一种多层软组织修复材料及其制备方法与流程
本发明涉及一种多层软组织修复材料及其制备方法。
背景技术:
软组织是人体重要的组织。但由于各种原因,如疾病、外伤等引起的软组织的损伤或缺损,已成为临床常见疾病之一,严重威胁人类健康。传统治疗软组织损伤或缺损的方法有自体移植和异体移植,但这些方法存在供体有限、二次伤害、免疫排斥等问题。寻找可靠、安全和有效的修复软组织损伤和缺损的方法具有重要意义。近年来,随之组织工程技术的发展,利用组织工程技术的方法修复、替代受损的软组织为治疗软组织损伤和缺损带来了新的思路。
小肠黏膜下层(smallintestinalsubmucosa,sis)为细胞外基质成分,富含多种生长因子,在特定的浓度及活性状态下能促进细胞生长、刺激血管新生,进而诱导组织再生。且sis具有无免疫原性、抗微生物活性、各向异性等特点,其安全性和疗效得到了临床实践验证。此外,sis来源丰富、易获取,不仅可避免传统器官移植中存在的供体有限问题,还能降低成本使更多的患者受益。因此sis成为研究较为广泛的软组织工程支架材料。专利cn107233630a、cn107854727a和cn107281552a均为对sis处理(如在sis上复合细胞或细胞外基质等)后得到软组织修复材料。但与其它生物天然修复材料一样,sis的机械性能不够理想、降解速度过快且不易控制。
高分子合成材料具有可塑形性优异、降解性能和力学性能可控等优点。聚氨酯(polyurethane,pu)是一类常用的医用高分子合成材料,将pu与sis结合,有望改善生物天然修复材料的力学性能和降解性能,得到更加理想的软组织修复材料。专利cn104341608a将pu与sis混合、交联后得到了回弹性能、力学性能优良,生物相容性良好的软组织修复材料(pu/sis)。但此pu/sis修复材料为单层修复材料,无法很好的模拟、修复具有双层或多层结构的软组织,如子宫壁、皮肤等。
因此寻找一种有效修复软组织的多层材料具有重要意义。
技术实现要素:
本发明的目的在于提供一种多层软组织修复材料及其制备方法。
本发明提供了一种多层软组织修复材料,它是由聚氨酯粘接的双层结构;其中,一层为小肠黏膜下层膜,另一层为聚氨酯与小肠黏膜下层粉末混合后交联而成的聚氨酯/小肠黏膜下层复合材料层;所述小肠黏膜下层膜与聚氨酯/小肠黏膜下层复合材料层的厚度比为1:10~1:1000;所述聚氨酯乳液的用量为0.02~1ml/cm2;所述聚氨酯为水性聚氨酯乳液。
进一步地,所述水性聚氨酯乳液的固含量为9wt%~40wt%。
进一步地,所述小肠黏膜下层膜的制备方法如下:
取小肠,除肌层和浆膜层,脱脂,脱细胞,去垢,冻干,灭菌,即得;
其中,所述脱脂是在体积比1:1的三氯甲烷和甲醇的混合溶液中浸泡10~14h,去离子水清洗;
和/或,所述脱细胞采用酶消化,4℃条件下,在浓度为0.25%的胰酶溶液浸泡过夜,生理盐水冲洗去除胰酶;
和/或,所述去垢是在浓度为0.5%的十二烷基硫酸钠溶液浸泡4~6h,去离子水清洗;
和/或,所述灭菌是利用环氧乙烷灭菌。
进一步地,所述聚氨酯/小肠黏膜下层复合材料制备方法如下:
将聚氨酯乳液与小肠黏膜下层粉末共混,混悬溶液导入模具后,冻干成型,交联,洗涤,冷冻干燥,即得;
其中,所述聚氨酯乳液与小肠黏膜下层粉末的质量比为5:1~9:1;
和/或,所述冻干为-40℃低温冰箱预冻24h后,冷冻干燥;
和/或,所述交联溶液为1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和n-羟基琥珀酰亚胺的混合溶液;
和/或,所述交联为在37℃条件下,避光反应30~40h。
进一步地,所述聚氨酯乳液的制备方法如下:
(1)预聚:取羟基供体、异氰酸酯与催化量的催化剂,预聚;
其中,羟基供体与异氰酸酯的摩尔比为1:0.9~1:2;优选地,羟基供体与异氰酸酯的摩尔比为1:1.05~1:1.85;
(2)扩链:加入扩链剂进行扩链;
其中,扩链剂与步骤(1)中羟基供体的摩尔比为1:8~8:1;优选地,扩链剂与羟基供体的摩尔比为1:1;
(3)中和乳化:加入中和剂,搅拌均匀,将反应体系滴加至20%(v/v)的丙酮水溶液中,高速搅拌1~2h,除去小分子有机物残留,即得;
其中,中和剂与步骤(2)中扩链剂的摩尔比为:0.5:1~3:1;优选地,中和剂与扩链剂的摩尔比为1.5:1。
进一步地,步骤(1)中,所述羟基供体为聚四氢呋喃醚、1,2-戊二醇、聚乙二醇、丙三醇中任意一种或多种;优选地,所述羟基供体为聚四氢呋喃醚;
和/或,步骤(1)中,所述异氰酸酯为异氟尔酮二异氰酸酯、甲基二异氰酸酯、1,6-己二异氰酸酯中任意一种或多种;优选地,所述异氰酸酯为异氟尔酮二异氰酸酯;
和/或,步骤(1)中,所述催化剂为辛酸亚锡或二月桂酸二丁基锡;优选地,所述催化剂为辛酸亚锡;
和/或,步骤(1)中,预聚为在74℃的油浴锅中,反应2.5h~3.5h;
和/或,步骤(2)中,所述扩链剂为2,2-二羟甲基丙酸、甲基二乙醇胺、二羟基甲基丙酸、硫酸丁二醇、乙二胺基乙磺酸钠中任意一种或多种;优选地,所述扩链剂为2,2-二羟甲基丙酸;
和/或,步骤(2)中,扩链的温度为50~55℃;扩链的时间为3~4h;
和/或,步骤(3)中,所述的中和剂为三乙胺;
和/或,步骤(3)中,除去小分子有机物残留的方法为旋转蒸发和透析;
和/或,步骤(3)中,高速搅拌的转速为1300rpm。
进一步地,所述小肠黏膜下层粉末的制备方法如下:
取小肠,除肌层和浆膜层,脱脂,脱细胞,去垢,冻干,低温粉碎,灭菌,即得;
其中,所述脱脂是在体积比1:1的三氯甲烷和甲醇的混合溶液中浸泡10~14h,去离子水清洗;
和/或,所述脱细胞采用酶消化,4℃条件下,在浓度为0.25%的胰酶溶液浸泡过夜,生理盐水冲洗去除胰酶;
和/或,所述去垢是在浓度为0.5%的十二烷基硫酸钠溶液浸泡4~6h,去离子水清洗;
和/或,低温粉碎为液氮冷却后,使用球磨仪进行低温粉碎,并过200目筛;
和/或,所述灭菌是利用环氧乙烷灭菌。
本发明还提供了一种制备前述的修复材料的方法,它由如下方法制备:
在小肠黏膜下层膜上涂一层聚氨酯乳液后将其粘贴在聚氨酯/小肠黏膜下层复合材料上,37℃下烘干,即得;
其中,所述聚氨酯乳液的用量为0.02~1ml/cm2。
本发明还提供了前述多层软组织修复材料在制备软组织修复材料中的用途。
进一步地,所述软组织修复材料为子宫壁软组织修复材料。
本发明制备的多层软组织修复材料具有下述有益效果:
(1)本发明通过水性聚氨酯乳液将sis膜和pu/sis复合材料牢固的粘接在一起;其中,pu/sis复合材料作为肌层,并对sis膜层起支撑作用,sis膜作为内膜层,更利于细胞的粘附与生长,可促进软组织的修复;根据现有文献的报道,未经改性的水性聚氨酯粘接性能较差,通常很难用于粘接,而经过改性后的聚氨酯材料则存在毒性强、生物相容性差的问题。然而,本发明却意外的发现,对于本发明特定的sis膜和pu/sis复合材料,可以采用未经改性的水性聚氨酯有效粘结,既达到了有效粘结,又保证了无毒性、生物相容性优良。
(2)本发明制备的pu/sis复合材料层为孔洞互相贯通的不规则多孔结构,孔隙率较高,可达94%,孔径均匀,大小在20~100μm间,此孔径大小适合细胞粘附,利于诱导细胞和毛细血管长入,促进细胞增殖;
(3)本发明制备的pu/sis复合材料层具有良好的力学性能,其压缩强度和拉伸强度均较高,与纯pu接近,克服了现有技术中sis力学强度差的缺点,可满足软组织修复材料对力学性能的要求;
(4)本发明制备的pu/sis复合材料层具有良好的回弹性,第50次循环后,其回弹性及最大压缩/拉伸应力变化与pu组在对应循环次数数据接近,仍能满足软组织修复材料对回弹性能的要求;
(5)本发明制备的sis-pu/sis复合支架具有良好的血液相容性和组织相容性,植入体内肌肉组织后,能与周围细胞和组织紧密结合,促进细胞向材料内部迁移和增殖,促进血管新生;
(6)本发明制备的sis-pu/sis复合支架植入肌肉组织后缓慢降解,可满足组织生长需要。
综上,本发明制备的多层软组织修复材料,通过水性聚氨酯乳液将sis膜和pu/sis复合材料牢固的粘接在一起;其不仅具有良好的力学性能和回弹性能,还具有良好的生物活性和生物相容性,在体内中可缓慢降解,也克服了现有软组织修复材料缺乏弹性和机械完整性、体内降解过快、修复部位的再细胞化效率低以及术后炎症反应重等缺点,具有良好的应用前景。
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出更多其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
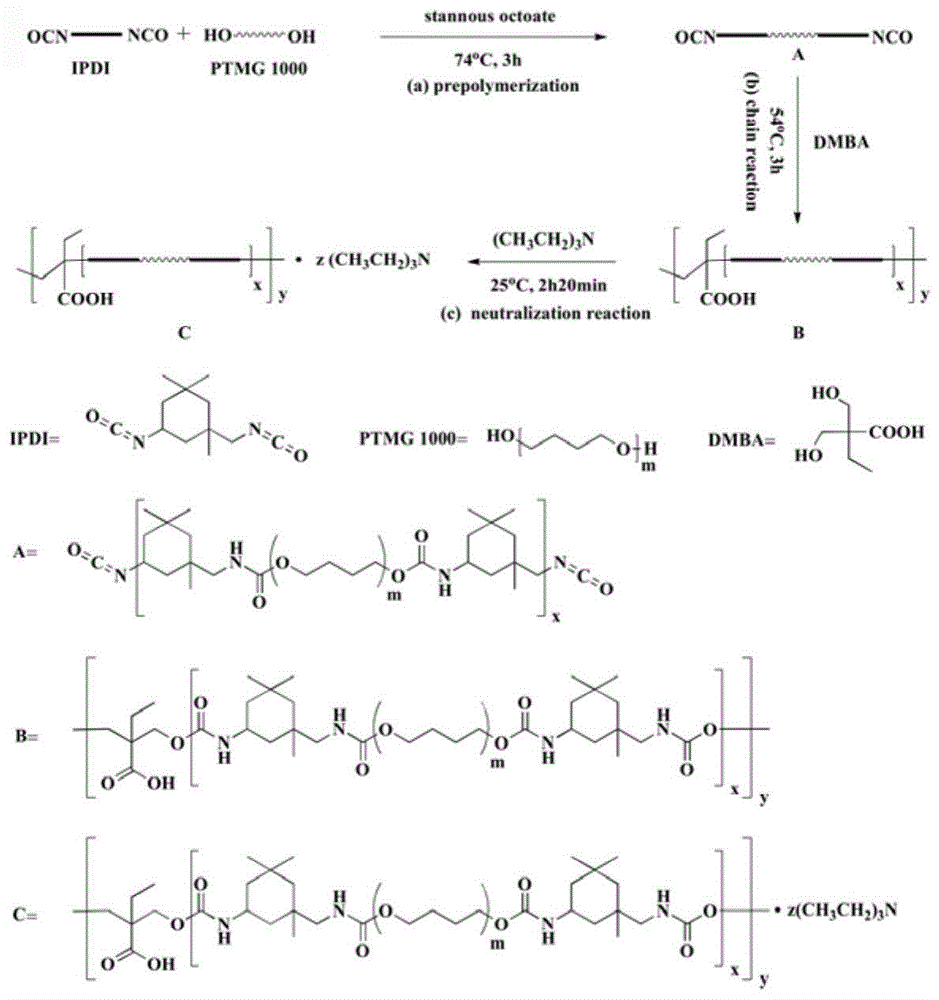
图1扩链剂为dmba的pu合成路线图。
图2pu的1h-nmr图。
图3sis、交联前后的pu和pu/sis复合材料的ftir分析结果图。
图4pu/sis复合材料的形态结构观察与孔径统计图:
a-c:pu/sis1、pu/sis2和pu/sis3的sem结果图;d-f:pu/sis1、pu/sis2和pu/sis3用甘油封片的he染色结果图(标尺:100μm);g-i:pu/sis1、pu/sis2和pu/sis3的平均孔径分布情况。
图5pu和pu/sis复合材料的压缩强度和弹性模量结果图,*表示与pu组相比差异有统计学意义,p<0.05:
a:压缩强度;b:弹性模量。
图6pu和pu/sis复合材料的压缩循环应力-应变曲线:
a-d:分别为pu、pu/sis1、pu/sis2和pu/sis3压缩循环应力-应变曲线。
图7pu和pu/sis复合材料的拉伸性能结果图,*表示与pu组相比差异有统计学意义,p<0.05:
a:拉伸强度;b:弹性模量;c:断裂伸长率;d-f:分别为pu、pu/sis1和pu/sis2的拉伸循环应力-应变曲线。
图8pu和pu/sis复合材料用中性树胶封片的he染色结果图(标尺:100μm)。
图9sps复合材料的制备流程图。
图10pp材料和sps材料肌肉埋植1周的desmin免疫荧光染色结果图(标尺:50μm)。
图11pp材料和sps材料肌肉埋植4周的desmin免疫荧光染色结果图(标尺:50μm)。
图12pp材料和sps材料肌肉埋植12周的desmin免疫荧光染色结果图(标尺:50μm)。
具体实施方式
缩略词:
sis:小肠黏膜下层,pu:聚氨酯,sds:十二烷基硫酸钠,ptmg:聚四氢呋喃醚,ipdi:异氟尔酮二异氰酸酯,dmba:2,2-二羟甲基丙酸,tea:三乙胺,edc:1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,nhs:n-羟基琥珀酰亚胺。
实施例1一种多层软组织修复材料的制备方法
1、sis膜的制备
1)分离出小肠黏膜下层:先将清洗干净的猪小肠剪成长度为15cm左右的肠段,再将小肠的肌层和浆膜层刮去,生理盐水洗涤并滤干后得到小肠黏膜下层;
2)脱脂:将制备的小肠黏膜下层浸入脱脂溶液(chcl3:ch3oh=1:1,v/v)中12h后,反复清洗至无异味;
3)脱细胞:4℃条件下,将已脱脂的小肠黏膜下层浸入浓度为0.25%的胰酶溶液中过夜,漂洗至无泡沫后,将其浸入浓度为0.5%的十二烷基硫酸钠溶液中4h,去离子水洗涤后冻干;
4)环氧乙烷灭菌备用。
2、sis粉末的制备
1)分离小肠黏膜下层:将清洗干净的猪小肠剪成长度为15cm左右的肠段,刮去小肠的肌层和浆膜层,生理盐水洗涤并滤干后得到小肠黏膜下层;
2)脱脂:将制备的小肠黏膜下层浸入脱脂溶液(chcl3:ch3oh=1:1,v/v)中12h后,反复清洗至无异味;
3)脱细胞:4℃条件下,将已脱脂的小肠黏膜下层浸入浓度为0.25%的胰酶溶液中过夜,漂洗至无泡沫后,将其浸入浓度为0.5%的sds溶液中4h,去离子水洗涤后冻干;
4)低温粉碎:剪碎的sis经液氮冷却后,使用球磨仪进行低温粉碎并过200目筛,消毒灭菌备用。
3、pu乳液的制备
1)预聚:将ptmg1000(33.33g,33.33mmol)、ipdi(24.18g,96.67mmol)和辛酸亚锡(0.02ml)加入三口烧瓶中,于74℃油浴锅中反应3h;
2)扩链:加入dmba(4.94g,33.33mmol)在54℃下反应3h,为使反应平稳进行,必要时可使用丙酮调节体系粘度;
3)中和乳化:加入tea(7ml),在室温下搅拌20min,将反应产物缓慢滴加至丙酮水溶液(丙酮:去离子水=1:4)中,1300rpm搅拌2h得到pu乳液,通过旋转蒸发和透析除去小分子有机物残留。
扩链剂为dmba的pu合成路线图如图1所示。
对制备的pu乳液分别进行核磁共振氢谱分析(1h-mnr)和傅里叶红外光谱(ftir)分析,结果分别如图2和图3所示。
由图2可知,合成的pu中ptmg含量最大,化学位移δ=1.63和3.45ppm分别对应l,k的核磁共振峰面积也较大。δ=2.85ppm为ipdi中j的核磁峰。δ=7.03~6.93ppm对应-nh-co-上h的核磁峰。δ=4.21和4.03~4.04ppm分别对应dmba中c和d的核磁峰。由于ipdi和dmba的含量较低,其核磁共振峰面积也较小。δ=1.31~1.30ppm处的三重峰和3.22~3.18ppm处的四重峰分别为tea中o和p的核磁峰。在δ=3.20ppm处未出现与-nco相连的ch2上的质子对应的核磁峰,表明ipdi中的自由-nco已反应完全。通过计算c、k和p处的的核磁峰面积,得到体系中dmba:ptmg:tea的摩尔比为10:40:1。
由图3可知,pu的ftir曲线中,3316cm-1处的吸收峰为氨基甲酸酯基中n-h的伸缩振动峰,2940cm-1和2855cm-1分别对应甲基和亚甲基中的c-h伸缩振动峰,1698cm-1处的吸收峰为氨基甲酸酯基中c=o的伸缩振动峰。1539cm-1处为pu中c-n的伸缩振动峰与n-h的弯曲振动峰的叠加。ipdi中-nco对应的吸收峰在2240~2270cm-1处,谱图中该峰基本消失,说明得到pu中不存在-nco,与1h-nmr的结果一致。
ftir和1h-nmr的测试结果均表明实验成功合成了pu乳液。
4、pu/sis复合材料的制备
1)共混:将1gsis粉末加入到5g固含量为15wt%的pu乳液中,室温下搅拌2.5h,得到pu/sis混悬溶液;
2)冻干:将制备的共混体系倒入模具中,于-40℃低温冰箱中预冻后,冷冻干燥24h;
3)交联:将冷冻干燥后的材料浸泡在edc/nhs溶液中,37℃下避光反应36h,pbs洗涤5次,每次30min,再用去离子水洗涤3次,经冷冻干燥得到pu/sis复合材料。
对制备的pu/sis复合材料进行ftir分析,分析结果如图3所示。
由图3可知,交联后的pu/sis复合材料的ftir曲线在1666cm-1处出现的吸收峰为复合材料经edc交联形成的酰胺键的特征吸收峰,表明pu/sis在交联剂edc的作用下发生了化学反应。
5、sis-pu/sis复合材料的制备
在sis膜上涂一层pu乳液后将其粘贴pu/sis复合材料上,37℃下烘干即得sis-pu/sis(sps)复合材料,pu乳液的用量为0.5ml/cm2。
实施例2一种多层软组织修复材料的制备方法
1、sis膜的制备
与实施例1相同。
2、sis粉末的制备
与实施例1相同。
3、pu乳液的制备
与实施例1相同。
4、pu/sis复合材料的制备
1)共混:将1gsis粉末加入到7g固含量为21wt%的pu乳液中,室温下搅拌2.5h,得到pu/sis混悬溶液;
2)冻干:将制备的共混体系倒入模具中,于-40℃低温冰箱中预冻后,冷冻干燥24h;
3)交联:将冷冻干燥后的材料浸泡在edc/nhs溶液中,37℃下避光反应36h,pbs洗涤5次,每次30min,再用去离子水洗涤3次,经冷冻干燥得到pu/sis复合材料。
对制备的pu/sis复合材料进行ftir分析,分析结果如图3所示。
由图3可知,交联后的pu/sis复合材料的ftir曲线在1666cm-1处出现的吸收峰为复合材料经edc交联形成的酰胺键的特征吸收峰,表明pu/sis在交联剂edc的作用下发生了化学反应。
5、sis-pu/sis复合材料的制备
与实施例1相同。
实施例3一种多层软组织修复材料的制备方法
1、sis膜的制备
与实施例1相同。
2、sis粉末的制备
与实施例1相同。
3、pu乳液的制备
与实施例1相同。
4、pu/sis复合材料的制备
1)共混:将1gsis粉末加入到9g固含量为27wt%的pu乳液中,室温下搅拌2.5h,得到pu/sis混悬溶液;
2)冻干:将制备的共混体系倒入模具中,于-40℃低温冰箱中预冻后,冷冻干燥24h;
3)交联:将冷冻干燥后的材料浸泡在edc/nhs溶液中,37℃下避光反应36h,pbs洗涤5次,每次30min,再用去离子水洗涤3次,经冷冻干燥得到pu/sis复合材料。
对制备的pu/sis复合材料进行ftir分析,分析结果如图3所示。
由图3可知,交联后的pu/sis复合材料的ftir曲线在1666cm-1处出现的吸收峰为复合材料经edc交联形成的酰胺键的特征吸收峰,表明pu/sis在交联剂edc的作用下发生了化学反应。
5、sis-pu/sis复合材料的制备
与实施例1相同。
以下用实验例的方式来证明本发明的有益效果:
实验例1pu/sis复合材料的形态学分析
1、实验方法
取实施例1~3制备的直径为6mm的pu/sis复合材料pu/sis1、pu/sis2和pu/sis3,切成300μm厚的薄片,冻干、喷金处理,利用扫描电子显微镜(sem)观察材料的横切面结构。并对pu/sis复合材料进行he染色,观察其形貌。利用ipp软件对pu/sis复合材料的孔径进行统计学分析。
2、实验结果
pu/sis复合材料的形态结构观察与孔径统计如图4所示。
由图4可知,pu/sis1、pu/sis2和pu/sis3的三维结构均为孔洞相互贯通的不规则多孔结构。pu/sis1、pu/sis2和pu/sis3的孔径主要在20~120μm、20~100μm和20~80μm区间分布,平均孔径分别为136.89±165.96μm、103.82±108.77μm和68.85±51.40μm。
实验结果说明本发明制备的pu/sis复合材料具有贯通的三维多孔结构,其孔径均匀,大小满足细胞生长需求,利于细胞长入。
实验例2pu/sis复合材料的压缩性能测试
1、实验方法
取实施例1~3制备的pu/sis复合材料pu/sis1、pu/sis2和pu/sis3及pu材料。利用方形模具将pu、pu/sis1、pu/sis2和pu/sis3制备成10×10×10mm的立方体,pbs浸泡20min后,每组取三个样本采用万能力学测试系统进行测试。实验环境为室温,测试速度为10mm/min,试样的压缩形变量是其厚度的50%,循环次数为50次,记录样品的最大应力、压缩模量和循环应力-应变曲线。计算样本在第10次循环、第30次循环和第50次循环的压缩回弹性(r压缩)和样本在第30次循环和第50次循环与第10次循环相比的最大压缩应力变化。样品压缩回弹性的计算公式如下:
r压缩=wunload/wload
wunload:压缩功
wload:回复功
2、实验结果
pu/sis复合材料的压缩性能如图5、图6、表1和表2所示。
表1pu和pu/sis复合材料在第10次循环、第30次循环和第50次循环的压缩回弹性。
*表示与pu/sis2相比差异有统计学意义,p<0.01。
表2pu和pu/sis复合材料在第30次循环和第50次循环与第10次循环相比的最大压缩应力变化。
*表示与pu/sis2相比差异有统计学意义,p<0.05。
由图5可知,制备的pu/sis复合材料pu/sis1、pu/sis2和pu/sis3的压缩强度分别为47.96±3.369kpa、151.8±6.489kpa和270.6±22.75kpa,弹性模量分别为213.9±14.60kpa,967.3±192.8kpa和1394±185.7kpa。较天然sis材料相比,制备的pu/sis复合材料强度显著性提高,且pu/sis2和pu/sis3的压缩强度和弹性模量与pu组接近。由图6可知,pu和pu/sis复合材料的压缩循环应力-应变曲线均出现滞后环。
由表1和表2可知,本发明制备的pu/sis复合材料均有良好的压缩回弹性能,且实施例2制备的pu/sis复合材料在第10次循环、第30次循环与第50次循环的回弹性数据均与pu组接近,在第30次循环和第50次循环与第10次循环相比的最大压缩应力分别减少了9.630±0.35%和14.36±0.78%,与pu组接近。
实验结果说明本发明制备的pu/sis复合材料具有优良的压缩强度和压缩回弹性能。
实验例3pu/sis复合材料的拉伸性能测试
1、实验方法
取实施例1~2制备的pu/sis复合材料pu/sis1和pu/sis2及pu材料。利用哑铃形模具将pu、pu/sis1和pu/sis2制备成75×4×2mm大小的哑铃型试样。pbs浸泡20min后,每组取三个样本采用万能力学测试系统测试各样本的拉伸强度、拉伸模量和断裂伸长率。实验环境为室温,拉伸速率为30mm/min,夹持长度为30mm。
将pu、pu/sis1和pu/sis2制备成75×4×2mm大小的哑铃状试样,浸泡在pbs中20分钟后,每组取三个样本采用万能力学测试系统进行测试。实验环境为室温,拉伸速率为60mm/min,夹持长度为30mm,试样应变量为试样夹持长度的100%,循环次数为50次,记录样品的最大应力和循环应力-应变曲线。计算样本在第10次循环、第30次循环和第50次循环的拉伸回弹性和样本在第30次循环和第50次循环与第10次循环相比的最大拉伸应力变化。样品数为3个,取其平均值。样品拉伸回弹性的计算公式如下:
r拉伸=wunload/wload
wunload:拉伸功
wload:回复功
2、实验结果
pu/sis复合材料的拉伸性能如图7、表3和表4所示。
表3pu和pu/sis复合材料在第10次循环、第30次循环和第50次循环的拉伸回弹性。
*表示与pu/sis2相比差异有统计学意义,p<0.05。
表4pu和pu/sis复合材料在第30次循环和第50次循环与第10次循环相比的最大拉伸应力变化。
*表示与pu/sis2相比差异有统计学意义,p<0.05。
由图7可知,制备的pu/sis复合材料pu/sis1和pu/sis2的拉伸强度分别为336.7±25.17kpa和406.7±11.55kpa,弹性模量分别为143.3±11.55kpa和133.3±5.774kpa,断裂伸长率分别为452.8±11.97%和861.4±49.69%。虽然制备的pu/sis复合材料拉伸强度和断裂伸长率低于pu组,但弹性模量高于pu组,且与天然sis材料相比,显著性提高。pu和pu/sis复合材料的拉伸循环应力-应变曲线均出现滞后环。
由表3和表4可知,本发明制备的pu/sis复合材料均有良好的拉伸回弹性能。其中,实施例2制备的pu/sis复合材料(pu/sis2)在第10次循环、第30次循环与第50次循环的回弹性数据分别为0.7281±0.0069,0.7269±0.0053和0.7248±0.0071,与pu组在对应循环次数的回弹性数据接近。pu/sis2在第30次循环和第50次循环与第10次循环相比的最大压缩应力分别减少16.72±0.31%和24.92±0.68%,均与pu组接近。
实验结果说明本发明制备的pu/sis复合材料具有优良的拉伸强度和拉伸回弹性能。
实验例4pu/sis复合材料的形貌分析
1、实验方法
利用he染色观察sis在pu/sis复合材料中的分布情况。
2、实验结果
pu和pu/sis复合材料以中性树胶为封片剂的he染色结果如图8所示。与pu组相比,pu/sis复合材料的孔径较小且孔径分布更为均匀。其原因可能是预冻时形成的冰晶的形状与溶液的组分有关,在sis加入后体系形成的冰晶大小更为均一,冻干后材料的孔径分布也就更为均一。pu与中性树胶接触后出现了部分溶解和褪色,因此在pu/sis组和pu组均可观察到pu组分边缘变得圆润且有溶解迹象。sis在与中性树胶接触后的颜色和形态则未发生变化,因此sis组分在pu/sis复合材料中呈现深粉色,而pu在体系中为浅粉色。sis颗粒被pu相包裹且在体系中分布均匀。
实验结果说明本发明可成功制备sis均匀分散的pu/sis复合材料。
实验例5本发明多层软组织修复材料肌肉埋植与体内降解情况观察
1、实验方法
(1)实验动物
健康成年雄性新西兰大白兔9只,体重2~3kg,购于四川省动物养殖中心,伦理备案号为2017093a。四川大学动物实验中心饲养一周后待用。
(2)实验材料及分组
制备大小为1cm×0.2cm×0.2cm块状的pp和sps复合材料,环氧乙烷消毒灭菌,备用。手术前,将材料浸泡于生理盐水中12小时,使材料充分水化。sps复合材料植入组作为实验组,pp材料植入组作为对照组。
(3)手术操作
将新西兰大白兔仰卧固定,采用浓度为3%的戊巴比妥腹腔注射麻醉。剃光其背部毛发并将其俯卧固定于手术板上,碘伏消毒皮肤后,铺好手术巾。无菌条件下,在新西兰大白兔背部脊柱两侧旁25~50mm处选择植入点,切开皮肤与筋膜,用止血钳沿肌纤维长轴轻轻分离肌肉,将对照品pp和试样sps植入深度为10~20mm的肌肉里,一侧植入试样,一侧植入对照品,逐层缝合肌膜和皮肤切口,待新西兰大白兔苏醒。
(4)术后大体观察
观察术后新西兰大白兔的生存状况如饮食、活动等,以及伤口的愈合情况:如缝线有无脱落、材料有无露出以及术区有无明显的渗液、流脓及红肿等。
(5)组织学观察
于术后1周、4周和12周,每个实验组随机选取3只新西兰大白兔,取出植入材料及周围组织。标本用oct冷冻切片包埋剂包埋,冰冻切片机切片。对切片进行desmin免疫荧光染色,在显微镜下观察骨骼肌细胞迁移情况。
desmin免疫荧光染色方法如下:
a.冰冻切片:分别在术后1周、4周和12周,每个实验组随机选取3只新西兰大白兔,取出植入材料及周围组织。标本用oct冷冻切片包埋剂包埋,冰冻切片机切片切成6μm厚的薄片,立即用10%中性甲醛室温固定5min后pbs清洗干净。
b.封闭:滴加山羊血清,置于37℃孵箱中封闭30min。
c.加一抗:吸水纸吸掉多余封闭液,一抗desmin以1:200稀释后直接滴加于细胞材料复合物上并放入湿盒,4℃孵育过夜。
d.清洗:取出细胞材料复合物,室温复温30min,pbs清洗3次。
e.加二抗:吸水纸吸掉多余液体,滴加新鲜配制好的山羊抗小鼠二抗(1:200)工作液于材料上并放入湿盒,37℃孵育60min后pbs洗涤3次。
f.dapi染色:滴加dapi避光孵育3min后用pbs清洗干净。
g.封片:用含抗荧光淬灭剂的封片液封片并在荧光显微镜下采集图像。
2、实验结果
(1)实验动物观察
术后各组新西兰大白兔全部存活,在手术当天即开始进食且活动正常,与术前相比无明显变化。术后伤口愈合良好,且未见红肿、感染、缝线脱落、移植物排出等不良反应发生。
(2)组织学观察
desmin免疫荧光染色观察:观察pp和sps复合材料在不同时间点于新西兰大白兔骨骼肌组织中desmin+(骨骼肌细胞)的迁移情况。desmin阳性的细胞在荧光显微镜下显示红色,材料的自发荧光在fitc通道显示为绿色。细胞核及材料在荧光显微镜下显示蓝色。由图10、图11和图12可知,术后1周,pp组与周围骨骼肌组织连接疏松,且在骨骼肌组织以外的区域未见到明显的desmin阳性的细胞。sps组与周围骨骼肌组织的连接比pp组紧密。sps组可观察到desmin阳性的细胞向sps复合材料的sis层迁移和聚集。术后4周,sps组与周围骨骼肌组织的连接更为紧密。pp组和sps组中骨骼肌细胞均开始向材料内部迁移,其中sps复合材料中的desmin阳性的细胞较多。术后12周,pp组和sps组与周围骨骼肌组织的连接均比第1周、第4周更为紧密。骨骼肌细胞进一步向材料内部迁移和增殖。
实验结果说明本实验制备的sps复合材料具有良好的组织相容性。
综上所述,本发明制备的多层软组织修复材料(sps复合材料),通过水性聚氨酯乳液将sis膜和pu/sis复合材料牢固的粘接在一起;既具有良好的力学性能和回弹性能,满足软组织对机械性能的要求;又具有可供细胞粘附、生长和增殖的多孔结构,可诱导和促进细胞长入和组织的再生和修复;还具有良好的组织相容性。克服了现有软组织修复材料缺乏弹性和机械完整性、体内降解过快、修复部位的再细胞化效率低以及术后炎症反应重等缺点,具有良好的应用前景。
- 还没有人留言评论。精彩留言会获得点赞!