冬凌草甲素立方液晶纳米粒及其制备方法与流程
本发明涉及药物制剂领域,特别是涉及一种冬凌草甲素立方液晶纳米粒及其制备方法。
背景技术:
冬凌草甲素是唇形科植物冬凌草中分离出来的一种贝壳杉烯二萜类天然有机化合物,它对多种肿瘤细胞均具有较强的抗肿瘤作用。临床上,它主要作为一种抗癌新药或抗癌辅助药,用于肝癌、食管癌、胰腺癌等消化系统癌症的治疗。
冬凌草甲素为7,20-环氧型对映贝壳杉烷二萜化合物,可溶于甲醇、乙醇、丙二醇、乙酸乙酯、丙酮等有机溶剂,难溶于水,为生物药剂学分类系统中的IV类药物,在口服给药过程中存在溶解屏障和渗透屏障。口服给药后体内的血药浓度低,而肿瘤细胞周围的冬凌草甲素浓度则更低,很难达到临床有效的治疗浓度,从而严重影响冬凌草甲素的抗癌效果。目前市场上主要采用有机溶剂加表面活性物质增加冬凌草甲素的溶解度,静脉滴注易引起血管炎、疼痛等不良反应,而口服制剂仅有冬凌草甲素粗提取物片剂,药物含量少,生物利用度低。因此,有必要研发新的给药剂型以提高冬凌草甲素的生物利用度。
技术实现要素:
基于此,本发明提供了一种冬凌草甲素立方液晶纳米粒,可显著提高冬凌草甲素的口服利用度。
具体技术方案如下:
一种冬凌草甲素立方液晶纳米粒,由以下重量百分比的原料制备而成:
所述两亲性脂质材料为单油酸甘油酯或植烷三醇;
所述稳定剂选自脂肪酸甘油酯、多元醇型非离子表面活性剂、聚氧乙烯型非离子表面活性剂、泊洛沙姆、卵磷脂中的至少一种;
所述溶剂选自乙醇、丙二醇、二甲基亚砜、聚乙二醇400、N-甲基吡咯烷酮和N,N-二甲基乙酰胺中的至少一种;
所述液晶材料与稳定剂的质量比为1∶0.01-0.30。
在其中一些实施例中,所述冬凌草甲素立方液晶纳米粒由以下重量百分比的原料制备而成:
在其中一些实施例中,所述两亲性脂质材料与稳定剂的质量比为1∶0.05-0.25。
在其中一些实施例中,所述冬凌草甲素立方液晶纳米粒由以下重量百分比的原料制备而成:
在其中一些实施例中,所述甘油单油酸酯与泊洛沙姆407的质量比为1∶0.12-0.15。
在其中一些实施例中,所述冬凌草甲素立方液晶纳米粒由以下重量百分比的原料制备而成:
在其中一些实施例中,所述植烷三醇与泊洛沙姆407的质量比为1∶0.09-0.12。
本发明还提供了一种上述冬凌草甲素立方液晶纳米粒的制备方法。
具体技术方案如下:
一种上述的冬凌草甲素立方液晶纳米粒的制备方法,包括以下步骤:
将冬凌草甲素溶解于溶剂中,得药物溶液;
向所述药物溶液中缓慢加入熔融的两亲性脂质材料和稳定剂的混合液,混合均匀,再加入超纯水,混合均匀,于室温下避光平衡40-56h,即得冬凌草甲素立方液晶凝胶;
将所述冬凌草甲素立方液晶凝胶置于水中,进行超声分散,得到冬凌草甲素立方液晶粗分散溶液;
将冬凌草甲素立方液晶粗分散溶液进行高压均质,即得所述冬凌草甲素立方液晶纳米粒。
在其中一些实施例中,所述高压均质的条件为:均质压力500-1500bar,均质次数3-13次。
在其中一些实施例中,所述高压均质的条件为:均质压力1100-1200bar,均质次数8-10次。
在其中一些实施例中,所述熔融温度为40-70℃。
在其中一些实施例中,将所述冬凌草甲素立方液晶凝胶置于水中,进行超声分散的步骤中,水的用量与所述冬凌草甲素立方液晶凝胶的配比为:8-12ml:1g。
在其中一些实施例中,所述超声分散的参数为:分散时间为10-15min,功率为150-400w,工作时间为4-6s,间断时间为8-12s。
立方液晶是一种新型脂质药物载体,是由两亲性脂质分散在水性环境中自发形成各种几何形态构成的一个中介相形态(Mesophase)。因其独特的三维网络结构而具有良好的药物传递性能。其三维网络结构主要由亲水域中的双连续水通道和亲脂域中的脂质双分子层构成的晶格单元在空间上延伸折叠,堆叠成具有三维,循环排列和最小表面积特点的紧密结构。难溶性药物具有较强的疏水性,与脂质结构极性相近,在亲脂域中具有较高的溶解度。
本发明的冬凌草甲素立方液晶纳米粒及其制备方法具有以下优点和有益效果:
本发明的发明人通过大量实验研究发现将冬凌草甲素与特定的液晶材料、溶剂、稳定剂以特定比例与特定比例的水配合做为制备原料可制备得到药物包封率高、粒径较小且分布均匀的载有冬凌草甲素的立方液晶纳米粒。药物颗粒大小,药物与溶出介质的接触面积,以及溶出层与介质之间的药物浓度差是影响难溶性药物溶出速率的关键参数。本发明的冬凌草甲素的立方液晶纳米粒,其粒径较小(小于250nm),且粒径均一性好,有利于肠上皮细胞内吞并转运到细胞内;冬凌草甲素包载进入立方液晶结构中后,冬凌草甲素由晶体颗粒转变成无定型的分子态,均匀分布于亲脂域中,双连续水通道与脂质双分子层的巨大交界面积显著增加了药物的溶出表面积;同时药物在亲脂域中有较大的溶解度,在溶出层和介质之间形成较大的药物浓度差,可进一步增大药物的溶出程度,有效克服药物溶解屏障;此外,立方液晶的脂质双分子层,具有类细胞结构,与细胞有很强的亲和性,具有良好的生物相容性,能够有效地跨越口服吸收过程中的小肠粘膜上层细胞的渗透屏障;因而,本发明的冬凌草甲素的立方液晶纳米粒能够显著提高冬凌草甲素的口服生物利用度。
本发明的冬凌草甲素的立方液晶纳米粒,能够有效的将难溶性药物冬凌草甲素包埋于液晶结构中,药物的包封率高,克服了其口服生物利用度低的缺点,同时纳米粒溶液也有利于口服,能够提高患者的顺应性。
本发明通过对各原料用量的进一步优选使制备得到的冬凌草甲素的立方液晶纳米粒的粒径更小且更均一,更有利于肠上皮细胞的内吞和转运,同时使立方液晶结构保持得更好,利用立方液晶独特的结构、生物亲和性及自稳定特质,有效克服冬凌草甲素在口服吸收过程中的溶解屏障和渗透屏障,显著提高了冬凌草甲素的口服相对生物利用度,同时能使冬凌草甲素缓慢释放,具有24小时缓释的效果,为冬凌草甲素提供了一种能够缓慢释放、吸收良好、生物利用度高的新型口服制剂。
本发明的制备方法,采用溶剂诱导的热熔凝胶分散技术,采用“从大到小”的立方液晶纳米粒制备思路,制备方法工艺简单,包封率高,使制备得到的冬凌草甲素的立方液晶纳米粒溶液粒径分布均匀,工艺的重复性良好,生产成本较低,有利于工业化生产,具有良好的应用前景。
附图说明
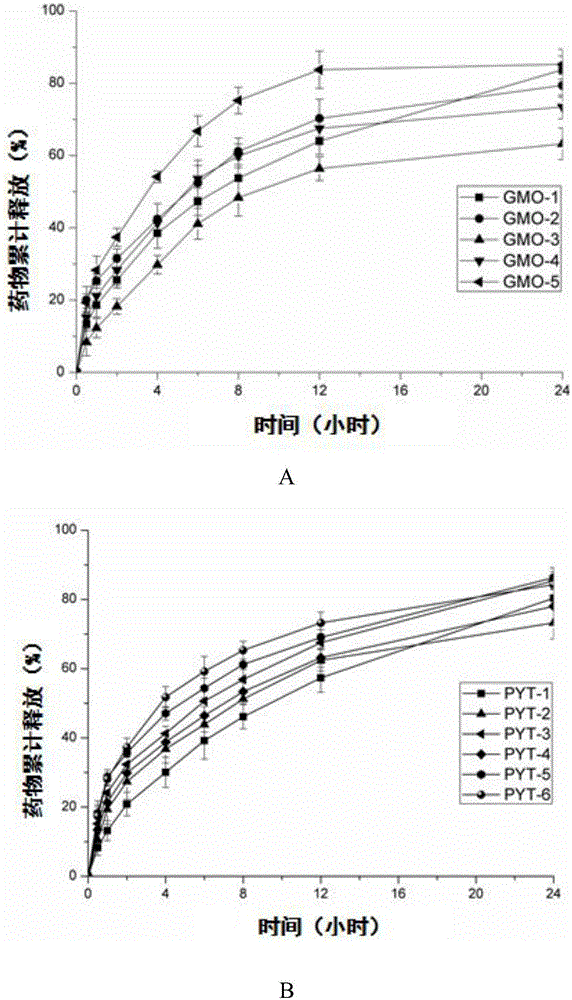
图1为实施例1中不同的体系的冬凌草甲素立方液晶纳米粒的药物释放曲线图,其中,A为GMO体系,B为PYT体系;
图2为实施例2中不同F127含量的冬凌草甲素立方液晶纳米粒的液晶结构的SAXS图谱,其中,A为GMO体系,B为PYT体系;
图3为实施例2中不同F127含量的冬凌草甲素立方液晶纳米粒的药物释放曲线图,其中,A为GMO体系,B为PYT体系;
图4为实施例3中不同载药量的冬凌草甲素立方液晶纳米粒的液晶结构的SAXS图谱,其中,A为GMO体系,B为PYT体系;
图5为实施例4中不同的均质条件下制备得到的冬凌草甲素立方液晶纳米粒的粒径分布图,其中,A为GMO体系,B为PYT体系;
图6为实施例5中冬凌草甲素立方液晶纳米透射电镜表征的结果示意图其中,A为GMO体系,B为PYT体系;
图7为实施例6中不同体系的冬凌草甲素立方液晶纳米粒冻干粉末的XRD表征图,其中,A为GMO体系,B为PYT体系;
图8为实施例6中不同体系冬凌草甲素立方液晶纳米粒冻干粉末的DSC表征图,其中,A为GMO体系,B为PYT体系;
图9为实施例7中不同体系的冬凌草甲素立方液晶纳米粒与对照组的体外药物释放曲线图;
图10为实施例8中不同批次的冬凌草甲素立方液晶纳米粒释放曲线图,其中,A为GMO体系,B为PYT体系;
图11为实施例9中36h内不同体系的冬凌草甲素立方液晶纳米粒与对照组的体内药物动力学曲线图;
图12为实施例9中前2h内不同体系的冬凌草甲素立方液晶纳米粒与对照组的体内药物动力学曲线图。
具体实施方式
以下通过具体实施例以及附图对本发明的冬凌草甲素立方液晶纳米粒及其制备方法做进一步详细的说明。
实施例1冬凌草甲素立方液晶纳米粒的制备及不同液晶体系的考察
本实施例的冬凌草甲素立方液晶纳米粒的制备方法,包括如下步骤:
将20mg冬凌草甲素溶于丙二醇中,得药物溶液;按表1中的各处方(均为质量百分比)称取甘油单油酸酯或植烷三醇以及稳定剂F127(泊洛沙姆407)于50mL塑料离心管中,于60℃水浴熔融,再将其缓慢加入上述药物溶液中,涡旋充分混合5min后,加入处方量的超纯水,涡旋震荡5min,所得体系于室温下避光平衡48h后,即得透明的冬凌草甲素立方液晶凝胶。在冰浴条件下,使用超声波破碎仪,将所得冬凌草甲素立方液晶凝胶在50mL水中超声分散10min,所用超声功率为200W,超声工作时间5s,间断时间10s。超声后得到冬凌草甲素立方液晶粗分散溶液,经高压均质(均质压力为1200bar,循环次数为9次)即可得到粒径均匀的载冬凌草甲素的立方液晶纳米粒溶液。
表1冬凌草甲素立方液晶纳米粒的原料处方(体系总质量为5g)
对本实施例制备得到的冬凌草甲素立方液晶纳米粒,进行体外释放实验,方法如下:冬凌草甲素体外释放度测定按中国药典2010年版(二部)附录XC第三法进行,以pH6.8磷酸盐缓冲液200mL为释放介质,转速为100r/min,温度为37±0.5℃,基于冬凌草甲素能自由通过透析袋,纳米粒不能通过透析袋,故采用透析袋法(截留分子量14000)进行体外释放的实验。精密量取适量本实施例中所制备的冬凌草甲素立方液晶纳米粒溶液6mL置于透析袋中,立即扎紧,以线圈宽松缠绕在搅拌桨下端,于不同时间点:0.5、1、2、4、6、8、12、24h取样5mL,并迅速补加同温新鲜介质,经0.22μm微孔滤膜滤过后采用高效液相色谱法测定冬凌草甲素含量。色谱条件:岛津SIL-20A色谱系统;色谱柱为Ultimate(C18,5μm,4.6mm×250mm);流动相为甲醇-水(60:40);柱温为30℃;检测波长为237nm;流速为1.0mL·min-1,进样量为20μL。根据测定的冬凌草甲素含量,计算冬凌草甲素累积释放百分率,绘制体外释放曲线。
不同体系对冬凌草甲素体外释放的影响结果见图1。由结果可知,随着所制备体系含水量的增加,GMO或PYT含量下降,载体中脂质成分下降,释药速率加快;由于投药量一定,丙二醇比例较小的体系,可能会出现药物包封不完全,使得药物在前期突释,如图中GMO-2、PYT-5体系;从图中可以发现GMO-3出现药物释放不完全现象,可能是由于体系中GMO含量较大,药物紧密地包封在脂质层中,整个体系的粘度较其他体系大,使得药物释放不完全或推迟释放。其中,发现GMO-1、PYT-1体系具有更好的24h缓释作用。
实施例2冬凌草甲素立方液晶纳米粒中不同质量比的F127与脂质对液晶相态以及释药行为的影响
只有将冬凌草甲素液晶进一步制备成纳米粒溶液后才能口服,才能够穿过胃肠道细胞,被人体吸收;但是将冬凌草甲素液晶进一步通过高压均质制备纳米粒时,纳米粒容易聚集沉淀,加入稳定剂,可以支撑维持液晶晶格单元的网络架构,防止在高压均质的过程中纳米粒聚集沉淀,能维持液晶纳米粒在制备和存储过程中的稳定性。
(1)本实施例对不同质量比(0%、9%、12%、15%、20%)的F127与脂质(GMO或PYT)的冬凌草甲素立方液晶纳米粒样品进行考察,其制备方法同实施例1,F127与脂质(GMO或PYT)的总量同实施例1中的GMO-1、PYT-1体系,其它原料以及用量分别同实施例1的GMO-1、PYT-1体系。采用小角X射线散射法(测试参数:X射线波长为发射功率为50kV×40mA 37℃,散射角测量范围为q=0.05~6nm-1,液体样品测量时间为60min,凝胶样品测量时间为20min。)表征所得冬凌草甲素立方液晶纳米粒样品的相态。
晶相结果见图2。结果表明,当F127与甘油单油酸酯的质量比达到20%时,冬凌草甲素立方液晶纳米粒(甘油单油酸酯体系)谱图中的谱峰消失,并在谱峰前面出现一个小的谱峰(见图2A),表明当F127与甘油单油酸酯的比例大于等于20%时,对冬凌草甲素立方液晶纳米粒内部结构产生破坏,而立方液晶的晶格结构是克服冬凌草甲素口服给药过程中溶解屏障和吸收屏障的关键,方液晶纳米粒内部结构的破坏会导致亲脂性的冬凌草甲素无法有效地分散在晶格单元的脂质双分子层中,因此无法起到增加生物利用度的作用。当F127与甘油单油酸酯的质量比为9-15%时对冬凌草甲素立方液晶内部结构无影响,谱图中四个谱峰均无明显变化。而对于植烷三醇体系,当F127与植烷三醇的质量比达到15%时,可以从SAXS谱图上明显看到谱峰发生了明显的位移,且谱峰消失(见图2B),说明在以植烷三醇为液晶材料的冬凌草甲素立方液晶纳米粒中,当F127与植烷三醇的质量比达到15%时,立方液晶的结构已经发生破坏。
由此说明,稳定剂(F127)的加入会影响冬凌草甲素立方液晶纳米粒的液晶相态,当稳定剂加入量过大时,冬凌草甲素立方液晶纳米粒的立方液晶的结构会发生破坏。立方液晶中三维网络结构主要由亲水域中的双连续水通道和亲脂域中的脂质双分子层构成的晶格单元在空间上延伸折叠,堆叠成具有三维,循环排列和最小表面积特点的紧密结构。难溶性药物具有较强的疏水性,与脂质结构极性相近,在亲脂域中具有较高的溶解度,因而倾向于以稳定的无定型的分子态分散在晶格单元的脂质双分子层,如果晶格遭到破坏,那么将大大降低药物分子在晶格中的分散,从而影响其溶解性,影响其生物利用度。
(2)对不同质量比(0%,9%、12%、15%、20%)的F127与脂质(GMO或PYT)的冬凌草甲素立方液晶纳米粒的药物释放行为进行研究,药物释放行为考察方法同实施例1。
释药行为结果见图3。结果表明,当F127的含量为0时,冬凌草甲素药物体外总体释放程度比较低,随着F127与脂质的质量比的升高,两种体系中的冬凌草甲素释放速率先加快,升高到一定比例后释放速率变慢。在GMO的冬凌草甲素立方液晶纳米粒中(图3A),当F127与GMO的质量比为9%时,药物在24h累计释放率小于80%,当F127与GMO的质量比为15%时,药物累计释放达到最高,而当F127与GMO的质量比达到20%时,药物释放明显小于其他三个比例,这是因为过高含量的稳定剂F127对冬凌草甲素立方液晶晶格造成了一定的影响;在PYT的冬凌草甲素的立方液晶纳米粒中(图3B),当F127与PYT的质量比为9%时,药物累计释放率为71.38%,而与GMO体系相同,当稳定剂含量过高,F127与PYT的质量比超过15%时,药物释放明显变慢,制剂生物利用度明显下降;当F127与GMO的质量比为15%,F127与PYT的质量比为12%时,两体系药物均无突释现象,且24小时累计释放率在80%以上,符合缓释制剂的要求。
(3)采用动态光散射法测定本实施例的冬凌草甲素立方液晶纳米粒的粒径。所制备样品用超纯水稀释一定倍数后,移液枪精密移取1mL,加入到样品池中,25℃条件下平衡3分钟,分散黏度为0.8872CP,使用马尔文纳米粒径测定仪(Zetasizer Nano ZS90)测定样品的粒径分布及电位。其中,多分散系数(PDI) 是反映粒径分布均匀性的指标,PDI越小表示粒径分布越集中,PDI越大表示粒径分布越不均匀,粒径大小差异较大。
粒径结果如表2所示,体系中没有稳定剂的存在会导致纳米粒整体粒径偏大,而在GMO体系中,随着处方中稳定剂含量的增加,所形成的液晶纳米粒稳定性增大,粒径逐渐减小,PDI减小,而当稳定剂的含量过高时(20%),纳米粒粒径增大,这是因为纳米粒的晶格结构遭到了破坏,这与药物释放和晶格结构的所得结果一致。在PYT体系中同样有一样的现象出现,不同的是,稳定剂的含量在15%就达到了“过高”的水平,引起这一现象的原因可能是GMO的相对分子质量(356)比PYT的(330)高所造成的。
实施例3:冬凌草甲素立方液晶纳米粒中载药量对立方液晶纳米粒粒径和药物包封率的影响
不同结构、不同含量的药物会影响立方液晶纳米粒的内部结构,因此,本实施例制备不同载药量的冬凌草甲素立方液晶纳米粒(原料处方如表2所示),制备方法如实施例1所示。
表2冬凌草甲素立方液晶纳米粒的原料处方
测定本实施例的不同载药量的冬凌草甲素立方液晶纳米粒的小角X射线散射图谱(测试参数同实施例2)、粒径分布以及包封率。
采用动态光散射法测定冬凌草甲素立方液晶纳米粒的粒径。测试方法同实施例2。
包封率的测定采用超滤离心法。精密移取所制备载冬凌草甲素的立方液晶纳米粒溶液0.5mL于超滤离心管的内管中,4000r/min离心20min,收集外管中游离的冬凌草甲素溶液,用甲醇洗涤后,并用甲醇定容至10mL容量瓶中,采用高效液相色谱方法(同实施例1)测定游离的冬凌草含量Cfree。同时,精密移取载冬凌草甲素的立方液晶纳米粒溶液0.5mL于10mL容量瓶中,加入甲醇破乳并定容,同法测定立方液晶纳米粒总药量Ctotal'。使用下列包封率计算公式,计算两种立方液晶纳米粒的包封率(EE%)。
所得粒径和包封率结果见表3,小角X射线散射图谱见图4。在甘油单油酸酯的冬凌草甲素立方液晶纳米粒体系中,随着所加入的冬凌草甲素药量由0.2%增加到0.5%过程中,粒径呈现先增大、再降低再增大的趋势,包封率始终大于85%。植烷三醇的冬凌草甲素立方液晶纳米粒也出现类似的现象。小角X射线散射图谱的结果显示:两种体系当所加入的药量达到0.5%时,SAXS谱图发生变化,甘油单油酸酯体系的谱峰消失(见图4A),植烷三醇体系中的谱峰的强度明显减弱(见图4B),这一现象提示当冬凌草甲素药量达到0.5%时,对两种体系的冬凌草甲素立方液晶纳米粒的立方液晶结构均有一定程度的破坏,药物包封率有所下降且粒径增大,因此本实施例两种体系的冬凌草甲素立方液晶纳米粒中的冬凌草甲素的最优载药量为0.4%。
表3不同药量对冬凌草甲素立方液晶纳米粒样品的粒径分布、包封率的影响(n=3)
实施例4:制备工艺条件对冬凌草甲素立方液晶纳米粒的影响
按实施例1中的GMO体系1与PYT体系1的原料处方,改变高压均质条件(均质压力分别为600bar,1200bar,1500bar,均质次数分别为3次,6次,9次),其它制备步骤同实施例1,得到不同均质条件下的冬凌草甲素立方液晶纳米粒。
采用动态光散射法(同实施例3)测定本实施例的冬凌草甲素立方液晶纳米粒的粒径,结果见图5。结果表明,两种体系的冬凌草甲素立方液晶纳米粒的PDI均随着均质压力的增大而减小。当均质压力为1200bar,循环次数为9次时,两种体系的冬凌草甲素立方液晶纳米粒的粒径分布均很集中,平均粒径大小分别为220nm(图5A)和250nm(图5B)左右。当高压均质压力增加到1200bar后,继续增大均质压力,发现立方液晶纳米粒的粒径并未减少,且PDI基本不再变化,因此,选择均质压力1200bar,循环次数为9次为最优均质条件。
即载冬凌草甲素的甘油单油酸酯立方液晶纳米粒的最优原料处方为:甘油单油酸酯37.80wt%、F1275.67wt%、丙二醇16.20wt%、超纯水39.93wt%、冬凌草甲素0.40%,F127与甘油单油酸酯的质量比为15%。载冬凌草甲素的植烷三醇立方液晶纳米粒的最优原料处方为:植烷三醇50.40wt%、F1276/05wt%、丙二醇12.60wt%、超纯水30.55wt%、冬凌草甲素0.40wt%,F127与植烷三醇的质量比为12%。最优均质条件为:均质压力1200bar,循环次数9次。
实施例5:冬凌草甲素液晶纳米粒的粒径、电位测量与形态表征
对于同一体系的冬凌草甲素立方液晶纳米粒的粒径大小是影响口服吸收的重要参数,它影响着粒子在肠上皮细胞的摄取,从而会导致不同的口服吸收效果,主要因为粒径小的粒子能被肠上皮细胞内吞转运到细胞内,而粒径大的粒子不易被细胞内吞。取实施例4在均质压力为1200bar,循环次数为9次的均质条件下制备的冬凌草甲素立方液晶纳米粒进行粒径、电位测定和形态表征。(粒径测量方法同实施例3)。形态表征采用透射电镜法:取冬凌草甲素立方液晶纳米粒4μL滴加到铜网上,用滤纸吸去多余的液体,磷钨酸负染,使用透射电镜(JEM1400)观察冬凌草甲素立方液晶纳米粒的形态,并拍照。
粒径测定结果数据汇总见表4。结果表明,所制备的载冬凌草甲素的甘油单油酸酯立方液晶纳米粒和植烷三醇立方液晶纳米粒,平均粒径分别为220.8±4.9nm和250.9±5.6nm,粒径分布集中,PDI较小,平均电位分别为-22.1±3.4mv和-14.0±2.1mv,该平均电位值可以使纳米粒之间形成排斥力,避免巨大的比表面积引起的聚集,从而使纳米粒能够克服制备和储存过程中存在的稳定性问题。
表4冬凌草甲素立方液晶纳米粒的粒径分布、电位(n=3)
透射电镜结果见图6。结果表明,甘油单油酸酯立方晶纳米粒的粒径分布约为220nm左右(图6A),植烷三醇立方液晶纳米粒约为250nm左右(图6B),与马尔文粒径测定仪测得的粒径相符。本实施例制备的两种冬凌草甲素立方液晶纳米粒形态规整,粒径分布均一,并且纳米粒互相没有粘连。
实施例6:冬凌草甲素在立方液晶纳米粒体系中的晶型测定
对两种体系的冬凌草甲素立方液晶纳米粒进行粉末X射线衍射和差示扫描量热的测定以表征两种冬凌草甲素立方液晶纳米粒中冬凌草甲素的存在状态。测试样品采用冻干法制备成粉末样品:分别取2mL实施例4在均质压力为1200bar,循环次数为9次的均质条件下制备的冬凌草甲素立方液晶纳米粒溶液,分装于10mL棕色西林瓶中,加入甘露醇作为冻干保护剂,浓度为5%,置于-20℃冰箱预冻24h后,迅速转移至冷冻干燥机搁板上,于搁板上冷冻干燥48小时,所采用的温度为10℃,冷阱温度为-50℃,真空度为0.1Mbar。所得即为冬凌草甲素立方液晶纳米粒冻干粉。
粉末X射线衍射测定条件如下:采用Cu靶Ka射线,电压:40kv,电流:40mA,衍射角度:5-50°,步长:0.1秒。
甘油单油酸酯的冬凌草甲素立方液晶纳米粒体系:对冬凌草甲素原料药(oridonin)、甘油单油酸酯(GMO)、泊洛沙姆407(Poloxamer407)、甘露醇(mannitol)、冬凌草甲素与辅料的物理混合物(physical mixture)和本实施例中所制备的载冬凌草甲素的GMO立方液晶纳米粒冻干粉进行XRD分析。
植烷三醇的冬凌草甲素立方液晶纳米粒体系:对冬凌草甲素原料药(oridonin)、植烷三醇(PYT)、泊洛沙姆407(Poloxamer407)、甘露醇(mannitol)、冬凌草甲素与辅料的物理混合物(physical mixture)和本实施例中所制备的载冬凌草甲素的PYT立方液晶纳米粒冻干粉进行XRD分析。
两种体系的X射线衍射法表征结果见图7,结果表明,冬凌草甲素原料药在多个角度出现特征的衍射峰,表明冬凌草甲素原料药是以晶体形式存在。
图7A结果表明,对于GMO体系,在原辅料的物理混合物中,冬凌草甲素在7.5°处的特征衍射峰仍然存在,其余角度的特征衍射峰被甘露醇的强衍射峰所掩盖,表明辅料的存在不影响冬凌草甲素的晶型。而在GMO立方液晶纳米粒冻干粉的XRD图谱中,甘露醇的特征图谱峰仍然存在,但是被减弱,泊洛沙姆407的两个特征衍射峰被甘露醇在同一角度的特征衍射峰所掩盖,而冬凌草甲素在7.5°处的特征衍射峰完全消失,表明在所制备GMO立方液晶纳米粒冻干粉中,冬凌草甲素是以无定形态存在的。由Noyes-Whitney方程可知,药物与溶出介质的接触面积是影响难溶性药物溶出速率的关键参数。药物包载进液晶结构中后,药物由晶体颗粒转变成无定型的分子态,均匀分布于亲脂域中,双连续水通道与脂质双分子层的巨大交界面积显著增加了药物的溶出表面积。因此,冬凌草甲素是以无定形态存在有利于其均匀分布于亲脂域中,增加与溶出介质的接触面积,提高生物利用度。
图7B结果表明,对于PYT体系,在原辅料的物理混合物中,冬凌草甲素在7.5°处的特征衍射峰也仍然存在,表明辅料的存在不影响冬凌草甲素的晶型。而在PYT立方液晶纳米粒冻干粉的XRD图谱中,甘露醇的特征图谱峰仍然存在,只是被减弱,泊洛沙姆407的两个特征衍射峰被甘露醇在同一角度的特征衍射峰所掩盖,而冬凌草甲素在7.5°处的特征衍射峰完全消失,表明在所制备PYT立方液晶纳米粒冻干粉中,冬凌草甲素也是以无定形态存在的,有利于其均匀分布于亲脂域中,增加与溶出介质的接触面积,提高生物利用度。
对本实施例中的两个体系分别进行差示扫描量热法(DSC)测定条件如下:参比物为空铝坩,升温速度为10K/min,温度设置范围40~300℃,测定气体为氮气。
差示扫描量热法(DSC)表征图谱结果见图8。
在图8A中,冬凌草甲素在261.0℃有一个明显的吸热峰,GMO是以无定形态存在,泊洛沙姆407在55℃有吸收峰,甘露醇(mannitol)在171.5℃有一个明显的吸热峰,对于GMO体系中物理混合物的冬凌草甲素(259.3℃)、甘露醇(170.6℃)、泊洛沙姆407(46.1℃)均出现特征的吸热峰,表明辅料的存在没有影响药物的结晶型,而在甘油单油酸酯立方液晶纳米粒冻干粉有甘露醇的特征吸热峰(167.2℃),泊洛沙姆的特征吸热峰(43.2℃)发生了位移,冬凌草甲素的特征吸收峰消失,这一结果可作为本实施例中XRD表征结果的补充,证明冬凌草甲素在所制备甘油单油酸酯立方液晶纳米粒中的原晶型已不存在,药物以无定形的形式存在,有利于其均匀分布于亲脂域中,增加与溶出介质的接触面积,提高生物利用度。
在图8B中,PYT也是以无定形态存在,对于PYT体系中物理混合物的冬凌草甲素(258.2℃)、甘露醇(170.3℃)、泊洛沙姆407(46.1℃)均出现特征的吸热峰,表明辅料的存在没有影响药物的结晶型,而在甘油单油酸酯立方液晶纳米粒冻干粉有甘露醇的特征吸热峰(167.2℃)和泊洛沙姆407的特征吸热峰(51.2℃),而冬凌草甲素的特征吸收峰消失,这一结果也可作为本实施例中XRD表征结果的补充,充分证明冬凌草甲素在植烷三醇立方液晶纳米粒中的原晶型已不存在,药物也以无定形的形式存在,有利于其均匀分布于亲脂域中,增加与溶出介质的接触面积,提高生物利用度。
实施例7:冬凌草甲素立方液晶纳米粒的释放行为及释放模型
对实施例4在均质压力为1200bar,循环次数为9次的均质条件下所制备的冬凌草甲素立方液晶纳米粒进行体外药物释放行为研究。冬凌草甲素体外释放试验对照组制备:精密冬凌草甲素5mg,并用丙二醇溶解制备为含药量为0.45wt%的混悬液。分别取等量含药立方液晶纳米粒和含药混悬液置于透析袋中,其余步骤同实施例1,计算冬凌草甲素累积释放百分率,绘制体外释放曲线。
体外释放结果见图9。结果表明,在PH6.8PBS中释放前2小时内,冬凌草甲素溶液释放21.43%,而载冬凌草甲素的GMO立方液晶纳米粒和PYT立方液晶纳米粒分别释放13.41%和8.35%。冬凌草甲素丙二醇溶液6小时内释放91.87%,证明透析袋对冬凌草甲素的释放无明显阻滞作用。24小时内,80%以上的冬凌草甲素从两种立方液晶体系中释放出来,具有明显的24h缓释作用。
为了阐明冬凌草甲素从两种立方液晶纳米粒中释放的机制,本实施例对冬凌草甲素GMO立方液晶纳米粒和冬凌草甲素PYT立方液晶纳米粒的释药曲线分别按照零级方程、一级方程、Higuchi方程和Weibull方程进行拟合。以相关系数R2值,确定释药曲线的最佳拟合模型。各模型公式如下:
零级释放模型:y=k1t+a1
一级释放模型:ln(100-y)=k2t+a2
Higuchi方程:y=k3t0.5+a3
Weibull方程:
上述公式中,y表示药物累计释放率,t为取样时间,a1,a2,a3和a4为常数,k1,k2,k3和k4为方程的释放速率常数。
两种体系的释药曲线拟合结果见表5和6。释药模型拟合结果表明,对于两种体系来说,Higuchi方程进行拟合相关系数R2值最大,体外释放度曲线符合Higuchi方程,GMO体系:y=17.5130t0.5+1.8128(R2=0.9924);PYT体系:y=16.9450t0.5+2.4840(R2=0.9972),表明两种冬凌草甲素液晶纳米粒的释放机制均以扩散为主,冬凌草甲素从PYT立方液晶纳米粒体系的释药速率慢于GMO立方液晶纳米粒体系,体外释放度结果表明,两种载药纳米粒缓释作用明显,均能持续缓释24h。
表5冬凌草甲素GMO立方液晶纳米粒各种释药模型拟合结果
R:相关系数
表6冬凌草甲素PYT立方液晶纳米粒多种释药模型拟合结果
R:相关系数
实施例8:冬凌草甲素立方液晶纳米粒处方工艺的重复性
为考察处方工艺的重复性,本实施例依照实施例4中的最优原料处方以及最优工艺参数分别制备了三批冬凌草甲素立方液晶纳米粒,并以体外释放度(方法同实施例1)为指标,考察处方工艺的重复性。根据体外释放度试验的每个时间点的累计释放百分率,计算相似因子f2值,两两比较释药曲线的批间差异,f2值计算公式如下:
f2=50lg{[1+1/n∑Wt(Rt-Tt)2]-0.5×100}
其中,f2为相似因子;Rt为参比样品t时间的累积释放百分率;Tt为试验样品t时间的累积释放百分率;n为释放度试验取样点数;Wt为权重因子。
一般认为,当50≤f2≤100时,制剂具有相似的体外释放度。f2值越大,表明两种制剂体外释放度越接近;当f2为100时,体外释放度完全相同;当f2值越趋近于0,表明释放度差异越大。
结果见图10。图10A结果表明,所制备三个批次的GMO立方液晶纳米粒两两比较f2值,其结果分别分别为82.29(batch 1vs batch 2),75.02(batch 2vs batch 3),90.42(batch 1vs batch 3),各批次间具有相似的体外释放度,批间差异小,制剂释放度重现性良好。图10B结果表明,所制备三个批次的PYT立方液晶纳米粒两两比较f2值,其结果分别分别为85.22(batch 1vs batch 2),75.35(batch 2vs batch 3),87.49(batch 1vs batch 3),各批次间具有相似的体外释放度,批间差异较小,制剂释放度重现性良好。
实施例9:冬凌草甲素立方液晶纳米粒的药代动力学研究
本实施例的体内药代动力学研究采用昆明种小鼠,数量165只,平均分为5组,实验前禁食12h以上,自由饮水。经灌胃给予GMO体系和PYT体系最优处方制备的载冬凌草甲素的立方液晶纳米粒(分别为实验组1与实验组2)、实施例2中的F127/GMO=20%与F127/PYT=20%制备的载冬凌草甲素的立方液晶纳米粒(分别为实验组3与实验组4)以及冬凌草甲素混悬液(对照组,由于目前市售的冬凌草甲素均为复方制剂,无法作为参照,因此本实施例采用冬凌草甲素混悬液作为参比制剂),所用剂量为20mg·kg-1,于给药后0.083、0.25、0.5、1、1.5、2、3、6、9、12、24h时摘除眼球取血(每个时间点3只小鼠)。
所取血样置于置肝素化离心管中,4000r·min-1离心10min;在无日光照射,采用较暗灯光照明的情况下,精密吸取血浆样品100μL,置于5mL肝素钠管中,精密量取乙酸乙酯2mL,涡旋混合器充分混合5分钟,4000r·min-1离心10min,吸取上层有机相1.5mL置于另一5mL塑料离心管中,使用氮吹仪吹干,加入甲醇100μL,再用涡旋混合器充分混合5分钟,4000r·min-1离心10min。取上清液20μL进行HPLC分析测定血药浓度,所用色谱条件为:Ultimate(C18,4.6mm×250mm,5μm);预柱:Eclipse,XDB-C8(4.6mm×12.5mm,5μm);流动相:60%(v/v)甲醇,40%(v/v)水;流速:1.0mL·min-1;检测波长:237nm;进样体积:20μL。
测得的血药浓度和时间数据用Winnolin药代动力学软件进行非房室模型拟合,血药浓度时间曲线如图11和12所示,药代动力学参数如表7所示。
由于两种制剂与参比制剂的给药剂量相同,载冬凌草甲素的甘油单油酸酯立方液晶纳米粒和植烷三醇立方液晶纳米粒分别相对于冬凌草甲素混悬液的相对生物利用度按下式计算:
冬凌草甲素的植烷三醇立方液晶纳米粒的半衰期T1/2为11.65±3.73h,冬凌草甲素的甘油单油酸酯立方液晶纳米粒的半衰期T1/2为8.51±2.29h,与参比制剂冬凌草甲素混悬液相比,半衰期明显延长,表明冬凌草甲素经立方液晶纳米粒包封后,延长了药物在体内的作用时间,且植烷三醇立方液晶纳米粒的缓释作用更为明显,这一结果经统计分析具有显著性差异(p<0.01);两种载冬凌草甲素的立方液晶纳米粒与参比制剂冬凌草甲素混悬液的Cmax、AUC0-t进行统计学检验可知,p<0.01,表明均具有显著性差异。经计算得,实验组1的冬凌草甲素的甘油单油酸酯立方液晶纳米粒的相对生物利用度为311.33%,实验组2的冬凌草甲素的植烷三醇立方液晶纳米粒的相对生物利用度为460.05%,表明两种立方液晶纳米粒相比原料药混悬液对照组的相对生物利用度有明显的提高,且植烷三醇立方液晶纳米粒相比甘油单油酸酯立方液晶纳米粒,相对生物利用度提高更为显著。另外,实验组3的生物利用度低于实验组1,实验组4的生物利用度低于实验组2,这是因为实验组3与实验组4的冬凌草甲素立方液晶纳米粒中的稳定剂含量分别高于实验组1和实验组2,稳定剂含量过高会破坏部分纳米粒中的液晶结构,药物的包封、释放和吸收均受到一定影响,从而影响生物利用度,这与实施例2中的结果是一致的。
表7单剂量口服载冬凌草甲素甘油单油酸酯立方液晶纳米粒、载冬凌草甲素的植烷三醇立方液晶纳米粒及冬凌草甲素混悬液20mg/kg后的药代动力学参数(n=6)
注:*p<0.05;**p<0.01
T1/2:消除半衰期,Tmax:血药浓度达峰时间,Cmax:血浆药物峰浓度,AUC:药时曲线下面积,MRT:平均滞留时间。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!