药物制剂的制作方法
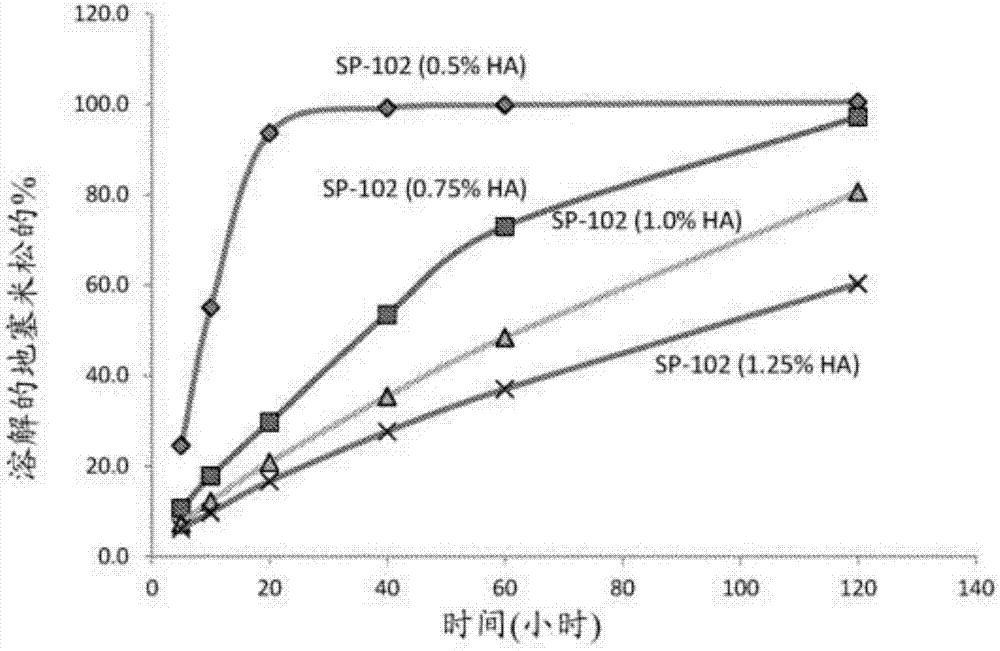
相关申请的交叉引用本申请要求2015年1月21日提交的美国临时专利申请号62/106,045的优先权,所述专利申请的公开内容据此以引用的方式整体并入本文。发明领域本申请涉及包含可溶性皮质类固醇和增粘剂的药物组合物。药物组合物适用于局部施用,诸如硬膜外注射、关节内注射或病灶内注射。发明背景在脊柱中,硬膜外腔(epiduralspace)(也称为“硬膜外腔(extraduralspace)”或“硬膜外腔(periduralspace)”)是脊管的最外侧部分。它是位于硬脑膜(其包围蛛网膜、蛛网膜下腔、脑脊液和脊髓)外的管(由周围的椎骨形成)内的腔。在人类中,硬膜外腔含有淋巴管、脊神经根、松散脂肪组织、小动脉和被称为硬膜外静脉丛的大型薄壁血管网络。硬膜外类固醇注射是一种微创手术,其可以帮助减轻由发炎的脊神经引起的个体的颈部、手臂、背部和腿部疼痛。例如,可以进行硬膜外类固醇注射以在个体中减轻由椎管狭窄、脊椎滑脱或椎间盘突出引起的疼痛。药物通过硬膜外腔(脊髓的保护性覆盖层(硬脑膜)与椎骨之间的区域)递送到脊神经。皮质类固醇注射可以减少炎症,并且可以在直接递送到个体的疼痛区域时有效。硬膜外类固醇注射的目标是将药剂放置在脊柱内的损伤和/或病理区域附近。层间注射、尾部注射和经椎间孔注射通常用于硬膜外类固醇注射。通过将针放置在硬膜外腔中的椎骨之间,然后注射药物来进行层间硬膜外注射。尾部注射是进入到硬膜外腔的最低部分中,进入到含有脊液的膜与椎骨间最厚的韧带之间的区域中的注射。经椎间孔注射是进入到位于神经根离开的脊柱的一侧处的开口(也称为孔)中的注射。泼尼松龙是具有主要糖皮质激素活性和低盐皮质激素活性的皮质类固醇药物,可用于治疗各种各样的炎症和自身免疫病状,诸如哮喘、葡萄膜炎、坏疽性脓皮病、类风湿性关节炎、溃疡性结肠炎、颞动脉炎和克罗恩氏病、贝尔氏麻痹、多发性硬化、丛集性头痛、血管炎、急性淋巴母细胞性白血病和自身免疫性肝炎、系统性红斑狼疮、川崎病和皮肌炎。甲泼尼龙通常因为其抗炎效果而被使用。甲泼尼龙指定的医学病状的列表相当长,并且与其他皮质类固醇(诸如泼尼松龙)相似。常见的用途包括关节炎疗法以及由于各种呼吸系统疾病引起的支气管炎症或急性支气管炎的短期治疗。它用于自身免疫疾病(最值得注意地系统性红斑狼疮)的急性期治疗和长期管理。它也用作多发性硬化症的治疗。地塞米松是糖皮质激素类的类固醇药物的有效合成成员。它用作抗炎剂和免疫抑制剂。地塞米松用于治疗许多炎症和自身免疫病状,诸如类风湿性关节炎和支气管痉挛。地塞米松也可用于治疗特发性血小板减少性紫癜,其是由于免疫问题引起的血小板数量减少。曲安奈德是具有明显的抗炎作用的合成皮质类固醇。-10注射剂(曲安奈德可注射悬浮液,usp)是无菌水性悬浮液中的曲安奈德,其适用于病灶内注射和关节内注射,并且不适用于静脉内、肌内、眼内、硬膜外或鞘内使用。每ml无菌水性悬浮液提供10mg曲安奈德,其中具有氯化钠用于等渗、0.9%(w/v)苄醇作为防腐剂、0.75%羧甲基纤维素钠和0.04%聚山梨醇酯80;可以加入氢氧化钠或盐酸以将ph调节到5.0与7.5之间。现有的药物组合物可能对缓解疼痛具有即时的或短期的效果。这可能足以用于短期施用,如此以致克服疼痛的急性发作或加重。然而,此类制剂可能需要重复施用,特别是对于持续疼痛或慢性疼痛。此外,对于局部疼痛,导致活性成分扩散到靶区域之外的硬膜外注射可能是不需要的,并且可能增加对总体较高剂量的需要以确保靶区域暴露于有效剂量。此外,促成组合物的非预期放置的药物组合物和施用方法可以导致不需要的效果,诸如由硬膜外注射引起的蛛网膜炎。存在对改进的药物组合物的需要,所述改进的药物组合物可以提供快速的局部起效以及长期的持久效果;具有便于注射到身体各个部位中的物理特征;并且是耐储存的。具体地说,需要适合于硬膜外注射、关节内注射或病灶内注射的稳定、长效的药物组合物。这种需要的一种解决方案是以包含在水中呈可溶性形式和不溶性形式的皮质类固醇的药物组合物的形式提出(pct国际公布号wo2014/116876)。皮质类固醇的可溶性形式提供立即减轻,而不溶性形式提供更持久的效果。然而,在2014年,美国食品与药品管理局(fda)发出警告,将皮质类固醇注射到脊柱的硬膜外腔可导致罕见但严重的不良反应,诸如失明、中风、瘫痪和死亡。作为回应,批准了由多学会疼痛工作组(multi-societypainworkgroup)(mpw)提出的17项安全建议,包括颗粒类固醇不应用于经椎间孔注射的建议。经椎间孔注射是用于施用疼痛药剂的有吸引力的途径,因为注射部位位置最接近假定的炎症部位。因此,存在对改进的药物组合物的需要,所述改进的药物组合物可以提供快速的起效和长期的持久效果并且被批准用于所有三种注射途径(经椎间孔、层间和尾部)。发明概述在一方面,本申请公开了包含可溶性皮质类固醇和至少一种增粘剂的水性药物组合物,其中药物组合物的粘度位于1kcp与200kcp之间。在一个实施方案中,本申请公开了水性药物组合物,其中可溶性皮质类固醇选自由以下组成的组的盐和酯:地塞米松、甲泼尼龙、泼尼松龙和曲安奈德。在另一个实施方案中,可溶性皮质类固醇选自由以下组成的组:地塞米松磷酸钠、甲泼尼龙琥珀酸钠、泼尼松龙琥珀酸钠和曲安奈德磷酸酯。在又另一个实施方案中,可溶性皮质类固醇为地塞米松磷酸钠。在一个实施方案中,至少一种增粘剂选自由以下组成的组:透明质酸钠、透明质酸、交联透明质酸、聚乙烯吡咯烷酮、羟丙基甲基纤维素、羟丙基纤维素、羟乙基纤维素和甘油。在另一个实施方案中,至少一种增粘剂为透明质酸钠。在一些实施方案中,可溶性皮质类固醇为地塞米松磷酸钠,并且至少一种增粘剂为透明质酸钠。在一些实施方案中,水性药物组合物包含小于2%w/v的增粘剂。在再其他实施方案中,水性药物组合物还包含防腐剂和/或麻醉剂。在另一方面,本申请提供了用于在有需要的个体中治疗炎症和/或疼痛的方法,所述方法包括将本文公开的水性药物组合物注射到个体中。在一个实施方案中,药物组合物被注射到硬膜外腔中。在另一实施方案中,使用小于20n的力以约0.5”/分钟的速率将药物组合物注射到硬膜外腔中。在又另一个实施方案中,个体每1至12周用制剂注射一次。在一个实施方案中,可溶性皮质类固醇选自由以下组成的组的盐和酯:地塞米松、甲泼尼龙、泼尼松龙和曲安奈德。在另一个实施方案中,可溶性皮质类固醇选自由以下组成的组:地塞米松磷酸钠、甲泼尼龙琥珀酸钠、泼尼松龙琥珀酸钠和曲安奈德磷酸酯。在又另一个实施方案中,可溶性皮质类固醇为地塞米松磷酸钠。在一个实施方案中,至少一种增粘剂选自由以下组成的组:透明质酸钠、透明质酸、交联透明质酸、聚乙烯吡咯烷酮、羟丙基甲基纤维素、羟丙基纤维素、羟乙基纤维素和甘油。在另一个实施方案中,至少一种增粘剂为透明质酸钠。在一些实施方案中,可溶性皮质类固醇为地塞米松磷酸钠,并且至少一种增粘剂为透明质酸钠。在一些实施方案中,制剂包含小于2%w/v的增粘剂。在又其他实施方案中,制剂还包含防腐剂和/或麻醉剂。在又另一方面,本申请提供了包含本文公开的水性药物组合物的注射器。附图简述图1示出了sp-102制剂在磷酸盐缓冲液(0.05m,ph7)和0.5%吐温80中的溶解曲线。图2示出了sp-102制剂在磷酸盐缓冲液(0.05m,ph7)中的溶解曲线。图3示出了在用对照(3a)和制剂sp-102(0.5%ha)(3b)和sp-102(1.25%ha)(3c)注射的动物#1中注射后立即记录的图像。图4示出了在用对照(4a)和制剂sp-102(0.5%ha)(4b)和sp-102(1.25%ha)(4c)注射的动物#1中注射后30分钟记录的图像。图5示出了在用对照(5a)和制剂sp-102(0.5%ha)(5b)和sp-102(1.25%ha)(5c)注射的动物#1中注射后60分钟记录的图像。图6示出了在用对照(6a)和制剂sp-102(1.25%ha)(6b)注射的动物#1中注射后120分钟记录的图像。图7示出了在用制剂sp-102(1.25%ha)注射的动物#1中注射后180分钟记录的图像。图8示出了在用制剂sp-102(1.0%ha)(8a)和sp-102(1.25%ha)(8b)注射的动物#2中注射后立即记录的图像。图9示出了在用制剂sp-102(1.0%ha)(9a)和sp-102(1.25%ha)(9b)注射的动物#2中注射后30分钟记录的图像。图10示出了在用制剂sp-102(1.0%ha)(10a)和sp-102(1.25%ha)(10b)注射的动物#2中注射后60分钟记录的图像。图11示出了在用制剂sp-102(1.0%ha)(11a)和sp-102(1.25%ha)(11b)注射的动物#2中注射后120分钟记录的图像。图12示出了在用制剂sp-102(1.25%ha)注射的动物#2中注射后180分钟记录的图像。图13示出了对于制剂sp-102(0.5%ha)、sp-102(1.0%ha)和sp-102(1.25%ha)的作为时间函数的可见造影染料的估计百分比的曲线图。图14示出了对于商业可注射类固醇产品的作为时间函数的可见造影染料的估计百分比的曲线图。发明详述本申请涉及包含可溶性皮质类固醇和增粘剂的药物组合物。药物组合物适用于局部施用,诸如硬膜外注射、关节内注射或病灶内注射。适用于本申请的皮质类固醇包括以下的盐或酯:甲泼尼龙、地塞米松、泼尼松龙和曲安奈德。本发明人已发现将可溶性形式的皮质类固醇与增粘剂组合在药物组合物中以用于局部注射的优点。增粘剂显著地延长了注射部位处的直接皮质类固醇暴露的持续时间。增粘剂还提供类固醇的缓释。释放的类固醇以持续和/或延长的方式在靶部位(诸如发炎神经和组织)上起效。较长的持久效果可允许类固醇周期性注射而不是每日注射,这是难以通过硬膜外或关节内施用做到的。先前已描述了可溶性类固醇和增粘剂的混合物以用于眼内施用(ep0244178)。然而,眼科制剂的粘度通常为25-50cps。为了实现通过硬膜外注射施用的可溶性类固醇的足够长的持久效果,注射溶液的粘度必须高得多(最小为2000-3000cps)。然而,高粘度溶液可能难以在没有过度的力的情况下进行注射。例如,麻醉药物布比卡因与1%(w/w)透明质酸的混合物不能通过硬膜外针或导管来施用(dollo,g.等intl.j.pharmaceutics,2004,272,109-119)。本发明提供了可溶性类固醇和增粘剂的混合物,其具有用于局部施用(诸如硬膜外注射、关节内注射或病灶内注射)的可注射性和注射能力的所需特征。制剂组分可溶性皮质类固醇。可溶性皮质类固的非限制性实例包括以下的盐或酯:地塞米松、甲泼尼龙、泼尼松龙和曲安奈德。可溶性皮质类固醇可具有一定范围的溶解度,但是它足够可溶以溶解在药物制剂中。皮质类固醇的溶解度部分地由其化学形式(诸如盐或酯)来决定。皮质类固醇的可溶性形式包括其盐,诸如钠盐、磷酸盐、琥珀酸盐以及其组合。可溶性皮质类固的非限制性实例包括地塞米松磷酸钠、甲泼尼龙琥珀酸钠、泼尼松龙琥珀酸钠和曲安奈德磷酸酯。应当理解,可溶性皮质类固醇凭借在施用至个体之后从增粘剂中释放可以提供快速和持久的效果。在一些实施方案中,将可溶性皮质类固醇注射到个体中,提供皮质类固醇的药效持续至少约1周、约2周、约3周、约4周、约5周、约6周、约7周、约8周、约9周、约10周、约11周或约12周。在一些实施方案中,皮质类固醇的药效提供有效减少或抑制炎症和/或疼痛的量。在一些实施方案中,皮质类固醇的药效提供有效抑制炎症和/或疼痛持续多达8周的量。在一些实施方案中,皮质类固醇的药效提供有效减少炎症和/或疼痛持续多达12周的量。应当理解,本文所述的药物组合物基本上不含不溶性皮质类固醇。在一些实施方案中,药物组合物完全不含不溶性皮质类固醇。增粘剂。增粘剂被包含在药物组合物中。增粘剂提供以下优点:当将药物组合物施用到靶部位(例如,个体的硬膜外腔)中时,由于粘性制剂在靶部位中的流通程度低而使制剂在靶部位中停留时间更长。增粘剂还可以促进活性药物与靶部位的结合,并且局部增强药物吸收和生物利用率。组合物的粘度还有助于药物组合物的稳定性。较高的粘度可改进保存期。组合物的粘度在很大程度上受增粘剂的量影响。与较低浓度相比,较高浓度的增粘剂产生较高的粘度。温度也影响粘度,其中与相同组合物的较高温度相比,较低温度产生更高的粘度。合适的增粘剂包括透明质酸钠、透明质酸、聚乙烯吡咯烷酮(pvp)、交联透明质酸、羟丙基甲基纤维素、羟丙基纤维素、羟乙基纤维素、甘油或其混合物。优选的增粘剂包括透明质酸钠、聚乙烯吡咯烷酮(pvp)、羟丙基纤维素钠和羧甲基纤维素。由于潜在的副作用,本发明制剂不包含聚乙二醇。增粘剂的量基于所使用的试剂,并且通常为约0.05%-30%(w/v)的量。在一些实施方案中,增粘剂的浓度为约0.1%w/v、约0.25%w/v、约0.5%w/v、约0.75%w/v、约1.0%w/v、约1.1%w/v、约1.15%w/v、约1.20%w/v、约1.25%w/v、约1.30%w/v、约1.35%w/v、约1.40%w/v、约1.45%w/v或约1.5%w/v。在一些实施方案中,增粘剂的浓度位于0.05%w/v与1.5%w/v之间;0.05%w/v与0.5%w/v之间;0.1%w/v与3.0%w/v之间;0.1%w/v与1.5%w/v之间;0.1%w/v与1.0%w/v之间;0.5%w/v与1%w/v之间;0.5%w/v与2.5%w/v之间;1.0%w/v与3.0%w/v之间;1.0%w/v与1.5%w/v之间;1.0%w/v与1.25%w/v之间;1.25%w/v与1.5%w/v之间;或1.5%w/v与3.0%w/v之间。在一些实施方案中,增粘剂的分子量位于500kda与5.0mda之间;500kda与3.0mda之间;500kda与2.0mda之间;500kda与1.0mda之间;500kda与2.0mda之间;1.0mda与3.0mda之间;1.0mda与2.5mda之间;1.0mda与2.0mda之间;以及1.2mda与1.8mda之间。在一些实施方案中,透明质酸钠的分子量为约711kda;约880kda;约1.56mda;约1.8mda以及约2.65mda。在一些实施方案中,分子量为数均分子量,而在其他实施方案中,分子量为重均分子量。在一些前述实施方案中,增粘剂为透明质酸钠。在一些实施方案中,增粘剂为透明质酸或透明质酸盐的药学上可接受的盐,诸如钠盐、磷酸盐或钙盐。在一些实施方案中,药物组合物的粘度为约300kcp、约250kcp、约200kcp、约150kcp、约140kcp、约130kcp、约120kcp、约110kcp、约100kcp、约90kcp、约80kcp、约70kcp、约40kcp、约30kcp、约25kcp、约20kcp、约15kcp、约10kcp、约5kcp、约4kcp、约3kcp、约2kcp或约1kcp。在一些实施方案中,组合物的粘度位于1kcp与300kcp之间;1kcp与100kcp之间;1kcp与50kcp之间;1kcp与10kcp之间;10kcp与50kcp之间;10kcp与100kcp之间;50kcp与100kcp之间;100kcp与300kcp之间;50kcp与200kcp之间;75kcp与180kcp之间;100kcp与150kcp之间;150kcp与200kcp之间;200kcp与250kcp之间;250kcp与300kcp之间。在一些实施方案中,药物组合物为凝胶。在替代实施方案中,药物组合物为水溶液。缓冲剂。用于与本文公开的药物组合物一起使用的合适的缓冲剂包括但不限于:有机酸盐,诸如柠檬酸、抗坏血酸、葡萄糖酸、碳酸、酒石酸、琥珀酸、乙酸或邻苯二甲酸的盐;三羟甲基氨基甲烷(tris)(三羟甲基氨基甲烷盐酸盐)或磷酸盐缓冲液。在一些实施方案中,缓冲剂为生理学上相容的。ph。制剂的ph可以固有地由制剂中存在的赋形剂提供;或者,可以采用ph调节剂。可以将ph调节剂(诸如缓冲剂或单纯的酸或碱)加入药物组合物中以将ph维持至6-8。例如,ph调节剂的量通常为0.1%-10%。在一些实施方案中,制剂的ph在生理范围内。渗透度。制剂的渗透度位于200mosm/kg与350mosm/kg之间、250mosm/kg与300mosm/kg之间、280mosm/kg与290mosm/kg之间。在一些实施方案中,制剂的渗透度在生理范围内。在一些实施方案中,药物组合物在人类中是等渗的。麻醉剂。在一个实施方案中,药物组合物还包含麻醉剂,诸如利多卡因、布比卡因或苯佐卡因。表面活性剂。本发明制剂优选不包含表面活性剂。然而,在一些实施方案中,药物组合物包含一种或多种非离子表面活性剂。包含表面活性剂使药物颗粒的溶解度和可湿性增加。合适的非离子表面活性剂包括聚山梨醇酯(例如,-80、-20)、泰洛沙泊、聚氧乙烯蓖麻油、泊洛沙姆(polaxamers)、聚乙二醇、辛酸甘油三酯、聚氧乙烯硬脂酸酯(例如氧化乙烯单硬脂酸酯)、聚氧乙烯化植物油和单硬脂酸甘油酯。优选的非离子表面活性剂为聚山梨醇酯,诸如-80。药物组合物中的非离子表面活性剂的量(如果存在的话)通常为药物组合物的0.001%-10%或0.01%-1%(w/v)。保存期。术语“保存期”是指药物组合物在不损失效力和/或性能概况的情况下可以储存的时间的量。在一些实施方案中,保存期是指药物组合物在不损失效力和/或性能的大于2%、5%、8%或10%的情况下可以储存的时间的量。本文提供的不含防腐剂的药物组合物被设计成具有至少12、24或36个月的保存期。在一些实施方案中,药物组合物的保存期位于12与24个月之间。在一些实施方案中,药物组合物在室温下储存并且保存稳定持续至少12、24或36个月。在一些实施方案中,药物组合物在低于室温下储存并且具有至少12、24或36个月的保存期。防腐剂。本发明制剂优选不包含防腐剂。然而,在一些实施方案中,药物组合物包含一种或多种防腐剂。包含防腐剂(诸如抗微生物防腐剂)使药物组合物的保存期增加。可以采用不与活性药物或任一赋形剂不利地相互作用的任何防腐剂。例如,防腐剂包括乙醇、苄醇、苯扎氯铵、苄索氯铵、苯甲酸、溴硝丙二醇、对羟基苯甲酸丁酯、西曲溴铵、氯己定。防腐剂的量范围可以为例如约0.01%-1%。示例性制剂在一个实施方案中,水性药物组合物包含可溶性皮质类固醇和至少一种增粘剂,其中水性药物组合物的粘度位于1kcp与200kcp之间。在一些实施方案中,水性药物组合物呈单位剂量并且体积为1ml、2ml、3ml、4ml、5ml、6ml、7ml、8ml或10ml。在一些实施方案中,增粘剂浓度位于0.05%w/v与1.5%w/v之间;0.05%w/v与0.5%w/v之间;0.1%w/v与1.5%w/v之间;0.1%w/v与1.0%w/v之间;0.5%w/v与1%w/v之间;0.5%w/v与2.5%w/v之间;1.0%w/v与1.5%w/v之间;1.0%w/v与1.25%w/v之间;或1.25%w/v与1.5%w/v之间。在一个实施方案中,药物组合物包含在水溶液(诸如水)中的可溶性甲泼尼龙琥珀酸钠和至少一种增粘剂,其中药物组合物的粘度位于1kcp与200kcp之间。在一些实施方案中,水性药物组合物呈单位剂量并且体积为1ml、2ml、3ml、4ml、5ml、6ml、7ml、8ml或10ml。在一些实施方案中,增粘剂浓度位于0.05%w/v与1.5%w/v之间;0.05%w/v与0.5%w/v之间;0.1%w/v与1.5%w/v之间;0.1%w/v与1.0%w/v之间;0.5%w/v与1%w/v之间;0.5%w/v与2.5%w/v之间;1.0%w/v与1.5%w/v之间;1.0%w/v与1.25%w/v之间;或1.25%w/v与1.5%w/v之间。在一些实施方案中,甲泼尼龙的每次注射的剂量范围为在1至10ml无菌溶液(诸如注射用水或盐水)中的20至120mg/剂量。在一个实施方案中,药物组合物包含在水溶液(诸如水)中的可溶性泼尼松龙琥珀酸钠和至少一种增粘剂,其中药物组合物的粘度位于1kcp与200kcp之间。在一些实施方案中,水性药物组合物呈单位剂量并且体积为1ml、2ml、3ml、4ml、5ml、6ml、7ml、8ml或10ml。在一些实施方案中,增粘剂浓度位于0.05%w/v与1.5%w/v之间;0.05%w/v与0.5%w/v之间;0.1%w/v与1.5%w/v之间;0.1%w/v与1.0%w/v之间;0.5%w/v与1%w/v之间;0.5%w/v与2.5%w/v之间;1.0%w/v与1.5%w/v之间;1.0%w/v与1.25%w/v之间;或1.25%w/v与1.5%w/v之间。在一些实施方案中,泼尼松龙的每次注射的剂量范围为在1至10ml无菌溶液(诸如注射用水或盐水)中的20至120mg/剂量。在一个实施方案中,药物组合物包含在水溶液(诸如水)中的可溶性地塞米松磷酸钠和至少一种增粘剂,其中药物组合物的粘度位于1kcp与200kcp之间。在一些实施方案中,水性药物组合物呈单位剂量并且体积为1ml、2ml、3ml、4ml、5ml、6ml、7ml、8ml或10ml。在一些实施方案中,增粘剂浓度位于0.05%w/v与1.5%w/v之间;0.05%w/v与0.5%w/v之间;0.1%w/v与1.5%w/v之间;0.1%w/v与1.0%w/v之间;0.5%w/v与1%w/v之间;0.5%w/v与2.5%w/v之间;1.0%w/v与1.5%w/v之间;1.0%w/v与1.25%w/v之间;或1.25%w/v与1.5%w/v之间。地塞米松的每次注射的剂量范围为在1至10ml无菌溶液(诸如注射用水或盐水)中的3至20mg/剂量。又在另一个实施方案中,药物组合物包含在水溶液(诸如水)中的可溶性曲安奈德磷酸酯和至少一种增粘剂,其中药物组合物的粘度位于1kcp与200kcp之间。在一些实施方案中,水性药物组合物呈单位剂量并且体积为1ml、2ml、3ml、4ml、5ml、6ml、7ml、8ml或10ml。在一些实施方案中,增粘剂浓度位于0.05%w/v与1.5%w/v之间;0.05%w/v与0.5%w/v之间;0.1%w/v与1.5%w/v之间;0.1%w/v与1.0%w/v之间;0.5%w/v与1%w/v之间;0.5%w/v与2.5%w/v之间;1.0%w/v与1.5%w/v之间;1.0%w/v与1.25%w/v之间;或1.25%w/v与1.5%w/v之间。曲安奈德的每次注射的剂量范围为在1至10ml无菌溶液(诸如注射用水或盐水)中的20mg至120mg/剂量。在另外的实施方案中,水性药物组合物包含可溶性地塞米松磷酸钠和透明质酸钠,其中水性药物组合物的粘度位于1kcp与200kcp之间。在一些实施方案中,透明质酸钠的分子量为500kda和2.0mda。在其他实施方案中,透明质酸钠的分子量为1.2mda和1.8mda。在一些实施方案中,透明质酸钠浓度位于0.05%w/v与1.5%w/v之间;0.05%w/v与0.5%w/v之间;0.1%w/v与1.5%w/v之间;0.1%w/v与1.0%w/v之间;0.5%w/v与1%w/v之间;0.5%w/v与2.5%w/v之间;1.0%w/v与1.5%w/v之间;1.0%w/v与1.25%w/v之间;或1.25%w/v与1.5%w/v之间。在一些实施方案中,水性药物组合物呈单位剂量并且体积为1ml、2ml、3ml、4ml、5ml、6ml、7ml、8ml、9ml或10ml。在另外的实施方案中,水性药物组合物包含可溶性地塞米松磷酸钠和透明质酸,其中水性药物组合物的粘度位于1kcp与200kcp之间。在一些实施方案中,透明质酸的分子量为500kda和2.0mda。在其他实施方案中,透明质酸的分子量为1.2mda和1.8mda。在一些实施方案中,透明质酸浓度位于0.05%w/v与1.5%w/v之间;0.05%w/v与0.5%w/v之间;0.1%w/v与1.5%w/v之间;0.1%w/v与1.0%w/v之间;0.5%w/v与1%w/v之间;0.5%w/v与2.5%w/v之间;1.0%w/v与1.5%w/v之间;1.0%w/v与1.25%w/v之间;或1.25%w/v与1.5%w/v之间。在一些实施方案中,水性药物组合物呈单位剂量并且体积为1ml、2ml、3ml、4ml、5ml、6ml、7ml、8ml、9ml或10ml。表1中的每种示例性制剂包含重量相当于5mg/ml的地塞米松的可溶性地塞米松磷酸钠和不同量的透明质酸钠。透明质酸钠的分子量为1.56mda。制剂还包含生理上相容的缓冲溶液,诸如15mmpbs溶液。每种制剂都被制备呈1ml、2ml、3ml、4ml、5ml、6ml、7ml、8ml、9ml和10ml单位剂量。表1.可溶性地塞米松磷酸盐和透明质酸的示例性制剂。表1中列出的每种制剂还任选地含有麻醉剂和/或防腐剂。在一些实施方案中,表1中公开的每种制剂的可溶性皮质类固醇可以用选自由以下组成的组的皮质类固醇来代替:甲泼尼龙琥珀酸钠、泼尼松龙琥珀酸钠和曲安奈德磷酸酯。包装和试剂盒。本发明制剂可以被包装在单位剂量小瓶或注射器中。它也可以被包装在双室小瓶或注射器中,其中可溶性类固醇和增粘剂各自在单独的隔室中。在一些实施方案中,单位剂量位于1ml与10ml之间;2ml与8ml之间;以及2ml与5ml之间。在一些实施方案中,单位剂量为约1ml、约2ml、约2.5ml、约3ml、约3.5ml、约4ml、约4.5ml、约5ml或约5.5ml。在任一前述实施方案中,单位剂量为凝胶药物组合物。在其他前述实施方案中,单位剂量为水性药物组合物。本公开还提供了包含本文公开的药物制剂和使用说明书的试剂盒。在一些前述实施方案中,药物组合物为无菌的。在一些前述实施方案中,药物组合物使用无菌技术来制备。例如,组合物的各种组分可以被单独地灭菌,并且然后在无菌条件下组合以提供无菌药物组合物。在一些前述实施方案中,药物组合物为最终灭菌的。方法本申请还提供了用于使用本文公开的任一水性药物组合物治疗炎症和/或疼痛(诸如与类风湿性关节炎、骨关节炎、下背痛、肌腱炎、椎管狭窄、椎间盘突出、脊神经根炎和慢性椎间盘疼痛相关的那些)的方法。在一个实施方案中,方法包括以下步骤:鉴定患有炎症和/或疼痛的个体,并且向个体的硬膜外腔注射本文公开的任一水性药物组合物。方法任选地包括向个体的硬膜外腔注射麻醉剂(诸如利多卡因、布比卡因或苯佐卡因)的步骤。麻醉剂可以单独注射的方式施用,或可与水性药物组合物组合并且一起注射。在另一个实施方案中,方法包括以下步骤:鉴定患有炎症和/或疼痛的个体,并且向个体的皮肤病灶注射本文公开的任一水性药物组合物。方法任选地包括向个体的皮肤病灶注射麻醉剂的步骤。麻醉剂可以单独注射的方式施用,或可与水性药物组合物组合并且一起注射。在另一个实施方案中,方法包括以下步骤:鉴定患有炎症和/或疼痛的个体,并且向个体的被疾病侵袭的关节注射本文公开的任一水性药物组合物。方法任选地包括向个体的被疾病侵袭的关节注射麻醉剂的步骤。麻醉剂可以单独注射的方式施用,或可与药物组合物组合并且一起注射。在一些实施方案中,所注射的类固醇的剂量基于类固醇的效力。在一些实施方案中,以单一剂量施用至个体的皮质类固醇的量位于2mg与20mg之间;5mg与15mg之间;以及5mg与10mg之间。在一些实施方案中,以单一剂量施用至个体的皮质类固醇的量为约2mg、5mg、8mg、10mg、15mg和20mg。在某些实施方案中,地塞米松的剂量为约3至20mg/剂量;甲泼尼龙的剂量为20至120mg/剂量,泼尼松龙的剂量为约20至120mg/剂量;曲安奈德的剂量为约20至120mg/剂量。在一些实施方案中,个体每1至12周;1至8周;1至4周;2至12周;4至12周;8至12周;2至8周;或2至4周用药物组合物注射一次。在一些实施方案中,个体约每1、2、4、6、8、10或12周用药物组合物注射。本文公开的方法和组合物可用于治疗哺乳动物(诸如人类、狗或猫)的个体。本文公开的方法和组合物具体地可用于治疗人类。其他用途。病灶内注射是药剂经皮直接递送到皮肤病灶中。病灶内注射被引入到病灶中或在病灶内进行。皮肤充当储存器,从而允许沉积在真皮中的药剂在一段时间内进行递送,产生延长的疗法同时避免或最小化全身疗法的不良反应。关节内注射是用于治疗炎性关节病状(诸如类风湿性关节炎、牛皮癣关节炎、痛风、肌腱炎、滑囊炎和偶尔的骨关节炎)的程序。将皮下注射针注射到被疾病侵袭的关节中,在其中所述皮下注射针递送抗炎剂,诸如皮质类固醇。本申请公开了具有一定范围粘度的药物组合物。粘度的选择部分地取决于注射的药物组合物在个体中的所需位置。例如,当需要药物组合物的局部量时,可以选择具有较高粘度的药物组合物。或者,如果需要药物组合物的更广泛的覆盖时,可以选择具有较低粘度的药物组合物。在一些实施方案中,方法包括通过经椎间孔注射施用药物组合物,其中药物组合物包含0.5%与1.5%之间、1.0%与1.5%之间或0.75%与1.25%之间的增粘剂。在一些实施方案中,方法包括通过层间注射施用药物组合物,其中药物组合物包含0.1%与1.5%之间、0.1%与1.0%之间、0.1%与0.75%之间、0.1%与0.5%之间、0.1%与0.25%之间、0.75%与1.5%之间、1.0%与1.5%之间或0.75%与1.25%之间的增粘剂。在一些实施方案中,方法包括通过尾部注射施用药物组合物,其中药物组合物包含0.1%与1.5%之间、0.1%与1.0%之间、0.1%与0.75%之间、0.1%与0.5%之间、0.1%与0.25%之间、0.75%与1.5%之间、1.0%与1.5%之间或0.75%与1.25%之间的增粘剂。在一些前述实施方案中,增粘剂为透明质酸或其盐。可注射性和注射能力。可注射性是可注射治疗剂在注射前从小瓶转移时容易穿过皮下注射针的能力。可注射性包括诸如抽出容易性、堵塞和起泡倾向以及剂量测量准确性等因素。注射能力是指注射期间制剂的性能。注射能力包括注射所需的压力或力、流动的均匀度以及免受堵塞(即,注射器针头没有堵塞)。可注射性和注射能力部分地受药物组合物的粘度、注射或转移流速以及针特征(诸如长度和容量)影响。注射能力的所需特征包括,例如在没有过度的力情况下平滑连续的注射。这种注射允许人员施用注射以在不引起过度应变的情况下维持对程序的连续控制。本申请公开了易于注射和/或可注射到个体中的组合物。本申请还公开了用于使用药物组合物对个体进行注射的方法,其中注射是容易的并且提供药物组合物的连续流动。在一些实施方案中,方法包括向注射器施加5n与90n之间、5n与50n之间、50n与100n之间、5n与25n之间、25n与50n之间或10n与40n之间的注射力。在一些实施方案中,方法包括向注射器施加不大于5n、不大于7n、不大于10n、不大于15n、不大于17n、不大于21n、不大于27n、不大于29n、不大于33n、不大于38n、不大于39n、不大于46n、不大于59n、不大于70n、不大于78n或不大于90n的力。在一些实施方案中,方法包括向注射器施加约5n、约7n、约10n、约15n、约17n、约21n、约27n、约29n、约33n、约38n、约39n、约46n、约59n、约70n、约78n或约90n的力。在一些实施方案中,注射力导致药物组合物以约0.4”/分钟、约0.5”/分钟、约0.6”/分钟、约0.7”/分钟、约0.8”/分钟、约0.9”/分钟、约1.0”/分钟、约1.1”/分钟、约1.2”/分钟、约1.3”/分钟、约1.4”/分钟、约1.5”/分钟、约1.75”/分钟、约2.0”/分钟、约2.25”/分钟或约2.36”/分钟的速率(即挤出速率)注射。在任一前述实施方案中,注射器包括具有19、20、21、22、23、24或25的针规的针。在一个实施方案中,注射器包括具有25的针规的针。本申请公开了用于减少“拉丝效应(stringingeffect)”的注射方法。拉丝效应是指当向个体注射药物组合物完成时,注射中使用的针孔中的剩余组合物与个体接触的现象。例如,当针从靶部位抽出时,针孔中的剩余组合物由于组合物的粘性性质而被拉出并且像丝一样拉长。当针离开个体时,它可能留下组合物的拖曳,从而潜在地将非预期区域和组织暴露于组合物。组合物的非预期放置可以导致不需要的效果,诸如由硬膜外注射引起的蛛网膜炎。在一些情况下,在针抽出时,注射到靶部位中的组合物可拉长并且伸展,并且可与个体的非预期区域和组织接触。在一些实施方案中,本文公开的方法和组合物减少了拉丝效应的发生。在一些实施方案中,在从注射部位抽出时,没有本文公开的药物组合物离开针进入到个体中。在一些实施方案中,药物组合物仅在施加注射力时进入个体。在一些实施方案中,组合物实现在分离或分隔时具有非常小的拉丝的干净断裂。在一些实施方案中,本申请公开了用于在有需要的个体中治疗炎症和/或疼痛的方法,所述方法包括将本文公开的水性药物组合物注射到个体的硬膜外、病灶内或关节内的腔中;并且其中方法包括选自由以下组成的组的一个或多个步骤:1)施加小于5n、小于7n、小于10n、小于15n、小于17n或小于21n的力以便使水性药物组合物以约0.4”/分钟、约0.5”/分钟、约0.6”/分钟、约0.7”/分钟、约0.8”/分钟、约0.9”/分钟、约1.0”/分钟、约1.1”/分钟、约1.2”/分钟、约1.3”/分钟、约1.4”/分钟、约1.5”/分钟、约1.75”/分钟、约2.0”/分钟、约2.25”/分钟或约2.36”/分钟的速率注射;以及2)每1至12周注射一次水性药物组合物。在一些实施方案中,本申请公开了用于在有需要的个体中治疗炎症和/或疼痛的方法,所述方法包括将本文公开的水性药物组合物注射到个体的硬膜外、病灶内或关节内的腔中;并且其中方法包括选自由以下组成的组的一个或多个步骤:1)施加小于21n的力以便使水性药物组合物以约0.5”/分钟的速率注射;以及2)每1至12周注射一次水性药物组合物。在一些实施方案中,本申请公开了用于在有需要的个体中治疗炎症和/或疼痛的方法,所述方法包括将表1中的任一示例性制剂注射到个体的硬膜外、病灶内或关节内的腔中;并且其中方法包括选自由以下组成的组的一个或多个步骤:1)施加小于5n、小于7n、小于10n、小于15n、小于17n或小于21n的力以便使水性药物组合物以约0.4”/分钟、约0.5”/分钟、约0.6”/分钟、约0.7”/分钟、约0.8”/分钟、约0.9”/分钟、约1.0”/分钟、约1.1”/分钟、约1.2”/分钟、约1.3”/分钟、约1.4”/分钟、约1.5”/分钟、约1.75”/分钟、约2.0”/分钟、约2.25”/分钟或约2.36”/分钟的速率注射;以及2)每1至12周注射一次水性药物组合物。在另外的实施方案中,注射水性药物组合物的步骤约每1、2、4、6、8、10或12周发生一次。在一些实施方案中,本申请公开了用于在有需要的个体中治疗炎症和/或疼痛的方法,所述方法包括将表1中的任一示例性制剂注射到个体的硬膜外腔中;并且其中方法包括选自由以下组成的组的一个或多个步骤:1)施加小于21n的力以便使水性药物组合物以约0.5”/分钟的速率注射;以及2)每1至12周注射一次水性药物组合物。在另外的实施方案中,注射水性药物组合物的步骤约每1、2、4、6、8、10或12周发生一次。在一些实施方案中,本申请公开了如本文所述的水性药物组合物在制造用于在有需要的个体中治疗炎症和/或疼痛的制剂中的用途,其中制剂被注射到个体中。术语“和/或”包括替代的主题以及组合的主题。例如,“x和/或y”包括“x或y”以及“x和y”。术语“约”包括并且描述了值或参数本身。例如,“约x”包括并且描述了“x”本身。在某些实施方案中,当与测量相关使用或用于修改值、单位、常数或值的范围时,术语“约”是指+1%-10%的变化。在一些实施方案中,当与测量相关使用或用于修改值、单位、常数或值的范围时,术语“约”是指+5%的变化。在一些实施方案中,当与测量相关使用或用于修改值、单位、常数或值的范围时,术语“约”是指+10%的变化。术语“之间”包括并且描述了值或参数本身。例如,“x与y之间”包括并且描述了“x”和“y”本身。任一前述实施方案可以与本文公开的一个或多个其他实施方案组合。例如,通过组合本文公开的各种实施方案,本申请提供了包含在水溶液(诸如水)中的可溶性地塞米松磷酸钠、可溶性甲泼尼龙和透明质酸钠的药物组合物,其中药物组合物的粘度位于1kcp与200kcp之间。在另一个例子中,通过组合本文公开的各种实施方案,本申请提供了在有需要的个体中治疗炎症和/或疼痛的方法,所述方法包括注射包含在水溶液(诸如水)中的可溶性地塞米松磷酸钠、可溶性甲泼尼龙和透明质酸钠的药物组合物,其中药物组合物的粘度位于1kcp与200kcp之间。以下实施例进一步说明本申请的实施方案。这些实施例仅意在说明本申请的实施方案,而不应被解释为限制性的。实施例实施例1.地塞米松磷酸钠(sp-102)制剂测试样品的制备本实施例描述了用于实施例2-5中详述的物理和化学分析、溶解研究、体内研究和组织病理学研究中的地塞米松磷酸钠(sp-102)制剂。将地塞米松磷酸钠、na2hpo4·7h2o、nah2po4·h2o和nacl组合在hplc级水中。缓慢加入透明质酸钠(ha,1.56mda),并且将所得混合物搅拌过夜以得到透明无色凝胶。通常制备100ml的批量制剂。所使用的地塞米松磷酸钠的量相当于5.0mg/ml地塞米松。加入的透明质酸钠的量如表2所示来变化。表2.具有不同量的透明质酸钠的sp-102制剂的组成。本发明人先前描述了包含不溶性形式和可溶性形式的皮质类固醇与增粘剂(诸如透明质酸)组合的药物组合物(pct国际公布号wo2014/116876)。如上所述,新的安全性建议限制了经椎间孔注射中颗粒类固醇的使用。表3提供了本发明的制剂sp-102与颗粒制剂sp-101的比较。表3.制剂sp-101和sp-102的比较。实施例2.sp-102制剂的物理和化学分析本实施例描述了含有0.5%、0.75%、1.0%和1.25%透明质酸钠的sp-102制剂的物理和化学分析。样品的外观和测量的ph呈现于表4中。通过hplc对样品中的地塞米松磷酸钠的分析也呈现于表4中。表4.sp-102制剂的物理和化学分析。样品外观phhplc测定sp-102(0.5%ha)无色透明凝胶7.2未测量到sp-102(0.75%ha)无色透明凝胶7.2103.6%sp-102(1.0%ha)无色透明凝胶7.2107.7%sp-102(1.25%ha)无色透明凝胶7.2109.5%sp-102制剂的粘度使用带有锥板cp-52轴和20rpm的转子转速(5分钟,25℃)的布氏粘度计来确定。粘度结果总结在表5中。表5.sp-102制剂的粘度。样品粘度(cps)sp-102(0.5%ha)334sp-102(0.75%ha)1108sp-102(1.0%ha)2110sp-102(1.25%ha)3647实施例3.sp-102制剂的溶解研究本实施例描述了含有0.5%、0.75%、1.0%和1.25%透明质酸钠的sp-102制剂的溶解。使用具有桨的2型usp溶解装置进行溶解研究。将介质(0.05m磷酸盐缓冲液,ph7.0,0.5%-80)在25±2rpm(37±0.5℃)下搅拌。在各个时间点取出样品(2ml),并且通过hplc分析来分析地塞米松磷酸钠含量。所使用的hplc条件如下:柱:watersxterrarp18柱,3.5μm,4.6×150mm流动相(等度):0.1%磷酸水溶液:乙腈(70:30)柱温度:40℃自动进样器温度:环境温度检测:uv242nm流速:1ml/分钟注射体积:10μl运行时间:10分钟稀释剂:溶解介质溶解研究的结果呈现于表6和表7中。表6.sp-102制剂的溶解曲线。*在250rpm下15分钟无穷大。表7.sp-102制剂的溶解曲线(归一化为完全溶解的无穷大)。表6和表7中呈现的数据表明,样品sp-102(0.5%ha)在20分钟内释放大于90%的地塞米松磷酸钠。在sp-102制剂中,释放速率随着透明质酸钠的量增加而降低。每个样品在释放介质中以250rpm混合15分钟后释放所有的地塞米松磷酸钠。然而,在25rpm下2小时后,样品sp-102(1.25%ha)和sp-102(1.0%ha)中分别仅释放约60%和80%的地塞米松磷酸钠。数据还显示透明质酸钠含量对研究的释放曲线存在非线性依赖关系(图1)。当在缺乏0.5%-80的介质中进行溶解研究时,这些样品获得了相似的释放曲线,这指示表面活性剂(-80)对sp-102制剂的释放曲线没有显著影响(图2)。实施例4.sp-102制剂的稳定性研究本实施例描述了sp-102(1.25%ha)制剂的稳定性。在加速条件下进行稳定性研究。老化后,通过hplc分析样品并且评估杂质(已知杂质:地塞米松;未知杂质:rrt0.97、rrt0.89、rrt1.47)。稳定性结果作为对照(冷藏样品)的百分比呈现。所使用的hplc条件如下:柱:watersxterrarpc18柱,5μm,4.6×250mm流动相a(mpa):0.02m甲酸铵流动相b(mpb):乙腈柱温度:40℃检测:uv242nm梯度条件:时间(分钟)mpa(%)mpb(%)090102901020406024406024.19010309010注射体积:50μl运行时间:30分钟稀释剂:0.02m甲酸铵中的30%乙腈稳定性研究的结果呈现于表8中。表8.sp-102(1.25%ha)的稳定性概述。稳定性数据表明sp-102(1.25%ha)在环境室温下高达2.4个月或在50℃下至少34天是稳定的,其中降解小于2%。已知杂质(地塞米松)增加至1.1%,而未知杂质(rrt1.47)增加至1.7%。sp-102(1.25%ha)的稳定性与含有硫酸氢钠(抗氧化剂)和苄醇(防腐剂)的商业地塞米松磷酸钠的稳定性相似。实施例5.sp-102制剂的体内研究本实施例描述了含有0.5%、1.0%和1.25%透明质酸钠的sp-102制剂的体内研究。使用两只猪进行体内研究。在相同的解剖位置处进入两只动物的硬膜外腔:在l4椎骨与l5椎骨之间以及在最后的胸椎与l1椎骨之间。将几种测试制剂注射到硬膜外腔中。一些制剂单独地注射,而其他制剂在手术室中混合。在手术室中混合的制剂为商业地塞米松(4mg/ml)和呈液体(300)或粉末形式的造影剂的组合。在注射测试制剂后,以15分钟的间隔记录几个荧光镜电影,以便监测测试制剂在硬膜外腔中的扩散。随后将动物改变位置成仰躺卧,并且在右股动脉上接入塞尔丁格技术(seldingertechnique)之后,从左锁骨下动脉插入椎动脉。使用椎动脉递送测试制剂,同时在脑中记录荧光镜电影,特别关注脑干。在一只动物(动物#1)中使用右椎动脉,而在第二只动物(动物#2)中使用左椎动脉。硬膜外注射时间进程用三种单独的制剂注射动物#1。制剂如下:注射剂1(对照):2ml的地塞米松磷酸钠(4mg/ml)和647碘海醇(造影剂)。注射剂2:2mlsp-102(0.5%ha)和647mg碘海醇。注射剂3:2mlsp-102(1.25%ha)和647mg碘海醇。对于对照和对于制剂sp-102(0.5%ha)和sp-102(1.25%ha),在动物#1注射后立即记录的注射后图像示于图3a-3c中。在注射后30分钟和60分钟记录的对应的图像分别呈现于图4a-4c和图5a-5c中。对于对照和制剂sp-102(1.25%ha),在注射后120分钟记录的图像示于图6a-6b中。最后,对于制剂sp-102(1.25%ha)的注射后180分钟记录的图像示于图7中。用两种单独的制剂注射动物#2。制剂如下:注射剂1:2mlsp-102(1.0%ha)和647mg碘海醇。注射剂2:2mlsp-102(1.25%ha)和647mg碘海醇。对于制剂sp-102(1.0%ha)和sp-102(1.25%ha),注射后立即记录的注射后图像示于图8a-8b中。在注射后30分钟、60分钟和120分钟记录的对应的图像分别呈现于图9a-9b、图10a-10b和图11a-11b中。最后,对于制剂sp-102(1.25%ha)的注射后180分钟记录的图像示于图12中。硬膜外注射时间进程研究表明,sp-102(1.25%ha)定位至注射部位的时间段比商业产品显著更长。通过追踪作为时间函数的可见造影染料的估计百分比,用图形描绘了两只动物的注射后图像(图13)。显然,透明质酸钠的添加引起地塞米松的停留时间以剂量依赖性方式延长。sp-102(1.25%ha)的硬膜外停留半衰期为约110分钟。相比之下,商业可注射类固醇产品(诸如depo-medrol和decadron)的硬膜外停留半衰期为约15分钟(图14)。实施例6.sp-102制剂的组织病理学研究通过肉眼病理学和组织病理学来分析来自实施例4的商业地塞米松和sp-102(1.25%ha)的注射剂。简而言之,尸检包括检查身体的外表面、所有孔窍以及胸腔和腹腔,包括其内容物。还对大脑进行了肉眼检查。收集大脑并且将其浸入10%nbf(中性缓冲福尔马林)中至少24小时。随后,将切片切成4μm,用苏木精和曙红(h&e)染色,并且使用光学显微镜检查。出血和/或坏死/梗死的观察结果总结于表9中。病理学数据示出在注射商业地塞米松或sp-102(1.25%ha)后没有感染或出血迹象。表9.商业地塞米松和sp-102(1.25%ha)的组织病理学。*评分:0,不存在;1,最小;2,轻微;3,中等;4,严重。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1