功能基化的脂质体及其制备方法及应用与流程
本发明涉及一种功能基化的脂质体及其制备方法及应用,属于纳米材料及生物化学领域。
背景技术:
脂质体有生物兼容性,生物降解能力,表面性质易调整,改变药代动力学,持续释放,有能力克服多药耐药,增强药物稳定性,靶向特定组织的特点,使其成为在许多疾病治疗中最有前景的生物材料之一。脂质体包裹阿霉素(dox)作为第一款被fda批准的药物,目前已有超过12款材料应用到临床中。
为了提高脂质体的临床应用性,需要对脂质体做一些特定的功能基化。比如:用抗体修饰脂质体,来提高脂质体的靶向性。可直接利用抗体自身的化学官能团或利用具有活性反应基团的偶联剂对抗体上的基团进行修饰,再偶联到磷脂分子的官能团上。而抗体的提纯较为复杂,成本较高,这也造成脂质体临床应用的成本较高。
多肽分子是人机体内重要的具有生物活性的一类物质,多肽分子通常具有较低的分子量(约1kda),因此有良好的组织穿透能力,此外还具有靶向性和良好的生物相容性,能作为靶向配体应用于药物递送领域从而达到肿瘤的靶向治疗。目前脂质体功能基化的方法大都集中在将已具备生物功能的多肽的化学基团直接修饰到脂质体上,实现脂质体多功能性。最常用的便是介导细胞与细胞外基质或者细胞间粘附作用的rgd多肽以及细胞穿透肽cpp。目前尚无采用多肽插入脂双层通过对多肽构象调控实现脂质体功能化的方法。
技术实现要素:
本发明的目的之一在于提供一种功能基化的脂质体。
本发明的目的之二在于提供该脂质体的制备方法。
本发明的目的之三在于提供该脂质体在制备溶菌酶人工抗体药物中的应用。
本发明的目的之四在于提供该脂质体在制备癌症抗体靶向药物中的应用。
实现本发明所述目的,本发明将磷脂和多肽通过偶联和置换反应形成磷脂-多肽偶联物,通过核磁表征验证产物化学结构。将合成的磷脂-多肽偶联物与胆固醇,磷脂和磷脂-peg按配比制备脂质体,其中磷脂-多肽偶联物组分占磷脂质量比例为1%~10%,多肽在脂质体表面形成环状结构。
一种功能基化的脂质体,其特征在于该脂质体是先将多肽通过化学反应连接在磷脂分子上,然后将该磷脂-多肽偶联物与磷脂,胆固醇通过水化薄膜方法组成脂质体。
一种制备上述的功能基化的脂质体的方法,其特征在于该方法的具体给步骤如下:
a.将磷脂去盐酸化后在氯仿中与氮-琥珀星氩氨-3(2-吡啶二硫代)-酸酯等摩尔比混合,反应三天后,加入氯仿、甲醇与超纯水的体积比为65:35:8的混合溶剂,再加入与二硬脂酰基磷脂酰乙醇胺dspe摩尔数比例为1:2的多肽,反应三天后,旋蒸去除氯仿和甲醇,再加入水,透析两天,冻干,即得到多肽-磷脂聚合物;
b.将步骤a所得多肽-磷脂聚合物、聚乙二醇化二硬脂酰基磷脂酰乙醇胺dspe-peg2000、胆固醇和氢化大豆磷脂按照多肽:磷脂的摩尔比例为3%-5%溶于氯仿中,旋蒸除去氯仿,加入水,马上在温度为500c下水化20分钟,温度为500c下超声10-20分钟,用100nm的膜挤推,得到功能基化的脂质体。
上述的多肽的序列为cgstiyasyyesghgc、cgstiyasyyesghg或cytygishggsasyec。
上述的功能基化的脂质体在制备溶菌酶人工抗体药物中的应用。
上述的溶菌酶为溶菌酶、水解酶或转移酶。
上述的功能基化的脂质体在制备癌症抗体靶向药物中的应用。
通过酶活实验和表面等离子共振技术(spr,biacoret200)验证了环状多肽功能基化的脂质体具有生物医学功能,例如,通过多肽序列的选择,制备可以结合溶菌酶抑制酶活性成为溶菌酶人工抗体,或者特异性结合egfr在癌细胞抗体靶向治疗方面有潜在应用。
附图说明
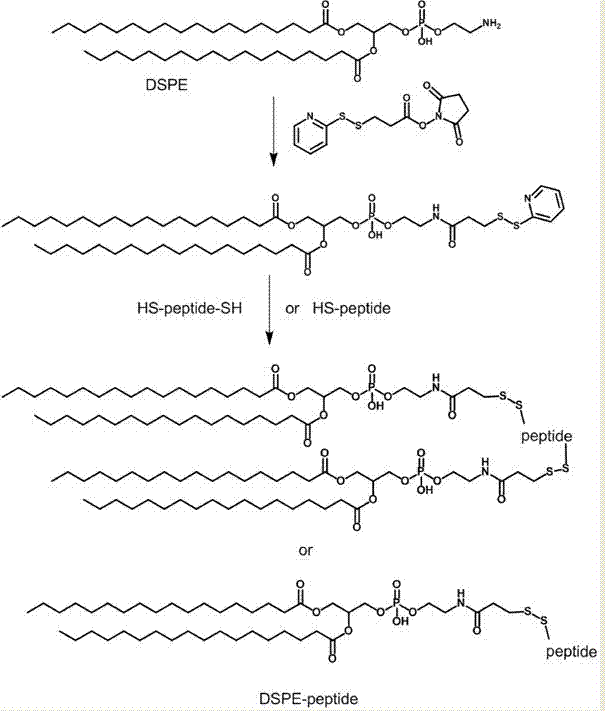
图1为本发明所得磷脂-多肽偶联物制备原理图;
图2为本发明所得磷脂-多肽偶联物合成脂质体原理图;
图3本发明所得(a)dspe-p1,(b)dspe-p1m和(c)dspe-p1s的核磁共振波氢谱图;
图4为本发明所得磷脂-多肽偶联物合成脂质体粒径(dls)和透射电子显微镜(tem)图;
图5为本发明所得通过调整脂质体表面的多肽密度来调控抗溶菌酶脂质体的活性图;
图6为本发明所得多肽修饰的脂质体lp-p1与lp-p1s活性对比图;
图7为本发明所得磷脂-多肽-磷脂合成脂质体特异性结合溶菌酶图;
图8为本发明所得(a)dspe-p2,(b)dspe-p2m和(c)dspe-p2s的核磁共振波谱图;
图9为本发明所得通过调整脂质体表面的多肽密度来调控靶向egfr脂质体的spr图;
图10为本发明所得磷脂-多肽合成脂质体特异性结合egfr图。
具体实施方式
下面结合附图,本发明的具体实施的过程和步骤以及结果分析如下:
实施例1
以发明溶菌酶抗体为例:
如图1显示先将磷脂去盐酸化,然后通过置换反应和偶联反应,制备形成磷脂-多肽-磷脂偶联物,最后制备的偶联物和磷脂合成脂质体(图2),即可得到多肽功能基化的脂质体,其特征在于该方法具有以下的工艺步骤:
a.磷脂去盐酸化:将10mgdspe加入2.6ml的氯仿中,加入0.05ml的三乙胺,在摇床上摇4h,温度45°c,待样品澄清。
b.磷脂-多肽(p1,p1m,p1s)-磷脂偶联物制备:磷脂去盐酸化后加入与dspe等摩尔数的氮-琥珀星氩氨-3(2-吡啶二硫代)-酸酯。反应三天后,加入1.4ml甲醇,0.32ml超纯水,加入与dspe摩尔数比例为1:2的多肽(p1,p1m,p1s),即多肽-磷脂聚合物,其中p1的序列为cgstiyasyyesghgc,来源于溶菌酶抗体部分序列,p1m的序列为(cgstiyasyyesghg),也就是比p1少了一个少了一个半胱氨酸,只能和一个磷脂反应,而p1s的序列为(cytygishggsasyec),是将p1序列打乱后形成的,反应三天后,旋蒸去除氯仿和甲醇,加入15ml水,透析两天,冻干。
c.多肽脂质体合成:将1.5nm的磷脂-多肽偶联物,3.75nmdspe-peg2000,30nm胆固醇,30.75nm氢化大豆磷脂溶于氯仿,旋蒸除去氯仿,加入2ml水,在温度为500c下水化20分钟,温度为500c下超声8分钟,停止超声5分钟,再超声8分钟,用100nm的膜在脂质体挤出器中挤推70次。
图3显示磷脂-多肽-偶联物的结构组分由核磁表征来决定,核磁表中7和6.6ppm的峰表示多肽中苯环的h峰,而1.2ppm的峰表示dspe中ch2的h峰,表明磷脂-多肽偶联物(dspe-p1,dspe-p1m,dspe-p1s)的成功合成。通过核磁图积分面积计算可以得到三组磷脂-多肽偶联物的反应率都在85%以上。
图4显示备脂质体的粒径大小和形貌用动态光散射粒度仪(dls)测量和透射电子显微镜(tem)来检测。tem表征显示脂质体的外貌均为球形,无聚集的脂质体尺寸较为均一。tem图显示平均粒径大小为70nm左右,比dls值略小,这可能是因为测dls时脂质体样品在溶液中,测的是脂质体的水合粒径;而在表征tem时,样品滴在铜网上并且等干燥后进行tem拍摄,所以tem图的样品的尺寸比测dls时要小。
合成的dspe-p1脂质体进行酶活实验,溶壁微球菌底物悬浊液每10mg溶解于30ml的pbs缓冲溶液(0.1m,ph=6.2)中,将溶菌酶(1.5×10-7m)溶解在10mm,ph=7.4的pbs缓冲液中。具体操作如下:取1ml的功能化的脂质体,其中多肽浓度为3.0×10-5m,加入0.5ml的上述溶菌酶溶液,震荡混合10s,水浴30min,之后1ml的溶壁微球菌底物悬浊液加入到混合物中,剧烈震荡后迅速转移至比色皿中,用紫外分光光度计测量在150s内,450nm处吸光度的变化。上述所有的测量过程均在250c恒温条件下进行。如图5显示当多肽在磷脂中的摩尔比例为4%时,其溶菌酶抑制率最高。图6显示游离的p1不能抑制溶菌酶,说明原有的基团并没有相对应的功能,通过p1修饰的脂质体(lp-p1)的溶菌酶脂质体能完全抑制溶菌酶,说明我们来源于溶菌酶天然抗体决定簇的cdr片段的多肽,功能化到脂质体表面能够同溶菌酶结构中裂开的位点处特异性的结合,从而抑制溶菌酶的活性。而lp,lp-p1m,lp-p1s拥有较低的活性说明实现脂质体功能基化需要两个条件有同溶菌酶特异性结合的正确序列和正确的构象。
将相同浓度均为0.04nm的lp-p1、lp-p1m和lp-p1s以及对应浓度的pep1,注入30μl/min的流动相缓冲溶液中,同芯片上三个通道上存在的溶菌酶、rnasea及bsa流过,测得的结合数据如图7。从图中可以观察到,除了lp-p1,其他多肽脂质体和游离的多肽(如自由的p1、lp-p1m及lp-p1s)与hewl之间只有较小的非特异性相互作用,而在在rnasea及bsa的通道,所有的多肽修饰脂质体包括lp-p1都表现出了微弱的非特异性结合。实验结果表明溶菌酶结构中裂开的位点处特异性的结合必须满足两个条件:1.有同溶菌酶特异性结合的正确序列,2.正确的构象。此外,我们构建的抗溶菌酶脂质体与溶菌酶结合是特异性的。
实施例2:以发明egfr抗体为例:
可将实施例2中的p1换成p2,其他操作方法参照实施例1,这样就可以得到一种新的偶联物以及靶向egfr脂质体。
如图1显示先将磷脂去盐酸化,然后通过置换反应和偶联反应,制备形成磷脂-多肽-磷脂偶联物,最后制备的偶联物和磷脂合成脂质体(图2),即可得到多肽功能基化的脂质体,其特征在于该方法具有以下的工艺步骤:
a.磷脂去盐酸化:将10mgdspe加入2.6ml的氯仿中,加入0.05ml的三乙胺,在摇床上摇4h,温度45°c,待样品澄清。
b.磷脂-多肽(p2,p2m,p2s)-磷脂偶联物制备:磷脂去盐酸化后加入与dspe等摩尔数的氮-琥珀星氩氨-3(2-吡啶二硫代)-酸酯。反应三天后,加入1.4ml甲醇,0.32ml超纯水,加入与dspe摩尔数比例为1:2的多肽(p2,p2m,p2s),即多肽-磷脂聚合物,其中p2(csawygtlyeydgc)来源于egfr天然抗体部分序列,p2m(csawygtlyeydg)也就是比p2少了一个少了一个半胱氨酸,只能和一个磷脂反应,p2s(cwsyagydgletyc)是将p2序列打乱后形成的,反应三天后,旋蒸去除氯仿和甲醇,加入15ml水,透析两天,冻干。
c.多肽脂质体合成:将1.5nm的磷脂-多肽偶联物,3.75nmdspe-peg2000,30nm胆固醇,30.75nm溶于氯仿,旋蒸除去氯仿,加入2ml水,在温度为500c下水化20分钟,温度为500c下超声8分钟,停止超声5分钟,再超声8分钟,用100nm的膜在脂质体挤出器中挤推70次。
图8显示磷脂-多肽-偶联物的结构组分由核磁表征来决定,核磁表中7和6.6ppm的峰表示多肽中苯环的h峰,而1.2ppm的峰表示dspe中ch2的h峰,表明磷脂-多肽偶联物(dspe-p1,dspe-p1m,dspe-p1s)的成功合成。通过核磁图积分面积计算可以得到三组磷脂-多肽偶联物的反应率都在85%以上(如表2.1)。
附图9中通过表面等离子技术(spr)显示我们通过改变多肽在磷脂中的摩尔比例来制备脂质体,通过spr检测来检测靶向egfr脂质体的最佳构象。从图中可以看到,多肽在磷脂中的摩尔比例为4%时结合曲线和信号处于最高值,依次为多肽在磷脂中的摩尔比例3.5%,3%,4.5%的结合曲线降低。也就是说多肽在脂质体表面的密度影响了靶向egfr脂质体同egfr的结合,最优化的靶向egfr脂质体条件为多肽在磷脂中的摩尔比例为4%。以及将相同浓度均为0.04nm的lp-p2、lp-p2m和lp-p2s以及对应浓度的pep1,注入30μl/min的流动相缓冲溶液中,同芯片上三个通道上存在的egfr、rnasea及bsa流过,测得的结合数据如图10。从图中可以观察到,除了lp-p2,其他多肽脂质体和游离的多肽(如自由的p2、lp-p2m及lp-p2s)与hewl之间只有较小的非特异性相互作用,而在在rnasea及bsa的通道,所有的多肽修饰脂质体包括lp-p2都表现出了微弱的非特异性结合。实验结果表明egfr结构中裂开的位点处特异性的结合必须满足两个条件:1.有同egfr特异性结合的正确序列,2.正确的构象。此外,我们构建的靶向egfr脂质体与egfr结合是特异性的。
序列表
<110>上海大学
<120>功能基化的脂质体及其制备方法及应用
<160>3
<170>siposequencelisting1.0
<210>1
<211>16
<212>dna
<213>人工序列()
<400>1
<210>2
<211>15
<212>dna
<213>人工序列()
<400>2
<210>3
<211>16
<212>dna
<213>人工序列()
<400>3
- 还没有人留言评论。精彩留言会获得点赞!