国专利5,538,877
美国专利5,538,880
美国专利5,543,158
美国专利5,550,318
美国专利5,563,055
美国专利5,580,859
美国专利5,589,466
美国专利5,591,616
美国专利5,610,042
美国专利5,633,016
美国专利5,641,515
美国专利5,656,610
美国专利5,702,932
美国专利5,736,524
美国专利5,739,169
美国专利5,780,448
美国专利5,789,215
美国专利5,798,339
美国专利5,801,005
美国专利5,824,311
美国专利5,824,348
美国专利5,830,880
美国专利5,846,225
美国专利5,846,233
美国专利5,846,945
美国专利5,925,565
美国专利5,928,906
美国专利5,935,819
美国专利5,945,100
美国专利5,981,274
美国专利5,994,624
Alcami和Smith,Cell.,71(1):153-167,1992.
Alcami等,J.Gen.Virol.,80:949-959,1999.
Alcami等,Sem.Virol.,5:419-427,1998.
Alcami等,Virology,74(23):11230-11239,2000.
Almendro等,J.Immunol.,157(12):5411-5421,1996.
Arap等,Cancer Res.,55(6):1351-1354,1995.
Arness等,Am.J.Epidemiol.,160:642-51,2004.
Austin-Ward和Villaseca,Revista Medica de Chile,126(7):838-845,1998.
Ausubel等,刊于《分子生物学最新实验方案》(In:Current Protocols in Molecular Biology),约翰威利父子公司(John,Wiley&Sons,Inc),纽约,16.15.1-16.18.10,1996.
Bajorin等,J.Clin.Oncol.,6(5):786-792,1988.
Bakhshi等,Cell,41(3):899-906,1985.
Blasco和Moss,J.Virology,66(7):4170-4179,1992.
Blasco等,J.Virology,67(6):3319-3325,1993.
Boyd等,Cell,79:341-351,1994.
Brizel,Semin.Radiat.Oncol.,8(4):237-246,1998.
Bukowski等,Clinical Cancer Res.,4(10):2337-2347,1998.
Caldas等,Cancer Res.,54:3568-3573,1994.
Carbonelli等,FEMS Microbiol.Lett.,177(1):75-82,1999.
Cebon等,Br.J.Haematol.,80(2):144-150,1992.
Chandler等,Proc.Natl.Acad.Sci.USA,94(8):3596-601,1997.
Chen和Okayama,Mol.Cell Biol.,7(8):2745-2752,1987.
Cheng等,Cancer Res.,54(21):5547-5551,1994.
Choi等,J.Clin.Oncol.,25(13):1753-1759,2007.
Christodoulides等,Microbiology,144(Pt 11):3027-3037,1998.
Cleary和Sklar,Proc.Natl.Acad.Sci.USA,82(21):7439-7443,1985.
Cleary等,J.Exp.Med.,164(1):315-320,1986.
Cocea,Biotechniques,23(5):814-816,1997.
Colamonici等,J.Biol.Chem.,270:15974-15978,1995.
Culver等,Science,256(5063):1550-1552,1992.
Curran,Seminars Radiation Oncol.,8(4Supp1):2-4,1998.
Davidson等,J.Immunother.,21(5):389-398,1998.
Dillman,Cancer Biother.Radiopharm.,14(1):5-10,1999.
Dobbelstein和Shenk,J.Virology,70:6479-6485,1996.
Durrant和Spendlove,Curr.Opin.Investig.Drugs,2(7):959-66,2001.
Eliopoulos等,Oncogene,11(7):1217-28,1995.
Erlandsson,Cancer Genet.Cytogenet.,104(1):1 18,1998.
Fechheimer等,Proc Natl.Acad.Sci.USA,84:8463-8467,1987.
Fraley等,Proc.Natl.Acad.Sci.USA,76:3348-3352,1979.
GenBank登录号NC001559
Gnant等,Ann Surg,230(3):352-360,1999.
Gnant等,Cancer Res.,59(14):3396-403,1999.
Gnant等,J.Natl.Cancer Inst.,91(20):1744-1750,1999.
Goebel等,Virology,179(1):247-266,517-563,1990.
Gopal,Mol.Cell Biol.,5:1188-1190,1985.
Graham和Van Der Eb,Virology,52:456-467,1973.
Graham等,Virology,229(1):12-24,1997.
Gross等,Genes Dev.,13(15):1899-911,1999.
Gross等,J.Biol.Chem.,274:1156–1163,1999.
Hanibuchi等,Int.J.Cancer,78(4):480-485,1998.
Harland和Weintraub,J.Cell Biol.,101(3):1094-1099,1985.
Heise等,Cancer Gene Ther.,6(6):499-504,1999.
Heise等,Cancer Res.,59(11):2623-2628,1999.
Hellstrand等,Acta Oncologica,37(4):347-353,1998.
Hermiston,J.Clin.Invest.,105:1169-1172,2000.
Ho等,J.Biol.Chem.,27:7765-7769,1998.
Homey等,Nature.Rev.Immunol.,2:175-184,2002.
Hui和Hashimoto,Infection Immun.,66(11):5329-5336,1998.
Hussussian等,Nat.Genet.,8(1):15-21,1994.
Ikeda等,Nat.Med.,5(8):881-7,1999.
Inouye和Inouye,Nucleic Acids Res.,13:3101-3109,1985.
Irie和Morton,Proc.Natl.Acad.Sci.USA,83(22):8694-8698,1986.
Irie等,Lancet.1(8641):786-787,1989.
Isaacs等,Proc.Natl.Acad.Sci.USA,89(2):628-32,1992.
Johnson和Hamdy,Oncol.Rep.,5(3):553-557,1998.
Ju等,Gene Ther.,7(19):1672-1679,2000.
Ju等,J.Neuropathol.Exp.Neurol.,59(3):241-250,2000.
Kaeppler等,Plant Cell Reports,9:415-418,1990.
Kamb等,Nat.Genet.,8(1):23-26,1994.
Kamb等,Science,2674:436-440,1994.
Kaneda等,Science,243:375-378,1989.
Kato等,J.Biol.Chem.,266:3361 3364,1991.
Kay等,Proc.Natl.Acad.Sci.USA,94(9):4686-4691,1997.
Kerr等,Br.J.Cancer,26(4):239-257,1972.
Kettle等,J.Gen.Virology,78:677-685,1997.
Kirn等,Nat.Med.,7:781-787,2001.
Kolmel,J.Neurooncol.,38(2-3):121-125,1998.
Kraus等,FEBS Lett.,428(3):165-170,1998.
Kulesh等,J.Clin.Microbiol.,42(2):601-609,2004.
Kyte和Doolittle,J.Mol.Biol.,157(1):105 132,1982.
Lareyre等,J.Biol.Chem.,274(12):8282-8290,1999.
Lee等,Biochem.Biophys.Res.Commun.,238(2):462-467,1997.
Levenson等,Hum.Gene Ther.,9(8):1233-1236,1998.
Liebermann,Oncogene,17(10):1189-94,1998.
Macejak和Sarnow,Nature,353:90-94,1991.
Magi-Galluzzi等,Anal.Quant.Cytol.Histol.,20(5):343-350,1998.
Mangray和King,Front Biosci.,3:D1148-1160,1998.
Marsters等,Recent Prog.Horm.Res.,54:225-234,1999.
Mastrangelo等,Cancer Gene Ther.,6(5):409-422,1999.
Mastrangelo等,Cancer Treat Res.,94:35-50,1998.
Mayer等,Radiat.Oncol.Investig.,6(6):281-288,1998.
McCart等,Gene Ther.,7(14):1217-23,2000.
Mitchell等,Ann.NY Acad.Sci.,690:153-166,1993.
Mitchell等,J.Clin.Oncol.,8(5):856-869,1990.
Mori等,Cancer Res.,54(13):3396-3397,1994.
Morton等,Arch.Surg.,127:392 399,1992.
Moss,刊于《费尔兹病毒学》中(In:Fields Virology.),费尔兹(编)(Fields(Ed.)),立品科特-瑞文出版社(Lippincott-Raven Publishers):费城,2637-2672,1996.
Mossman等,Virology,215(1):17-30,1996.
Mougin等,Ann.Biol.Clin.,(Paris)56(1):21-8,1998.
Mumby和Walter,Cell Regul.,2(8):589-98,1991.
Natoli等,Biochem.Pharmacol.,56(8):915-20,1998.
Nicolau和Sene Nobori等,1994
Nomoto等,Gene,236(2):259-271,1999.
Ochi等,Am.J.Gastroenterol.,93(8):1366-1368,1998.
Ohara,Gan To Kagaku Ryoho,25(6):823-828,1998.
Okamoto等,Proc.Natl.Acad.Sci.USA,91(23):11045-11049,1994.
Omirulleh等,Plant Mol.Biol.,21(3):415-428,1993.
Orlow等,Cancer Res,54(11):2848-2851,1994.
PCT申请WO 94/09699
PCT申请WO 95/06128
Pelletier和Sonenberg,Nature,334(6180):320-325,1988.
Pietras等,Oncogene,17(17):2235-2249,1998.
Puhlmann等,Cancer Gene Ther.,7:66-73,2000.
Qin等,Proc.Natl.Acad.Sci.USA,95(24):14411-14416,1998.
Ravindranath和Morton,Intern.Rev.Immunol.,7:303 329,1991.
《雷明顿药物科学》(Remington’s Pharmaceutical Sciences”)第15版,1035-1038和1570-1580,1990.
Rippe,等,Mol.Cell Biol.,10:689-695,1990.
Rosel等,J.Virol.,60(2):436-449,1986.
Rosenberg等,Ann.Surg.210(4):474-548,1989.
Rosenberg等,N.Engl.J.Med.,319:1676,1988.
Sambrook等,刊于《分子克隆:实验室手册》(Molecular cloning:a laboratory manual),第2版,冷泉港实验室出版社,纽约冷泉港,1989.
Saraiva和Alcami,J.Virology,75(1):226-33,2001.
Serrano等,Nature,366:704-707,1993.
Serrano等,Science,267(5195):249-252,1995.
Sinkovics和Horvath,J.Clin.Viro.,16:1-15,2000.
Smith等,Immunol.Rev.,159:137-154,1997.
Smith等,J.Clin.Oncol.,18:2046-2052,2000.
Smith等,Neuron.,20:1093-1102,1998.
Solyanik等,Cell.Prolif.,28(5):263-278,1995.
Spriggs等,Cell.,71(1):145-52,1992.
Stokke等,Cell.Prolif.,30(5):197-218,1997.
Symons等,Cell,81:551-560,1995.
Todo等,Cancer Res.,61:153-161,2001.
Tsujimoto和Croce,Proc.Natl.Acad.Sci.USA,83(14):5214-5218,1986.
Tsujimoto等,Nature,315:340-343,1985.
Tsujimoto等,Science,228(4706):1440-1443,1985.
Tsumaki等,J.Biol.Chem.,273(36):22861-22864,1998.
Upton等,Science,258(5086):1369-1372,1992.
Upton等,Virology,184(1):370-82,1991.
Vanderplasschen等,Proc.Natl.Acad.Sci.USA,95(13):7544-7549,1998.
Vicari和Caus,Cytokine Growth Factor Rev.,13:143-154,2002.
Vogelstein和Kinzler,Cell,70(4):523-6,1992.
Wallach等,Annu.Rev.Immunol.,17:331-367,1999.
Wold等,J.Virol..52:307-313,1984.
Wold等,Trends Microbiol.,2:437-443,1994.
Wong等,Gene,10:87 94,1980.
Wu等,Biochem.Biophys.Res.Commun.,233(1):221-226,1997.
Zhao-Emonet等,Biochim.Biophys.Acta,1442(2-3):109-119,1998.
- 还没有人留言评论。精彩留言会获得点赞!