一类pH响应的两亲性棒状阿霉素聚合物前药的制备方法与流程
本发明涉及化学药物及生物应用领域,具体涉及一种ph刺激响应的两亲性棒状聚合前药及其制备及其用途。
背景技术:
阿霉素(doxorubicin,dox)是常见的抗癌药物之一,其cas号:23214-92-8,化学结构式:c27h29no11,相对分子量:543.52,它可抑制rna和dna的合成,对rna的抑制作用最强,阿霉素属周期非特异性药物,对多种肿瘤均有作用,因此,对各种生长周期的肿瘤细胞都有杀灭作用。阿霉素主要适用于急性白血病,对急性淋巴细胞白血病及粒细胞白血病均有一定的治疗作用,一般作为第二线药物,即在首选药物耐药时可考虑应用此药。其作用机理主要是该药物嵌入dna而抑制核酸的合成。
抗癌药物分子包括阿霉素最常见的问题是水溶性较差、分子尺寸较小以及非选择性,在药物递送过程中,存在血液循环稳定性差、药物生物利用度低、毒副作用较强以及医疗效率低等问题。为了早日实现临床治疗的应用,研究人员目前主要集中在将纳米技术与抗癌药物相结合的递送系统以便增强其水溶性,保护其生物药物活性,以及增大药物上载量,这对推动癌症治疗的发展具有重要的科学意义。
利用纳米技术与药物相结合的传输药物的递送系统中,刺激响应的两亲性聚合前药递送系统拥有可设计的结构与功能、药物生物利用度高等优势,可有效降低癌症化学治疗过程中药物毒副作用以及治疗效率低等劣势,近些年来一直都是国内外研究的热点。
例如,cn105943496a公开了一种具有邻基效应的ph响应半乳糖化壳聚糖-聚乙二醇聚合物键合阿霉素前药,它是通过具有ph响应的壳聚糖-聚乙二醇嫁接物通过化合键上修饰上阿霉素而获得的聚合前药,所制得的胶束具有良好的ph响应性及肝靶向性,可进一步研制开发为治疗肝癌的新型靶向制剂。此外,cn105251013a公开了一种具有氧化还原响应性可降解水溶性抗肿瘤聚合物前药,该聚合前药具有以聚丙交酯为主链,侧链上键接大量低聚聚乙二醇支链和抗肿瘤药物分子的特殊的梳型结构。此水溶性抗肿瘤聚合物前药载药量适中且可控,易降解,且具有氧化还原响应性。
因此,设计、构建集具备高药物上载量和高胶束稳定性等功能的聚合前药递送系统具有很强的必要性。
技术实现要素:
针对当前药物递送系统的胶束的不稳定性,较低的药物上载量,也难以保证药物的选择性可控释放等缺陷,本发明旨在提供一类ph刺激响应的超高载药量的两亲性棒状聚合前药的制备方法,此方法合成的两亲性棒状聚合前药具有较高药物释放量及细胞毒性,从而实现药物的有效递送及高效释放。
本发明的技术方案具体如下:
一类ph响应的两亲性棒状阿霉素聚合物前药,制备方法包括以下步骤:
(1)制备基于葡聚糖的棒状原子自由基聚合反应(atrp)引发剂葡聚糖溴(dex-br),其反应式如下所示,包括以下步骤:a)在氩气(ar,1-12pa)氛围条件下,将溶于离子液体(1-烯丙基-3-甲基氯化咪唑)的葡聚糖(dex)冷却至0℃,再加入n-甲基吡咯烷酮(nmp)与n,n-二甲基甲酰胺(dmf)的混合溶液,随后缓慢加入2-溴异丁酰溴(bibb),冰浴0.5-2h,然后升至室温(25℃)并避光反应12-72h,将所得的混合溶液滴加入去离子水中进行沉淀,用丙酮溶解所得沉淀,再重复提纯三次得到淡黄色中间产物,在真空干燥箱(25-30℃)里烘干,b)接着再将所得淡黄色中间产物溶于nmp中,冷却至0℃,随后缓慢加入bibb,冰浴0.5-2h,然后升至室温(25℃)并避光反应12-72h,接着沉淀于去离子水中,用丙酮溶解所得沉淀,重复提纯三次得到淡黄色粉末,真空条件下25-30℃烘干,得到产物dex-br;
(2)制备ph响应的单体2-甲氧基-2-氧乙基-丙烯酸甲酯(mgma),其反应式如下所示,包括以下步骤:在温度≤0℃和氩气(ar,1-12pa)氛围条件下,将溶解在无水二氯甲烷(dcm)的一定量的甲基丙烯酰氯的溶液缓慢滴加到分散在无水dcm中的乙醇酸甲酯和三乙胺(tea)混合溶液中,搅拌过夜,洗涤纯化,硅胶柱层析得到纯净的产物mgma;
(3)制备的棒状聚合前药(dex-p(mgma)),其反应式如下所示,包括以下步骤:在室温(25℃)和氩气(ar,1-12pa)氛围条件下,将步骤(1)得到的dex-br,步骤(2)得到的mgma与cubr溶解在二甲基亚砜(dmso)(或甲醇)溶剂中,冷冻-解冻循环三次,加入配体三(2-二甲氨基乙基)胺(me6tren)(或配体n,n,n′,n′′,n-五甲基二乙烯三胺(pmedta)),再冷冻-解冻循环两次,在室温下搅拌反应8-24h,得到的混合物用四氢呋喃(thf)稀释过al2o3柱子,浓缩,再沉淀于乙醚中,用thf溶解并再次沉淀于乙醚中,烘干,得到dex-(pmgma);其中dex-p(mgma)中m值范围为20-100,聚合度x值范围为1~100,得到的聚合物其分子量范围为15900~114900g·mol-1;
(4)制备两亲性棒状聚合前药(dex-p(mgma)-b-p(oegma),(dmo)),其反应式如下所示,包括以下步骤:在室温(25℃)和氩气(ar,1-12pa)氛围条件下,将步骤(3)得到的dex-p(mgma)、乙二醇甲基丙烯酸酯(oegma)和cubr溶解在dmf和dmso的混合溶剂中,冷冻-解冻循环三次,加入配体me6tren(或配体pmedta),再冷冻-解冻循环两次,在25℃下反应12-48h,得到的混合物用thf稀释后过al2o3柱子,浓缩,再用thf溶解并再次沉淀于乙醚中,烘干,得到dex-p(mgma)-b-p(oegma);其中dex-p(mgma)-b-p(oegma)中m值范围为20-100,x值的范围为1~100,n值可为2~9,聚合度y值的范围为1~50,得到的聚合物其分子量范围为18600~249900g·mol-1;
(5)制备ph响应的两亲性棒状聚合前药(dex-p(mgma-nh2)-b-p(oegma),dmo-hydrazide),其反应式如下所示,包括以下步骤:在室温(25℃)和氩气(ar,1-12pa)氛围条件下,将dex-p(mgma)-p(oegma)溶解在一定比例的无水甲醇和dmf的混合溶剂中,而后加入一定量的水合肼,在25℃下反应6-36h,得到的混合物用水透析两天,冷冻干燥,得到dex-p(mgma-nh2)-b-p(oegma);其中dex-p(mgma-nh2)-b-p(oegma)中m值范围为20-100,x值的范围为1~100,n值可为2~9,聚合度y值的范围为1~50;
(6)制备ph响应的两亲性棒状阿霉素聚合前药(dex-p(mgma-dox)-b-p(oegma),dmo@dox),其反应式如下所示,包括以下步骤:在室温(25℃)和氩气(ar,1-12pa)氛围条件下,将dex-p(mgma-nh2)-b-p(oegma)和阿霉素(dox)溶解在一定量的无水甲醇和dmf中,滴入2-3滴的三氟乙酸(tfa),在25℃下避光反应12-72h,得到的混合物用甲醇透析,冷冻干燥,得到产物dex-p(mgma-dox)-b-p(oegma);其中dex-p(mgma-dox)-b-p(oegma)中m值范围为20-100,x值的范围为1~100,n值可为2~9,聚合度y值的范围为1~50;
(7)制备ph响应的两亲性球状纳米胶束,包括以下步骤:在室温(25℃),取一定量步骤(6)得到的dex-p(mgma-dox)-b-p(oegma)材料溶于适量的dmf或dmso中,避光搅拌5~120min在搅拌下缓慢滴加到一定量的水中,搅拌5~60min,用水透析除去有机溶剂便得到球状的纳米胶束水溶液。
进一步的,所述步骤(1)a)中葡聚糖(dex)和离子液体(1-烯丙基-3-甲基氯化咪唑)的摩尔比为1:(10~1000),无水nmp和dmf溶液的体积比为1:(0.1~10),nmp和dmf的混合溶液的总体积为离子液体体积的2倍,所述步骤(1)b)中溶解在无水nmp中的淡黄色中间产物的摩尔浓度范围0.015~0.03mol·l-1。
进一步的,所述步骤(2)中溶于无水dcm中的乙醇酸甲酯和tea的摩尔浓度范围分别为0.2~10mol·l-1和0.3~20mol·l-1;溶于无水dcm中的甲基丙烯酰氯的摩尔浓度范围为0.5~18mol·l-1。
进一步的,所述步骤(3)中dex-br、mgma、cubr和配体me6tren(或配体pmedta)的摩尔比范围为1:(6~3000):(2~100):(4~400);其中dex-br的摩尔浓度为0.2~20mmol·l-1。
进一步的,所述步骤(4)中dex-p(mgma)、oegma、cubr和配体me6tren(或配体pmedta)的摩尔比范围为1:(6~2000):(3~100):(3~100);在dmf和dmso的混合溶剂中dmf:dmso的体积比为1:(0.1~5)。
进一步的,所述步骤(5)中溶解在无水甲醇和dmf中的dex-p(mgma)-b-p(oegma)的摩尔浓度范围为0.001~1mmol·l-1;水合肼、甲醇和dmf的体积比范围为1:(5~30):(1~15)。
进一步的,所述步骤(6)中dex-p(mgma-nh2)-b-p(oegma)与dox的摩尔浓度范围分别为0.001~0.5mol·l-1和0.001~1mol·l-1,无水甲醇与dmf的体积比为1:(0.1~10)。
进一步的,所述步骤(7)中所制得的阿霉素聚合前药两亲性分子的浓度范围为0.01mg·l-1~1000mg·l-1;有机溶剂和水的比例范围为1:(3~1000);用水透析后所得的球状纳米粒子其直径范围为5~1000nm。
主要优点:
1.针对目前药物递送系统存在的胶束不稳定以及低载药量等问题,本项目创造性地提出一类ph响应的两亲性棒状阿霉素聚合前药的递送系统,此聚合材料可以通过调节亲疏水比例,能够有效地提高药物上载量和胶束稳定性,以及调控纳米胶束的尺寸,解决当前药物递送系统存在的一些不足,使肿瘤的精确诊断治疗早日成为可能。
2.本发明中的两亲性棒状阿霉素聚合前药,具有ph刺激响应性,根据癌细胞内相对较低的ph值,可以促使材料中的腙键断裂并有效地快速释放抗癌药物,因此,实现对癌症的有效治疗。
附图说明
为了使本发明的目的、技术方案和有益效果更加清楚,本发明提供如下附图:
图1为实施例1中两亲性ph响应棒状阿霉素聚合前药(dex-p(mgma-dox)-b-p(oegma),dmo@dox)的合成线路示意图。
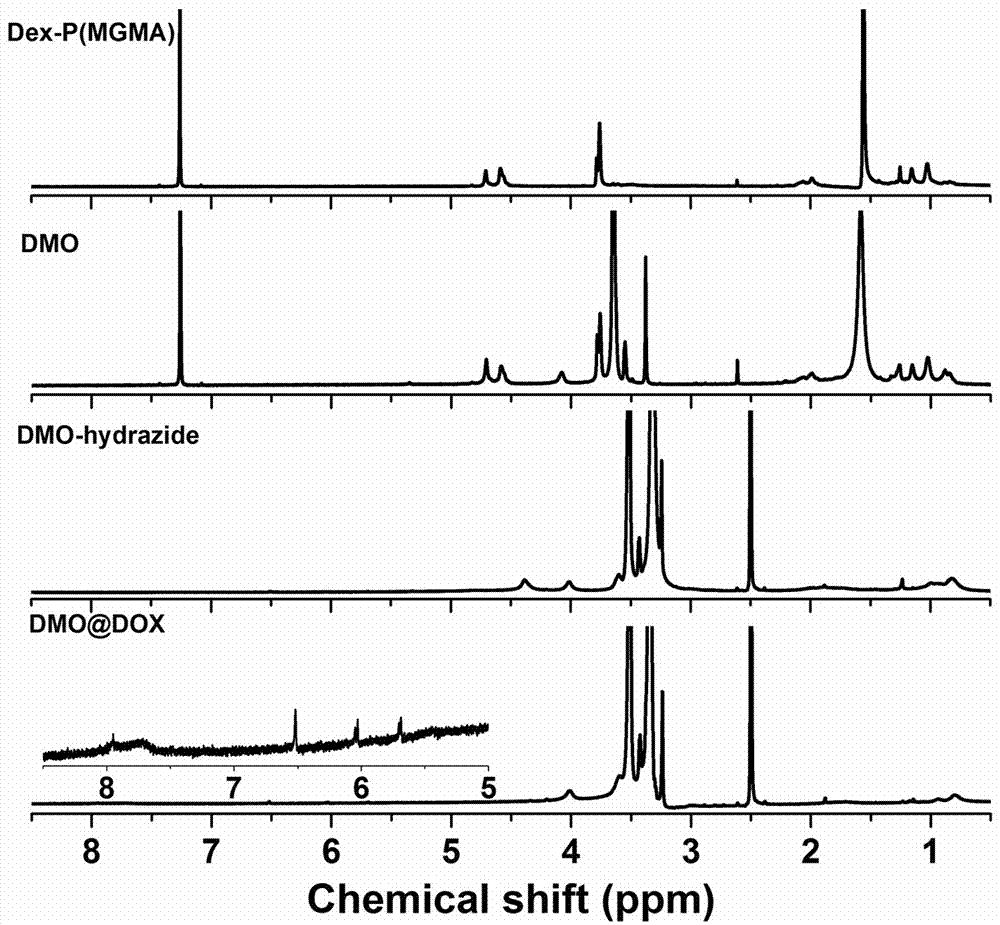
图2为实施例1中两亲性ph响应棒状阿霉素聚合物(dex-p(mgma-dox)-b-p(oegma),dmo@dox)的核磁示意图。
图3为实施例1中两亲性棒状聚合纳米胶束的tem和dls示意图。
图4为实施例1中两亲性ph敏感棒状聚合前药释放示意图。
图5为实施例1中两亲性ph响应棒状阿霉素聚合前药(dex-p(mgma-dox)-b-p(oegma),dmo@dox)的毒性示意图。
具体实施方式
以下实施例用于说明本发明,但不用来限制本发明的范围。
实施例1制备ph响应的的dex-p(mgma)-b-p(oegma)两亲性棒状聚合前药
(1)制备dex-br:在氩气氛围条件下,将溶于离子液体(1-烯丙基-3-甲基氯化咪唑)的葡聚糖(dex)冷却至0℃,再加入n-甲基吡咯烷酮(nmp)与n,n-二甲基甲酰胺(dmf)的混合溶液,随后缓慢加入2-溴异丁酰溴(bibb),冰浴0.5-2h,然后升至室温(25℃)并避光反应12-72h,接着沉淀于去离子水中,用丙酮溶解所得沉淀,再重复提纯三次得到淡黄色中间产物,在真空干燥箱(25-30℃)里烘干,接着将所得淡黄中间产物溶于nmp中,冷却至0℃,随后缓慢加入bibb,冰浴0.5-2h,然后升至室温(25℃)并避光反应12-72h,接着沉淀于去离子水中,用丙酮溶解所得沉淀,重复提纯三次得到淡黄色粉末,真空条件下25-30℃烘干,得到产物dex-br;
(2)制备mgma:在温度≤0℃和氩气(ar)氛围条件下,一定量的溶于无水dcm(60ml)的甲基丙烯酰氯(0.144mol)逐滴加入到乙醇酸甲酯(0.144mol)、tea(0.288mol)、无水dcm(100ml)溶液中,搅拌过夜,洗涤纯化,柱层析得到纯净的产物。
(3)制备dex-p(mgma):在室温和氩气(ar)氛围条件下,将dex-br(0.01mmol),mgma(8.4mmol),cubr(0.21mmol)溶解在dmso(5ml)溶剂中,冷冻-解冻循环三次,加入配体me6tren(0.42mmol),再冷冻-解冻循环两次,在25℃下反应8-24h。得到的混合物用thf稀释过al2o3柱子,浓缩,再沉淀于乙醚中,烘干。
(4)制备dex-p(mgma)-b-p(oegma)(dmo):在室温和氩气(ar)氛围条件下,将dex-p(mgma)(0.01mmol),oegma(5.25mmol),cubr(0.21mmol)溶解在dmf(3.0ml)和dmso(3.0ml)溶剂中,冷冻-解冻循环三次,加入配体me6tren(0.21mmol),再冷冻-解冻循环两次,在25℃下反应12-48h。得到的混合物用thf稀释过al2o3柱子,浓缩,再沉淀于乙醚中,烘干。如图2中的dmo的核磁图,图中3.38ppm和3.65ppm出现的峰可以看出dex-p(mgma)-b-p(oegma)的成功合成。
(5)制备ph响应的dex-p(mgma-nh2)-b-p(oegma)(dmo-hydrazide):在室温和氩气(ar)氛围条件下,将dex-p(mgma)-b-p(oegma)(0.005mmol)溶解在无水甲醇(10ml)和dmf(3.75ml)中,加入一定量的水合肼(1.5ml),在25℃下反应6-36h。得到的混合物用水透析,冷冻干燥。如图中2中的dmo-hydrazide的核磁图,图中mgma在3.76ppm处的特征峰的消失可以看出肼基成功取代了mgma的甲氧基,即是dex-p(mgma-nh2)-b-p(oegma)的成功合成。
(6)制备两亲性阿霉素聚合前药dex-p(mgma-dox)-b-p(oegma)(dmo@dox):在室温和氩气(ar)氛围条件下,将dex-p(mgma-nh2)-b-p(oegma)(0.128mmol)和dox(0.32mmol)溶解在无水甲醇(8ml)和dmf(8ml)中,滴入2-3滴的tfa,在25℃下避光反应12-72h。得到的混合物用甲醇透析,冷冻干燥。如图2中dmo@dox中8ppm附近和4-6ppm之间的峰表明dex-p(mgma-dox)-b-p(oegma)的成功合成。
(7)制备阿霉素聚合前药纳米胶束:取5mgdex-p(mgma-dox)-b-p(oegma)溶解在1ml的dmf或dmso中,分散到6ml的水中,用水透析48h制得胶束。胶束的尺寸大小如图3中的tem和水相及dmf相的dls图,可以看出胶束的尺寸大小为三十几纳米以及尺寸均匀,表明此胶束可以快速进入细胞。图4的药物释放图可以看出此胶束在72小时后释放量高达91%,图5的细胞毒性图可以看出药物作用在72小时后宫颈癌细胞(hela)和人乳腺癌细胞(mcf-7)的存活率低至20%和15%,说明此药物载体对癌细胞的毒性较大。
最后说明的是,以上优选实施例仅用以说明本发明的技术方案而非限制,尽管通过上述优选实施例已经对本发明进行了详细的描述,但本领域技术人员应当理解,可以在形式上和细节上对其作出各种各样的改变,而不偏离本发明权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!