光稳定性及溶出性优异的药物制剂的制作方法
【
技术领域:
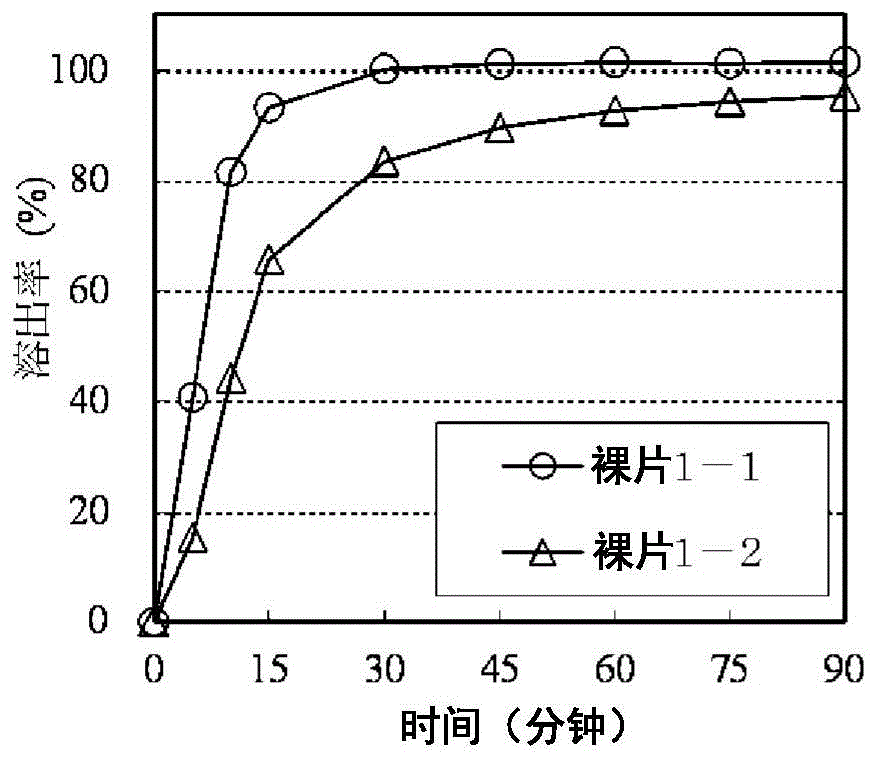
】本发明涉及含有多环性吡啶酮化合物的制剂。本发明涉及以光稳定化物质及聚合物包覆,即使经光照射亦不着色的含有多环性吡啶酮化合物的固体制剂,详言之,涉及含有选自由食用焦油色素、食用色淀化的焦油色素、食用天然色素、氧化铁、氧化钛及滑石构成的组的1种以上作为光稳定化物质,含有羟丙基甲基纤维素作为聚合物,即使经光照射也不会着色的含有多环性吡啶酮化合物的固体制剂。【现有技术】流行性感冒为以流行性感冒病毒感染为起因的急性呼吸器官感染症。于日本每个冬天都有数百万人的类流行性感冒患者的报告,流行性感冒伴随高患病率及死亡率。于婴幼儿、高龄者等高危险群为特别重要的疾病,于高龄者并发肺炎的合并率高,因流行性感冒造成死亡者高龄者占大多数。抗流行性感冒药公知有抑制病毒脱核步骤的欣美特(symmetrel;商品名:金刚烷胺(amantadine))或福玛丁(flumadine;商品名:金刚乙胺(rimantadine))、抑制病毒从细胞出芽/释出的为神经氨酸酶抑制剂的奥司他韦(oseltamivir;商品名:克流感(tamiflu))或扎那米韦(zanamivir;商品名:瑞乐莎(relenza))。然而,有耐性株出现或副作用的问题、病原性或致死性高的新型流行性感冒病毒世界性大流行等之虞,因此期待开发新的作用机制的抗流行性感冒药。由于属于源自流行性感冒病毒的酶的帽状结构(cap)依赖性内切核酸酶对于病毒增殖是必要的,且具有宿主所不具有的病毒特异性酶活性,因此认为可应用为抗流行性感冒药的标靶。就抑制帽状结构(cap)依赖性内切核酸酶的化合物而言,于专利文献1中记载有下述式(ii)表示的化合物,可作为具有抗病毒作用,尤其是具有抑制流行性感冒病毒增殖活性的化合物使用。于活体内给予(例如口服给予)式(ii)表示的化合物时,必须提供高效地被体内吸收而显示高药理效果的同时缩短流行性感冒患病期间的化合物,为了达到所述目的,提供为式(ii)表示的化合物的前药的式(i)表示的化合物。式(i)表示的化合物亦公开于专利文献1。然而,专利文献1没有公开式(i)表示的化合物的具体制剂。于专利文献5至7公开有改善溶出性的制剂。然而,于专利文献5至7使用的化合物与式(i)表示的化合物的化学构造有很大的差异,根据专利文献5至7记载的制剂处方对于是否可改善式(i)表示的化合物的溶出性则不明,亦未有公开或暗示。再者,通过改善溶出性的添加剂亦有可能生成多量的类似物质。另外,于专利文献2至4记载有通过对于经光照射而着色的制剂包覆氧化钛,可减轻制剂的着色。然而,制剂是否会因为光照射而着色因化合物而异,不清楚含有式(i)表示的化合物的制剂是否会着色,亦未有公开或暗示。[先前技术文献][专利文献][专利文献1]国际公开第2016/175224号说明书[专利文献2]日本特开2013-14610号公报[专利文献3]国际公开第2002/060446号说明书[专利文献4]国际公开第2007/052592号说明书[专利文献5]日本特开2010-270112号公报[专利文献6]国际公开第2004/052342号说明书[专利文献7]国际公开第2012/144592号说明书技术实现要素:[发明欲解决的课题]本发明的课题为发现即使经光照射亦不着色的含有式(i)表示的化合物的制剂,并提供从该制剂溶出式(i)表示的化合物的溶出性经改善的制剂。[解决课题的方法]本发明人等发现于含有式(i)表示的化合物的制剂经光照射时,制剂本体会着色。另外,发现式(i)表示的化合物溶解性低,要获得所期望的溶出性有困难。本发明人等为了解决上述课题,进行深入研究的结果发现,通过在含有式(i)表示的化合物的制剂包覆光稳定化物质及聚合物,则即使经光照射,亦几乎不会着色,因而完成本发明。另外,式(i)表示的化合物的溶解性低,要获得所期望的溶出性有困难,探讨种种崩解剂,发现类似物质少且可改善溶出性的崩解剂,因而完成本发明。以下,亦有将于本发明完成的制剂称为“本发明制剂”的情况。即,本发明涉及(1)一种含有式(i)表示的化合物或其药学上可接受的盐的固体制剂,其具有包含光稳定化物质及聚合物的包覆层,(2)如上述(1)所述的固体制剂,其中,该包覆层中的光稳定化物质为选自由食用焦油色素、食用色淀化的焦油色素、食用天然色素、氧化铁、氧化钛及滑石构成的组的1种以上,(3)如上述(1)所述的固体制剂,其中,该包覆层中的光稳定化物质为选自由食用赤色2号、食用赤色3号、食用赤色102号、食用赤色104号、食用赤色105号、食用赤色106号、食用黄色4号、食用黄色5号、食用绿色3号、食用青色1号、食用青色2号、食用赤色3号铝色淀、食用黄色4号铝色淀、食用黄色5号铝色淀、食用青色1号铝色淀、食用青色2号铝色淀、胭脂红、叶绿素铜钠盐、叶绿素铜、铁丹(日文:バンガラ)、氧化铁红、氧化铁黄、氧化铁黑、黄色铁氧化物、氧化钛及滑石构成的组的1种以上,(4)如上述(3)所述的固体制剂,其中,该包覆层中的光稳定化物质为选自由氧化铁红、氧化铁黄、氧化铁黑、黄色铁氧化物、氧化钛及滑石构成的组的1种以上,(5)如上述(4)所述的固体制剂,其中,该包覆层中的光稳定化物质为氧化钛和/或滑石,(6)一种含有式(i)表示的化合物或其药学上可接受的盐的固体制剂,其具有包覆层,该包覆层含有选自由食用赤色2号、食用赤色3号、食用赤色102号、食用赤色104号、食用赤色105号、食用赤色106号、食用黄色4号、食用黄色5号、食用绿色3号、食用青色1号、食用青色2号、食用赤色3号铝色淀、食用黄色4号铝色淀、食用黄色5号铝色淀、食用青色1号铝色淀、食用青色2号铝色淀、胭脂红、叶绿素铜钠盐、叶绿素铜、铁丹、氧化铁红、氧化铁黄、氧化铁黑、黄色铁氧化物、氧化钛及滑石构成的组的1种以上及聚合物,(7)如上述(6)所述的含有式(i)表示的化合物或其药学上可接受的盐的固体制剂,其中,该固体制剂具有包含氧化钛和/或滑石及聚合物的包覆层,(8)如上述(1)至(7)中任一项所述的固体制剂,其中,该包覆层中的聚合物为选自纤维素类聚合物、丙烯酸类聚合物及乙烯类聚合物的1种以上,(9)如上述(1)至(7)中任一项所述的固体制剂,其中,该包覆层中的聚合物为选自羟丙基甲基纤维素、羟丙基纤维素、羧甲基乙基纤维素、羟丙基甲基纤维素苯二甲酸酯、羟丙基甲基纤维素乙酸酯琥珀酸酯及乙基纤维素的1种以上的纤维素类聚合物,(10)如上述(9)所述的固体制剂,其中,该纤维素类聚合物为羟丙基甲基纤维素,(11)如上述(1)至(7)中任一项所述的固体制剂,其中,该包覆层中的聚合物为选自甲基丙烯酸共聚物、甲基丙烯酸氨基烷基酯共聚物e及甲基丙烯酸氨基烷基酯共聚物rs的1种以上的丙烯酸类聚合物,(12)如上述(1)至(7)中任一项所述的固体制剂,其中,该包覆层中的聚合物为选自聚乙烯醇、聚乙烯吡咯烷酮及聚乙烯醇-甲基丙烯酸甲酯-丙烯酸共聚物的1种以上的乙烯类聚合物,(13)如上述(12)所述的固体制剂,其中,该乙烯类聚合物为聚乙烯醇,(14)一种含有式(i)表示的化合物或其药学上可接受的盐的固体制剂,其具有包含氧化钛和/或滑石及羟丙基甲基纤维素的包覆层,(15)如上述(1)至(14)中任一项所述的固体制剂,该固体制剂还含有崩解剂,(16)一种固体制剂,其含有式(i)表示的化合物或其药学上可接受的盐及崩解剂,(17)如上述(15)或(16)所述的固体制剂,其中,该崩解剂为选自低取代羟丙基纤维素、羧甲基纤维素钙、交联羧甲基纤维素钠、交联聚维酮、部分α化淀粉及羧甲基淀粉钠的1种以上,(18)如上述(17)所述的固体制剂,其中,该崩解剂为低取代羟丙基纤维素或交联羧甲基纤维素钠,(19)如上述(18)所述的固体制剂,其中,该崩解剂为交联羧甲基纤维素钠,(20)如上述(19)所述的固体制剂,其中,该包覆层中的光稳定化物质为氧化钛和/或滑石,聚合物为羟丙基甲基纤维素,(21)如上述(1)至(20)中任一项所述的固体制剂,其中,在照射120万lux·hr的光时,色差δe为13以下,(22)一种固体制剂,其含有光稳定化物质及式(i)表示的化合物或其药学上可接受的盐,在照射120万lux·hr的光时,色差δe为13以下,(23)如上述(22)所述的固体制剂,其中,该光稳定化物质为选自食用焦油色素、食用色淀化的焦油色素、食用天然色素、氧化铁、氧化钛及滑石构成的组的1种以上,(24)如上述(23)所述的固体制剂,其中,该光稳定化物质为氧化钛和/或滑石,(25)如上述(1)至(24)中任一项所述的固体制剂,该固体制剂为铝箔泡罩的包装形态,(26)一种固体制剂,为铝箔泡罩的包装形态,且含有式(i)表示的化合物或其药学上可接受的盐,(27)如上述(1)至(26)中任一项所述的固体制剂,其中,该固体制剂为颗粒剂或片剂,(28)如上述(1)至(27)中任一项所述的固体制剂,其中,从溶出试验开始45分钟后式(i)表示的化合物的溶出率为80%以上,(29)一种分析含有式(i)表示的化合物或其药学上可接受的盐的固体制剂中的分解物的方法,其包括下列步骤:a)将含有式(i)表示的化合物或其药学上可接受的盐的固体制剂作为试样,对该试样进行色谱的步骤,式(i):及b)在以上述步骤获得的色谱数据中,获得关于式(ii)表示的化合物的含量或含有比例的数据的步骤,式(ii):(30)一种式(i)表示的化合物或其药学上可接受的盐的分解组成物,其含有式(ii)表示的化合物,式(ii):式(i):(31)如上述(1)至(28)中任一项所述的固体制剂,该固体制剂含有10mg、20mg、40mg或80mg的式(i)表示的化合物,(32)如上述(1)至(28)及(31)中任一项所述的固体制剂,该固体制剂用于缩短流行性感冒患病期间,(33)如上述(1)至(28)及(31)中任一项所述的固体制剂,该固体制剂用于减少流行性感冒病毒。[发明的效果]根据本发明,于含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含光稳定化物质及聚合物的包覆层,即使经光照射,制剂的颜色与试验初期相比几乎无变化。即,本发明制剂优选为色差δe为13以下的制剂。【附图说明】图1为使用低取代羟丙基纤维素作为崩解剂的片剂的溶出行为。图2为使用交联羧甲基纤维素钠作为崩解剂的片剂的溶出行为。【发明的实施方式】使用式(i)表示的化合物或其药学上可接受的盐作为本发明制剂中的有效成分式(i)表示的化合物或其药学上可接受的盐的制法公开于专利文献1。式(i)表示的化合物或其药学上可接受的盐于活体内转换为式(ii)表示的化合物,具有帽状结构(cap)依赖性内切核酸酶抑制作用。因此,式(i)表示的化合物或其药学上可接受的盐可作为流行性感冒的治疗剂和/或预防剂使用。式(i)表示的化合物或其药学上可接受的盐可用于通过流行性感冒病毒诱发的症状和/或疾病。例如对于伴随发热、恶寒、头痛、肌肉痛、全身倦怠感等类感冒症状或喉咙痛、流鼻涕、鼻塞、咳嗽、痰等气管炎症状,腹痛、呕吐、腹泻等胃肠症状,进一步地,伴随急性脑症、肺炎等二次感染的并发症的治疗和/或预防,对于症状的改善。即,本发明中使用的化合物有益于治疗和/或预防流行性感冒病毒感染症。式(i)表示的化合物或其药学上可接受的盐对于流行性感冒患病期间的缩短为有效者。例如,可缩短流行性感冒患病期间约20至40小时、约25至30小时。具体而言,可缩短“咳嗽”、“喉咙痛”、“头痛”、“鼻塞”、“轻微发烧或恶寒”、“肌肉或关节疼痛”、“疲劳感”至获得改善的时间。尤其有益于缩短“鼻塞”、“肌肉或关节疼痛”、“疲劳感”、“轻微发烧或恶寒”、“头痛”至获得改善的时间。更有益于缩短“鼻塞”及“肌肉或关节疼痛”至获得改善的时间。进一步地,本发明中使用的化合物(亲代化合物和/或前药)由于可以短期内使体内的流行性感冒病毒减少,可成为有益于流行性感冒病毒感染症的治疗和/或预防的优越药品。本发明中使用的化合物给予后于72小时以内,优选于48小时以内,更优选于24小时以内确认到体内流行性感冒病毒量的减少效果,与其他药剂相比,可期待更早期的治疗效果。式(i)表示的化合物或其药学上可接受的盐具备作为药物的有用性。式(i)表示的化合物或其药学上可接受的盐为前药,由于具有口服吸收性高,显示良好的生物利用度及清除率,肺移行性高等优点,可成为优越的药品。式(i)表示的化合物或其药学上可接受的盐代谢稳定性或口服吸收性高,显示良好的生物利用度及清除率。另外,式(i)表示的化合物或其药学上可接受的盐肺移行性高,半衰期长。进一步地,式(i)表示的化合物或其药学上可接受的盐具有非蛋白质结合率高,herg通道抑制或cyp抑制低,确认有cpe(cytopathiceffect、细胞变性作用)抑制效果,和/或于光毒性试验、ames试验、基因毒性试验显示阴性,无肝损害等毒性等优点。因此,本发明的药物组成物可成为优越的药品。式(i)表示的化合物或其药学上可接受的盐的给予量,依给予方法、患者的年龄、体重、状态及疾病的种类而异,通常,在口服给予时,成人每日给予约0.05mg至3000mg,优选约0.1mg至1000mg,更优选约10mg至80mg,最优选约10mg至40mg,必要时亦可分次给予。在非口服给予时,成人每日给予约0.01mg至1000mg,优选约0.05mg至500mg、约1mg至80mg。其可1日1次或分成数次给予。式(i)表示的化合物或其药学上可接受的盐以该化合物的作用的增强或该化合物的给予量的减低等为目的,可与其他药剂等(以下,简称为并用药剂)组合使用。例如,于流行性感冒的疾病可与神经氨酸酶抑制剂(例如,奥司他韦、扎那米韦、帕拉米韦(peramivir)及依那韦(inavir)等)、rna相关性rna聚合酶抑制剂(例如,法匹拉韦(favipiravir))、m2蛋白质抑制剂(例如,金刚烷胺)、pb2cap结合抑制剂(例如,vx-787)、抗ha抗体(例如,mhaa4549a)或免疫作用药(例如,硝唑尼特(nitazoxanide))组合使用。此时,本发明中使用的化合物与并用药剂的给予时期并无特别限制,对于给予对象,可同时给予,亦可以时间差给予。进一步地,与式(i)表示的化合物或其药学上可接受的盐的并用药剂,可作为含有各个活性成分的2种以上制剂给予,亦可作为含有所有活性成分的单一制剂给予。并用药剂的给予量可将临床上使用的用量作为基准适当选择。另外,式(i)表示的化合物或其药学上可接受的盐与并用药剂的调配比,可根据给予对象、给予途径、对象疾病、症状、组合等适当选择。例如,给予对象为人类时,对于式(i)表示的化合物或其药学上可接受的盐1重量份,可使用并用药剂0.01至100重量份。式(i)表示的化合物或其药学上可接受的盐对于帽状结构依赖性内切核酸酶的抑制活性高,由于为病毒特异性的酶,有选择性高等的效果,可成为副作用减轻的药品。以下,对于将式(i)表示的化合物或其结晶或式(ii)表示的化合物特定化的方法加以说明。若无特别说明,本说明书中及申请专利范围所述的数值为大约值。数值的变动因装置校正、装置错误、物质的纯度、结晶大小、试样大小、其他因素引起。本说明书中使用的“结晶”指构成固体的原子、离子、分子等具有规则正确排列构造,其结果具有如周期性、各向异性的构造。结晶形态的结晶化度可通过包含例如粉末x线衍射测定、动态水分吸附测定、差示扫描热量测定、示差热热重量同时测定、溶液比色测定、溶解特性的多种技术测定。化合物的nmr分析于300mhz进行,使用dmso-d6、cdcl3测定。粉末x线衍射模型的测定依照日本药典的一般试验法中记载的粉末x线衍射测定法,进行于各实施例中获得的结晶的粉末x线衍射测定。测定条件如以下所示。(装置)rigaku公司制造的miniflex600或rint-ttriii(操作方法)检测器:高速一维检测器(d/tecultra2)及可变刀口效应测定法:反射法光源的种类:cu管球使用波长:cukα线管电流:10ma或15ma管电压:30kv或40kv试样盘:铝或玻璃x线的入射角(θ):3-40°,取样宽度:0.01°,或x线的入射角(θ):4-40°,取样宽度:0.02°通常于粉末x线衍射中的衍射角度(2θ)产生±0.2°范围内的误差,因此,衍射角度的值亦包含±0.2°程度范围内的数值。因此,不仅是粉末x线衍射中的峰的衍射角度完全一致的结晶,峰的衍射角度以±0.2°程度误差一致的结晶亦包含于本发明。于本发明制剂中的式(i)表示的化合物或其药学上可接受的盐的调配量,对于制剂全量为1至80重量%,优选为5至75重量%,更优选为10至70重量%。本发明制剂含有光稳定化物质。本说明书中,作为光稳定化物质,只要是可使式(i)表示的化合物或其药学上可接受的盐对于光稳定化,此外可防止制剂变色的添加物即可,可使用日本药典、日本药典外药品规格、药品添加物规格及食品添加物公定书中所收载者。光稳定化物质为有遮蔽光的遮光效果的遮光物质或有吸收光的效果的光吸收物质。具体而言,可列举食用焦油色素、食用色淀化的焦油色素、食用天然色素、氧化铁、氧化钛、滑石等。优选可列举食用赤色2号、食用赤色3号、食用赤色102号、食用赤色104号、食用赤色105号、食用赤色106号、食用黄色4号、食用黄色5号、食用绿色3号、食用青色1号、食用青色2号、食用赤色3号铝色淀、食用黄色4号铝色淀、食用黄色5号铝色淀、食用青色1号铝色淀、食用青色2号铝色淀、胭脂红、叶绿素铜钠盐、叶绿素铜、铁丹、氧化铁红、氧化铁黄、氧化铁黑、黄色铁氧化物、氧化钛、滑石等。更优选可列举作为遮光物质的氧化钛、滑石;作为光吸收物质的氧化铁红、氧化铁黄、氧化铁黑、黄色铁氧化物;最优选作为遮光物质的氧化钛、滑石。本发明制剂的光稳定化物质可调配于制剂中,亦可包覆制剂的表面,优选为在包覆制剂的表面的所谓包覆层中含有光稳定化物质。若在制剂的包覆层中含有光稳定化物质,即可吸收、遮蔽来自制剂外的光,可提升制剂中含有的式(i)表示的化合物的光稳定性或防止制剂变色。本发明制剂中的光稳定化物质的含量只要是式(i)表示的化合物或其药学上可接受的盐对于光为稳定化的量即可。具体而言,光稳定化物质中如氧化钛或滑石的将光遮蔽的遮光物质对于制剂的表面积1mm2为0.00075至0.075mg,优选为0.001至0.05mg,更优选为0.0015至0.03mg。本发明制剂含有聚合物。本说明书中,聚合物可使用收载于日本药典、日本药典外药品规格、药品添加物规格及食品添加物公定书中者。具体而言,可列举羟丙基甲基纤维素(羟基丙基甲基纤维素)、聚乙烯醇、乙基纤维素、羧甲基乙基纤维素、羧甲基纤维素、羧甲基纤维素钠、羟乙基纤维素、羟乙基甲基纤维素、羟丙基纤维素、羟丙基甲基纤维素乙酸酯琥珀酸酯、羟丙基甲基纤维素苯二甲酸酯、富马酸/硬脂酸/聚乙烯缩醛二乙氨基乙酸酯/羟丙基甲基纤维素混合物等纤维素类聚合物,丙烯酸乙酯/甲基丙烯酸甲酯共聚物分散液、甲基丙烯酸氨基烷基酯共聚物、甲基丙烯酸共聚物、甲基丙烯酸2-甲基-5-乙烯吡啶酯/甲基丙烯酸共聚物、干燥甲基丙烯酸共聚物、甲基丙烯酸二甲基氨基乙基酯/甲基丙烯酸甲酯共聚物等丙烯酸类聚合物,聚乙烯吡咯烷酮、交联聚维酮、羧乙烯聚合物、聚乙烯缩醛二乙氨基乙酸酯、聚乙烯醇、聚乙烯醇-甲基丙烯酸甲酯-丙烯酸共聚物体及聚乙烯醇共聚物等乙烯类聚合物,巴西棕榈蜡,硬脂醇,虫胶,鲸蜡醇等,优选为羟丙基甲基纤维素(羟基丙基甲基纤维素)。本发明制剂的聚合物可调配于制剂中,亦可包覆制剂的表面,优选为包覆制剂的表面而形成包覆层的作为所谓的包衣剂使用。只要在制剂的包覆层中含有聚合物,则可与光稳定化物质同时将制剂表面包覆,可提升制剂中含有的式(i)表示的化合物的光稳定性或防止制剂变色。本说明书中的包覆层中的聚合物含量只要可将光稳定化物质包覆于制剂表面的量即可。本发明制剂亦可含有崩解剂。崩解剂可使用收载于日本药典、日本药典外药品规格、药品添加物规格及食品添加物公定书等中的崩解剂,根据崩解剂的种类亦有包含式(ii)表示的化合物的类似物质的量增加的情况。具体而言,可列举交联羧甲基纤维素钠、交联聚维酮、羧甲基纤维素钙、羧甲基淀粉钠、低取代羟丙基纤维素等,优选为交联羧甲基纤维素钠。本发明制剂中崩解剂的含量对于制剂全量为0.5至20重量%,优选为0.75至15重量%,更优选为1至10重量%。若比该量少,则固体制剂,尤其是片剂有可能不能充分崩解。本发明制剂亦可含有赋形剂。赋形剂可使用收载于日本药典、日本药典外药品规格、药品添加物规格及食品添加物公定书等中的赋形剂。具体而言,可列举d-甘露醇、木糖醇、山梨糖醇、麦芽糖醇、乳糖醇、寡糖醇等糖醇类,木糖、葡萄糖(glucose)、果糖(fructose)、麦芽糖(maltose)、乳糖(lactose)、蔗糖(sucrose)、异性化糖、发芽糖浆、精制白糖、白糖、精制白糖球状颗粒、无水乳糖、白糖/淀粉球状颗粒等糖类,半消化体淀粉、葡萄糖水合物、粉糖、结晶纤维素、微结晶纤维素、普鲁兰多糖(pullulan)、β-环糊精、氨基乙磺酸、粉饴、氯化钠、柠檬酸、柠檬酸钠、甘氨酸、葡萄糖酸钙、l-谷氨酸、酒石酸、酒石酸氢钾、碳酸铵、右旋糖酐40、糊精、乳酸钙、聚维酮、聚乙烯二醇(聚乙二醇)1500、聚乙烯二醇1540、聚乙烯二醇4000、聚乙烯二醇6000、柠檬酸酐、dl-苹果酸、磷酸氢钠、磷酸二氢钾、磷酸二氢钠、l-天冬氨酸、褐藻酸、羧甲基纤维素钠、含水二氧化硅、交联聚维酮、甘油磷酸钙、硅酸镁铝、硅酸钙、硅酸镁、轻质无水硅酸、合成硅酸铝、小麦粉、小麦淀粉、小麦胚芽粉、小麦胚芽油、米粉、米淀粉、乙酸苯二甲酸纤维素、氧化钛、氧化镁、二羟基铝氨基乙酸酯、磷酸三钙(tertiarycalciumphosphate)、滑石、碳酸钙、碳酸镁、沉降碳酸钙、天然硅酸铝、玉米淀粉、玉米淀粉造粒物、马铃薯淀粉、羟丙基纤维素、羟丙基淀粉、无水磷酸氢钙、无水磷酸氢钙造粒物、磷酸二氢钙等,优选为糖类、结晶纤维素,更优选为乳糖、结晶纤维素。本发明制剂中赋形剂的含量对于制剂全量为10至90重量%,优选为15至87.5重量%,更优选为20至85重量%。本发明制剂可含有粘合剂。粘合剂可使用收载于日本药典、日本药典外药品规格、药品添加物规格及食品添加物公定书等中的粘合剂。具体而言,可列举羟丙基纤维素、玉米淀粉、α化淀粉、部分α化淀粉、阿拉伯胶、阿拉伯胶末、明胶、琼脂、糊精、普鲁兰多糖、聚乙烯吡咯烷酮、聚乙烯醇、结晶纤维素、甲基纤维素、乙基纤维素、羧甲基乙基纤维素、羧甲基纤维素、羧甲基纤维素钠、羟乙基纤维素、羟乙基甲基纤维素、羟丙基纤维素、羟丙基甲基纤维素等,优选为聚乙烯吡咯烷酮。本发明制剂中粘合剂的含量对于制剂全量为0.1至20重量%,优选为0.25至15重量%,更优选为0.5至10重量%。本发明制剂亦可含有润滑剂。润滑剂可使用收载于日本药典、日本药典外药品规格、药品添加物规格及食品添加物公定书等中的润滑剂。具体而言,可列举硬脂酸金属盐、蔗糖脂肪酸酯、滑石、含水二氧化硅、硬脂酰富马酸钠等,优选为硬脂酰富马酸钠。润滑剂的含量对于制剂全量通常为0.05至10重量%,优选为0.075至7.5重量%,更优选为0.1至5重量%。为了有效率的进行聚合物的包衣作业,于本发明制剂包覆层的包衣剂中亦可含有塑化剂或凝集防止剂,可使用收载于日本药典、日本药典外药品规格、药品添加物规格及食品添加物公定书中者。具体而言,可列举柠檬酸三乙酯、甘油脂肪酸酯、蔗糖脂肪酸酯、蓖麻油、三乙酸甘油酯,滑石等。另一方面亦有调配聚乙烯二醇(聚乙二醇)的情况,亦有增加类似物质的情况。本发明制剂亦可含有色素或着色剂,可使用收载于日本药典、日本药典外药品规格或药品添加物规格等中的色素。色素可含于片剂中,亦可含于包覆层中。色素具体而言可列举氧化铁、焦油色素及天然色素等。氧化铁有氧化铁红、黄色铁氧化物、氧化铁黄、氧化铁黑等。焦油色素有食用黄色4号铝色淀、食用青色1号铝色淀、食用赤色3号铝色淀、食用青色1号、食用青色2号、食用黄色4号、食用黄色5号、食用赤色102号、食用赤色2号、食用赤色3号等。天然色素有姜黄萃取液、β-胡萝卜素、胡萝卜素液、叶绿素铜钠盐、叶绿素铜、裸麦绿叶萃取物粉末、裸麦绿叶青汁干燥粉末、裸麦绿叶萃取物、氧化钛、滑石等。色素亦包含于光稳定化物质使用者。本发明制剂必要时亦可含有上述以外的添加剂,可使用收载于日本药典、日本药典外药品规格、药品添加物规格及食品添加物公定书中的添加剂。另外,所述添加剂的含量可为任意比例。作为上述以外的添加剂具体而言可列举香料、流化剂、矫味剂等。香料具体而言可列举橘子香精、橘子油、焦糖、樟脑、桂皮油、绿薄荷油、草莓香精、巧克力香精、樱桃香料、甜橙精油、松油、薄荷油、香草香料、苦香精、水果香料、薄荷香精、混合香料、薄荷香料、薄荷醇、柠檬粉、柠檬油、玫瑰油等。流化剂具体而言可列举含水二氧化硅、轻质无水硅酸、结晶纤维素、合成硅酸铝、滑石等。矫味剂具体而言可列举阿斯巴甜、蔗糖素、甘氨酸、氯化钠、氯化镁、盐酸、稀盐酸、柠檬酸及其盐、柠檬酸酐、l-谷氨酰胺及其盐、琥珀酸及其盐、乙酸、酒石酸及其盐、碳酸氢钠、富马酸及其盐、苹果酸及其盐、冰醋酸、肌苷酸二钠、蜂蜜等。本发明制剂只要是固体制剂即可。具体而言只要是颗粒剂、细粒剂、片剂、散剂、胶囊剂、丸剂等即可,优选为颗粒剂或片剂。固体制剂中含有的式(i)表示的化合物的重量并无特别限制,具体而言优选为10mg、20mg、40mg或80mg。于该情况,10mg表示在9.0至11.0mg的范围,优选在9.5至10.5mg的范围,20mg表示在18.0至22.0mg的范围,优选在19.0至21.0mg的范围,40mg表示在36.0至44.0mg的范围,优选在38.0至42.0mg的范围,80mg表示在72.0至88.0mg,优选在76.0至84.0mg的范围。本发明制剂中的颗粒剂的制造方法并无特别限制,具体而言为将有效成分、崩解剂、赋形剂等添加剂混合,制造混合粉末后将该混合粉末造粒的方法,优选为添加水或含有粘合剂的水或溶剂进行造粒的湿式造粒法或压缩成形,未使用水的干式造粒法或熔融造粒法。作为混合有效成分或添加剂等的机械可使用v型混合机或容器掺混机。另外,作为造粒的机械可使用湿式挤出造粒机、流动层造粒机、搅拌造粒机、干式破碎造粒机或熔融挤出造粒机。湿式造粒的情况,造粒时的水分对于混合粉末为1至50%,优选为5至47.5%,更优选为10至45%。本发明制剂中的片剂的制造方法并无特别限制,具体而言可为根据上述方法制造颗粒,进一步地,对该颗粒混合崩解剂及润滑剂,再将该混合颗粒以压片机进行压片的压片法。作为混合有效成分或添加剂等的机械可使用v型混合机或容器掺混机。另外,压片机可使用单冲压片机、旋转式压片机等。本发明制剂于制造上述颗粒剂或片剂后,有将该颗粒剂或片剂以光稳定化物质及聚合物包覆,形成包覆层的情况。于颗粒剂形成包覆层时,可使用流动层造粒包衣机、流动层转动包衣机等。于片剂形成包覆层时可使用盘包衣机、通气式包衣机等。于制剂表面通过光稳定化物质及聚合物形成包覆层时,将光稳定化物质及聚合物溶解或悬浊于水或乙醇等溶剂中,调制包衣液。在包衣机中边将颗粒剂或片剂流动,边将该包衣液喷雾于该颗粒剂或片剂,干燥的,形成包覆层。另外,在溶出试验开始45分钟后,本发明制剂的式(i)表示的化合物的溶出率为70%以上,优选为75%以上,更优选为80%以上。在本发明制剂中,即使经光照射,类似物质从实验开始时起几乎未增加,制剂的色差也几乎没有变化,尤其是制剂的色差δe从实验开始几乎无变化。具体而言,将制剂放入曝光装置,作为总照射光量照射120万lux·hr的光时,制剂的色差为δ13以下。并且,本发明包含分析含有式(i)表示的化合物或其药学上可接受的盐的固体制剂中的分解物的方法,该方法包含下列步骤:a)将含有式(i)表示的化合物或其药学上可接受的盐的固体制剂作为试样,对该试样进行色谱的步骤;及b)于上述步骤获得的色谱数据,获得关于式(ii)表示的化合物的含量或含有比例的数据的步骤例如,可通过高效液相色谱测定含有式(ii)表示的化合物的类似物质的量(含量或含有比例)。此时,可使用式(ii)表示的化合物作为测定类似物质时的参考。式(ii)表示的化合物的含量或含有比例可从色谱数据的峰面积算出。例如,以色谱测定式(i)表示的化合物、式(ii)表示的化合物时,可使用260nm的波长作为测定波长。含有比例可使用对于制剂全体的比例、对于式(i)表示的化合物的比例、对于式(i)表示的化合物及式(ii)表示的化合物合计的比例等。进一步地,通过减低式(ii)表示的化合物的量有可能可减低制剂的色差δe。另外,式(i)表示的化合物的主要类似物质为以下的构造:固体制剂,尤其是片剂中式(i)表示的化合物的含量只要是患者容易服用,可制造片剂的含量即可,每1片为1至400mg,优选为1.25至350mg,更优选为2.5至300mg。具体而言,每1片的式(i)表示的化合物的含量为10mg、20mg、40mg或80mg。此时,10mg表示在9.0至11.0mg的范围,优选在9.5至10.5mg的范围,20mg表示在18.0至22.0mg的范围,优选在19.0至21.0mg的范围,40mg表示在36.0至44.0mg的范围,优选在38.0至42.0mg的范围,80mg表示在72.0至88.0mg的范围,优选在76.0至84.0mg的范围。本发明的含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含光稳定化物质及聚合物的包覆层。优选为使用遮蔽或吸收光的物质作为包覆层中的光稳定化物质的固体制剂。光稳定化物质优选为选自由食用焦油色素、食用色淀化的焦油色素、食用天然色素、氧化铁、氧化钛及滑石构成的组的1种以上的光稳定化物质。光稳定化物质优选为食用赤色2号、食用赤色3号、食用赤色102号、食用赤色104号、食用赤色105号、食用赤色106号、食用黄色4号、食用黄色5号、食用绿色3号、食用青色1号、食用青色2号、食用赤色3号铝色淀、食用黄色4号铝色淀、食用黄色5号铝色淀、食用青色1号铝色淀、食用青色2号铝色淀、胭脂红、叶绿素铜钠盐、叶绿素铜、铁丹、氧化铁红、氧化铁黄、氧化铁黑、黄色铁氧化物等光吸收物质,氧化钛、滑石、二氧化硅等光遮蔽物质。尤其是优选为选自由氧化铁红、氧化铁黄、氧化铁黑、黄色铁氧化物、氧化钛及滑石构成的组的1种以上。进一步优选为氧化钛和/或滑石。包覆层中的聚合物优选为选自由纤维素类聚合物、丙烯酸类聚合物及乙烯类聚合物构成的组的1种以上。纤维素类聚合物优选为选自由羟丙基甲基纤维素、羟丙基纤维素、羧甲基乙基纤维素、羟丙基甲基纤维素苯二甲酸酯、羟丙基甲基纤维素乙酸酯琥珀酸酯及乙基纤维素构成的组的1种以上。尤其是优选为羟丙基甲基纤维素。丙烯酸类聚合物优选为选自由甲基丙烯酸共聚物、甲基丙烯酸氨基烷基酯共聚物e及甲基丙烯酸氨基烷基酯共聚物rs构成的组的1种以上。乙烯类聚合物优选为选自聚乙烯醇、聚乙烯吡咯烷酮、交联聚维酮及聚乙烯醇-甲基丙烯酸甲酯-丙烯酸共聚物的1种以上。包覆层中的光稳定化物质优选为氧化钛及滑石,聚合物优选为羟丙基甲基纤维素。以下,记载优选的方面。含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含遮蔽或吸收光的光稳定化物质及聚合物的包覆层,更优选含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含氧化钛、滑石及聚合物的包覆层。作为另一方面,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含光稳定化物质及选自由纤维素类聚合物、丙烯酸类聚合物及乙烯类聚合物构成的组的1种以上的聚合物的包覆层,优选含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含遮蔽或吸收光的光稳定化物质及选自由纤维素类聚合物、丙烯酸类聚合物及乙烯类聚合物构成的组的1种以上的聚合物的包覆层,更优选地,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含选自由食用焦油色素、食用色淀化的焦油色素、食用天然色素、氧化铁、氧化钛及滑石构成的组的1种以上的光稳定化物质及选自由纤维素类聚合物、丙烯酸类聚合物及乙烯类聚合物构成的组的1种以上的聚合物的包覆层,特别优选地,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含氧化钛、滑石及选自由纤维素类聚合物、丙烯酸类聚合物及乙烯类聚合物构成的组的1种以上的聚合物的包覆层。作为另一方面,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含光稳定化物质及纤维素类聚合物的包覆层,优选地,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含遮蔽或吸收光的光稳定化物质及纤维素类聚合物的包覆层,更优选地,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含选自由食用焦油色素、食用色淀化的焦油色素、食用天然色素、氧化铁、氧化钛及滑石构成的组的1种以上的光稳定化物质及纤维素类聚合物的包覆层,更优选地,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含氧化钛、滑石及纤维素类聚合物的包覆层。作为另一方面,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含光稳定化物质及羟丙基甲基纤维素的包覆层,优选地,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含遮蔽或吸收光的光稳定化物质及羟丙基甲基纤维素的包覆层,更优选地,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含选自由食用焦油色素、食用色淀化的焦油色素、食用天然色素、氧化铁、氧化钛及滑石构成的组的1种以上的光稳定化物质及羟丙基甲基纤维素的包覆层,更优选地,含有式(i)表示的化合物或其药学上可接受的盐的固体制剂具有包含氧化钛、滑石及羟丙基甲基纤维素的包覆层。本发明制剂通过光的吸收物或遮蔽物包装时,作为总照射光量照射120万lux·hr的光时,制剂的色差为δ13以下。进一步地,将只含有式(i)表示的化合物的制剂通过光的吸收物或遮蔽物包装时,即使作为总照射光量照射120万lux·hr的光时,制剂的色差亦为δ13以下。该光的吸收物或遮蔽物有铝或着色膜等,包装形态有铝箔泡罩的包装形态等。片剂的形状可采用任何形状,具体而言可作成圆形、椭圆形、球形、棒状型、甜甜圈型的形状的片剂。另外,亦可为多层片、有核片等,优选为制造法简便的单层锭。亦可附有益于提升识别性的标记、文字等刻印及分割用的割线。[实施例]以下,列举实施例、比较例及参考例对本发明加以详细说明,然而,本发明不只限于以上。化合物ii可通过国际公开第2016/175224号说明书中公开的方法制造。实施例a化合物i的制造方法于化合物ii(4.0g,8.3mmol)中加入碳酸钾(1483.4mg,10.7mmol)及碘化钾(549.5mg,3.3mmol)、四氢呋喃(33.1g)、n,n-二甲基乙酰胺(3.8g)及水(80.3mg),搅拌的。升温至60℃,加入碳酸氯甲基酯甲酯(1758.9mg,14.2mmol)。于60℃搅拌9小时,冷却至20℃。加入乙酸(822.0mg)、2-丙醇(3.1g)及水(20.0g),以四氢呋喃(1.8g,8.9g)萃取2次。通过减压浓缩,蒸馏除去溶剂至液体重量为约32g。升温至45℃后加入2-丙醇(1.6g),冷却至20℃。加入通过乙酸钠(339.0mg)及水(46.0g)调制的乙酸钠水溶液后冷却至5℃。于5℃搅拌3小时后滤取产生的淡黄白色沉淀。将获得的固体以2-丙醇(4.7g)及水(6.0g)的混合液洗净后,以2-丙醇(6.3g)再次洗净固体。于获得的淡黄白色固体中加入二甲亚砜(30.9g),搅拌的。升温至60℃,加入二甲亚砜(2.2g)与水(4.8g)的混合液。再加入二甲亚砜(19.9g)与水(28.4g)的混合液,冷却至20℃。于20℃搅拌3小时后滤取产生的白色沉淀。获得的固体以二甲亚砜(8.0g)与水(4.8g)的混合液洗净后,以水(12.0g)再次洗净固体。通过将获得的固体干燥,获得白色结晶的化合物i(4.21g)。1h-nmr(dmso-d6)δ:2.91-2.98(1h,m),3.24-3.31(1h,m),3.44(1h,t,j=10.4hz),3.69(1h,dd,j=11.5,2.8hz),3.73(3h,s),4.00(1h,dd,j=10.8,2.9hz),4.06(1h,d,j=14.3hz),4.40(1h,d,j=11.8hz),4.45(1h,dd,j=9.9,2.9hz),5.42(1h,dd,j=14.4,1.8hz),5.67(1h,d,j=6.5hz),5.72-5.75(3h,m),6.83-6.87(1h,m),7.01(1h,d,j=6.9hz),7.09(1h,dd,j=8.0,1.1hz),7.14-7.18(1h,m),7.23(1h,d,j=7.8hz),7.37-7.44(2h,m)粉末x线衍射2θ(°):8.6±0.2°、14.1±0.2°、17.4±0.2°、20.0±0.2°、24.0±0.2°、26.3±0.2°、29.6±0.2°及35.4±0.2°(1)崩解剂的探讨a.调配性试验进行式(i)表示的化合物与崩解剂的调配性试验,评估经时保存品的类似物质量。将式(i)表示的化合物与崩解剂以1∶1混合,以水进行湿式调制后于40℃/75%相对湿度下保管2星期及1个月,测定含有式(ii)表示的化合物的总类似物质量。总类似物质测定法为如下所述。崩解剂使用低取代羟丙基纤维素(信越化学公司制造)、交联羧甲基纤维素钠(fmcbiopolymer公司制造)、淀粉乙醇酸钠(jrspharma公司制造)及交联聚维酮(basf公司制造)。(总类似物质量测定法)依照以下的方法、条件,以液相色谱测定类似物质。·检测器:紫外线吸光光度计(测定波长260nm)·管柱:xbridgec18,3.5μm,3.0×150mm·管柱温度:35℃左右的一定温度·流动相a:0.1%三氟乙酸/0.2mmedta溶液、流动相b:乙腈·流动相的送液:将流动相a及流动相b的混合比变更为如表1所示,调控浓度梯度。[表1]·流量:约0.6ml/min(式(i)表示的化合物的保持时间约16分钟)·注入量:5μl·试样冷却器温度:约5℃·自动注射器洗净液:乙腈/甲醇混液(1:3)·面积测定范围:试样溶液注入后50分钟·总类似物质量的计算式总类似物质的合计量(%)=σati/σat×100ati:试样溶液的各个类似物质的高峰面积σat:试样溶液高峰面积的合计(空白及系统峰除外)σati:试样溶液的各个类似物质的高峰面积的合计(结果)表2表示总类似物质量。其结果,相比于淀粉乙醇酸钠及交联聚维酮,低取代羟丙基纤维素及交联羧甲基纤维素钠中,总类似物质量有降低的倾向。[表2]b.式(i)表示的化合物的溶出性(裸锭的制造方法)选择总类似物质量少的低取代羟丙基纤维素及交联羧甲基纤维素钠作为崩解剂,制造含有所述崩解剂的片剂后进行该片剂的溶出试验。表3表示本发明制剂每1片剂的光稳定化物质及聚合物的包覆前裸片的处方。将式(i)表示的化合物、乳糖水合物(dmv-fonterra公司制造)、崩解剂以30网目的筛过筛,以高速混合机(深江工业公司制造,lfs-gs-2j型)造粒。造粒时使用聚乙烯吡咯烷酮(basf公司制造)的水溶液作为粘合剂。另外,造粒时的水分调整为约20%、40%。[表3]裸片1-1裸片1-2裸片2-1裸片2-2式(i)表示的化合物20.020.020.020.0乳糖水合物72.472.472.472.4低取代羟丙基纤维素11.011.0––交联羧甲基纤维素钠––11.011.0聚乙烯吡咯烷酮5.55.55.55.5结晶纤维素10.010.010.010.0硬脂酰富马酸钠1.11.11.11.1裸片计(mg)120.0120.0120.0120.0造粒水分(%(w/w))21.341.321.341.4造粒条件、溶出试验法如以下所述。(造粒条件)造粒机:lfs-gs-2j型高速混合机·搅拌器旋转数:333min-1·腔室器旋转数:2500min-1·液注加速度:20g/min将经造粒、干燥及整粒的颗粒、结晶纤维素及为润滑剂的硬脂酰富马酸钠(jrspharma公司制造)混合后,通过静态压缩机,以5kn压片,制造片剂。(溶出试验法)溶出试验根据第16版日本药典溶出试验法第2法(含有界面活性剂的溶出试验第2液,桨法,桨旋转数:50rpm,结果:2片的平均值)进行。(实验结果)分别于图1表示裸片1-1及1-2的溶出试验结果,图2表示裸片2-1及2-2的溶出试验结果。其结果,使用低取代羟丙基纤维素作为崩解剂时,与造粒水分约20%的裸片1-1制剂相比,造粒水分约40%的裸片1-2制剂的溶出行为较慢。另一方面,使用交联羧甲基纤维素钠作为崩解剂时,造粒水分约20%的裸片2-1制剂及造粒水分约40%的裸片2-2制剂的溶出行为几乎相同。因此,认为作为崩解剂,使用于调配性试验中总类似物质量少且即使造粒时的水分量变更,其溶出性几乎没变化的交联羧甲基纤维素钠最适当。(2)光稳定化物质、聚合物的选择a.光稳定化物质的影响为了研究光稳定化物质的影响,将光稳定化物质及聚合物包覆于裸片,测定制剂中的类似物质量及色差。表4表示经光稳定化物质及聚合物包覆的制剂的处方。使用氧化钛及滑石作为光稳定化物质,使用聚乙烯醇作为聚合物,使用聚乙二醇4000作为塑化剂。作为类似物质量,测定式(ii)表示的化合物的量及总类似物质量。另外,制剂的色差为曝光一定的光后测定制剂的色差。[表4]包覆制剂的制造方法、包覆条件、式(ii)表示的化合物的测定法及制剂的色差测定法如以下所述。(包覆制剂的制造方法)表4表示本发明制剂每1片剂的裸片及于裸片的光稳定化物质(氧化钛、滑石)及聚合物(聚乙烯醇)的包覆量。光稳定化物质及聚合物包覆于裸片表面,进行包覆的机械使用快速包衣机lab(freund公司制造)。以下为进行包覆的条件。(包覆条件)·进料量:约0.5kg·包衣机:labcoaterhc-labo(freund产业公司制造)·给气温度:60℃(设定温度)·给气风量:1.0m3/min·喷嘴径:1.0mm·喷嘴盖径:1.3mm·喷雾空气量:约40nl/min·包衣液速度:约2g/min·盘旋转数:25至32min-1(式(ii)表示的化合物的测定法)根据以下的方法、条件,以液相色谱测定式(ii)表示的化合物的量。·检测器:紫外线吸光光度计(测定波长260nm)·管柱:xbridgec18,3.5μm,3.0×150mm·管柱温度:35℃左右的一定温度·流动相a:0.1%三氟乙酸/0.2mmedta溶液、流动相b:乙腈·流动相的送液:将流动相a及流动相b的混合比变更为如表5所示,调控浓度梯度。[表5]·流量:约0.6ml/min·注入量:5μl·试样冷却器温度:约5℃·自动注射器洗净液:乙腈/甲醇混液(1:3)·面积测定范围:试样溶液注入后50分钟·式(ii)表示的化合物的量的计算式式(ii)表示的化合物的量(%)=atii/σat×100atii:试样溶液的式(ii)表示的化合物的峰面积σat:试样溶液的峰面积的合计(空白及系统峰除外)(色差的测定法)使用分光色差计(镜片直径4mm),依照下述的计算式,以各片剂的初始为基准,测定1至3片的色调(δe),以下述的式算出其平均值。然而,l表示亮度,a表示色度(+:红色度,-:绿色度),b表示色度(+:黄色度,-:蓝色度)。另外,表6表示通过色差计的外观判定基准。δe={(δl)2+(δa)2+(δb)2}1/2[表6]δe外观判定基准0至低于0.5微弱0.5至低于1.5稍微1.5至低于3.0被查觉的程度3.0至低于6.0引人注目的程度6.0至低于12.0多12.0以上很多(实验结果)表7表示于40℃/75%相对湿度下将容器开栓,于初始、2周后、1个月后的式(ii)表示的化合物的量(%),表8表示总类似物质量(%)。另外,表9表示120万lux·hr的光照射后的制剂的色差(δe)。结果,于40℃/75%相对湿度下,即使将容器开栓,式(ii)表示的化合物的量或总类似物质量,于实施例1至3的制剂或比较例1的制剂中从初始至实验开始1个月后为止几乎无变化。另一方面,关于制剂的色差,与经光稳定化物质包覆的实施例1至3的制剂比较,未包覆光稳定化物质的比较例1的制剂较大,为12以上,若根据表6的外观判定基准,明了为变化为“很大”的制剂的色差。[表7]式(ii)表示的化合物的量(%)实施例1实施例2实施例3比较例1初始0.090.080.100.102周后0.130.130.130.141个月后0.150.170.140.16[表8]总类似物质量实施例1实施例2实施例3比较例1初始0.500.570.550.612周后0.550.600.620.621个月后0.620.620.720.61[表9]制剂的色差(δe)实施例1实施例2实施例3比较例1120万lux·hr(曝光)0.760.721.0514.44c.光稳定化物质及聚合物的最适化为了最适化光稳定化物质及聚合物,通过于裸片包覆光稳定化物质及聚合物,测定制剂中的类似体量及制剂的色差。表10表示经光稳定化物质及聚合物包覆的制剂的处方。使用氧化钛及滑石作为光稳定化物质,使用聚乙烯醇、羟丙基纤维素、羟丙基甲基纤维素作为聚合物,使用聚乙二醇4000作为塑化剂。[表10](实验结果)表11表示于40℃/75%相对湿度下将容器开栓,于初始、2周后的式(ii)表示的化合物的量(%),表12表示总类似物质量(%)。另外,表13表示120万lux·hr的光照射后的制剂的色差(δe)。其结果,于40℃、75%相对湿度下,即使将容器开栓,式(ii)表示的化合物的量或总类似物质量,于实施例4至6的制剂从初始至实验开始2周后为至几乎无变化,尤其是于光稳定化物质为氧化钛及滑石、聚合物为羟丙基甲基纤维素的实施例6的制剂,总类似物质量几乎未增加。制剂的色差于经光稳定化物质包覆的实施例4至6的制剂几乎未变化,若根据表6的外观判定基准,变化为“微弱”或“稍微”制剂的色差的程度。[表11]式(ii)表示的化合物的量(%)实施例4实施例5实施例6初始0.080.090.112周后0.130.150.13[表12]总类似物质量实施例4实施例5实施例6初始0.570.550.612周后0.600.640.59[表13]制剂的色差(δe)实施例4实施例5实施例6120万lux·hr(曝光)0.720.470.93(临床试验)通过将安慰剂作为比较对象的随机分配的双盲比较试验,实施于流行性感冒病毒感染患者的临床试验用药(有效成分(式(i)表示的化合物、以下,亦称为“化合物ii-6”):10mg、20mg、40mg)的单次口服给予的有效性及安全性的评估。主要评估项目为对于流行性感冒患病期间(临床试验用药给予开始至7个流行性感冒症状(“咳嗽”、“喉咙痛”、“头痛”、“鼻塞”、“轻微发烧或恶寒”、“肌肉或关节疼痛”、“疲劳感”)获得改善的时间)通过被验者本人以4阶段[0:无,1:轻症,2:中程度,3:重症]的评估,评估临床试验用药相对于安慰剂的有效性。对象为选择全部符合下述基准的患者。(a)20岁以上、低于65岁的男性或女性患者。(b)亦符合以下的任一项,诊断为流行性感冒病毒感染症的患者。·通过鼻腔或咽头的分泌液的流行性感冒快速诊断[rapidantigentest(rat)]为阳性·有38℃以上的发热(腋温)·因流行性感冒病毒感染症引起的以下的全身症状及呼吸器官症状中各个有1项以上为中等程度以上的症状·全身症状(头痛,轻微发烧或恶寒,肌肉或关节疼痛,疲劳感)·呼吸器官症状(咳嗽,喉咙痛,鼻塞)(c)发病起至48小时的患者(登记时)。然而,发病的定义为以下中的任何一种。·体温开始上升的时点(正常体温至少上升1℃以上)·具有1项以上全身症状及呼吸器官症状中的任何一种症状的时点临床试验用药的给予方法(i)被验药化合物ii-6的10mg片:含有化合物ii-610mg的白色至淡黄白色的圆形膜包衣片(本发明制剂)。化合物ii-6的20mg片:含有化合物ii-620mg的白色至淡黄白色的椭圆形膜包衣片(本发明制剂)。(ii)安慰剂或对照药化合物ii-6的10mg片的安慰剂:与化合物ii-6的10mg片不能区分的片剂。化合物ii-6的20mg片的安慰剂:与化合物ii-6的20mg片不能区分的片剂。给予量及给予方法将判断为合格的被验者以1∶1∶1∶1的比率随机分配给化合物ii-6的10mg组、20mg组、40mg组或安慰剂组的任何一组。对于被验者,以由化合物ii-6片和/或安慰剂片构成的以下的表表示的组合,于第1日分别单次口服给予计3片。每个给予组的临床试验用药[表14]有效性的主要评估项目有效性的主要评估项目为至流行性感冒症状消失为止的时间(流行性感冒患病期间)。设成从开始给予的时点至流行性感冒症状消失为止的时间。流行性感冒症状的消失指记录被验者的患者日记中流行性感冒七种症状(咳嗽、喉咙痛、头痛、鼻塞、轻微发烧或恶寒、肌肉或关节疼痛、疲劳感)全都成为“0:无”或“1:轻症”的时点,该状态至少持续21.5小时(24小时-10%)。有效性的次要评估项目有效性的次要评估项目为如下所述。(1)至流行性感冒各症状消失为止的时间设成从开始给予时点至流行性感冒各症状消失为止的时间。症状的消失指作为对象的项目为“0:无”或“1:轻度”的时点,该状态至少持续21.5小时(24小时-10%)。主要评估项目的分析对于为主要评估项目的流行性感冒患病期间,记载主要分析及次要分析。主要分析增加itti(意向治疗感染(intention-to-treatinfected)集团,为了敏感性分析,亦于符合计划集(pps(perprotocolset))集团实施。除此之外的分析只于itti集团实施。(1)主要分析将itti集团作为对象,应答流行性感冒患病期间,以给予组的母数效果、为分配因子的现在有无抽烟及于给予前的基线时点流行性感冒七种症状的合计评分作为共变量的cox比例风险模型,对于安慰剂组算出各给予组的风险比值、其95%置信区间、p值。为了防止通过实施复数次检测第1种错误率增加,将p值以hommel法调整。(2)次要分析应答流行性感冒患病期间,以给予组的说明变量、为分配因子的给予前流行性感冒七种症状的合计评分的类别(11点以下、12点以上)及有无抽烟作为分层因子,以分层一般化wilcoxon检测比较安慰剂组与临床试验用药的各给予组。另外,对各组绘制kaplan-meier生存曲线,算出流行性感冒患病期间的中值及其95%置信区间。使用greenwood法算出置信区间。次要评估项目的分析(1)至流行性感冒各症状消失的时间设成于每个流行性感冒症状,应答于至各症状消失的时间,实施与主要评估项目相同的分析。此时,将给予前的症状为“0:无”或“1:轻度”的病例从分析对象除去。(1)关于主要评估项目的结果(流行性感冒的患病期间)于随机筛选的400人的患者中完成389人(10mg给予组98人(98%)、20mg给予组95人(95%)、40mg给予组99人(99%)及安慰剂组97人(97%))的试验。ittipopulation(给予临床试验用药,且确认为流行性感冒病毒感染的病例)对于主要评估项目,以400人的患者构成。于符合计划集(pps)病例以368人(10mg给予组89人(89%)、20mg给予组92人(92%)、40mg给予组96人(96%)及安慰剂组91人(91%))构成。对于各个群的ittipopulation,从快速抗原检测试验,75%至79%的患者感染a型流行性感冒病毒,21%至25%的患者感染b型流行性感冒病毒。以下的表表示分析结果。[表15]该试验的主要评估项目,即至症状减轻的时间的中值于10mg给予组为54.2小时(95%ci:47.7,66.8),于20mg给予组为51.0小时(95%ci:47.7,66.8),于40mg给予组为49.5小时(95%ci:44.5,64.4),于安慰剂组为77.7小时(95%ci:67.6,88.7)。(2)至七种症状中的各者减轻的时间下述的表表示至7种各个流行性感冒症状(“咳嗽”、“喉咙痛”、“头痛”、“鼻塞”、“轻微发烧或恶寒”、“肌肉或关节疼痛”、“疲劳感”)改善的时间的分析结果。(i)至“鼻塞”的症状缓和的时间[表16](ii)至“肌肉或关节疼痛”的症状缓和的时间[表17](iii)至“疲劳感”的症状缓和的时间[表18](iv)至“轻微发烧或恶寒”的症状缓和的时间[表19](v)至“头痛”的症状缓和的时间[表20](vi)至“咳嗽”的症状缓和的时间[表21](vii)至“喉咙痛”的症状缓和的时间[表22]a对于安慰剂的分层一般化wilcoxon检测。分层因素:抽烟习惯、于基线的复合症状的评分。b对于安慰剂的cox比例风险模型。共变量:抽烟习惯,于基线的复合症状的评分。于基线的症状的评分为“中程度”或“重症”的患者的子集ci:置信区间于40mg给予组,使用cox比例风险模型进行分析时看到与安慰剂组比较,对于以下的5症状:“鼻塞”、“肌肉或关节疼痛”、“疲劳感”、“轻微发烧或恶寒”、“头痛”,至症状改善的时间显著的减少。例如对于“鼻塞”及“肌肉或关节疼痛”的2症状,至症状改善的时间的中值,40mg给予组比安慰剂组各别缩短21.0小时、16.4小时。于10mg给予组或20mg给予组,以下显示的症状:“肌肉或关节疼痛”、“鼻塞”、“轻微发烧或恶寒”,看到统计学上显著差异。(临床试验(ph3:成人及青少年))通过将奥司他韦75mg1日2次,给予5日或安慰剂作为比较对象的随机分配的双盲比较试验实施于流行性感冒病毒感染患者的临床试验用药(有效成分(化合物ii-6):40mg、80mg)单次口服给予的有效性及安全性的评估。主要评估项目为对于至流行性感冒患病期间(临床试验用药给予开始的7个流行性感冒症状(“咳嗽”、“喉咙痛”、“头痛”、“鼻塞”、“轻微发烧或恶寒”、“肌肉或关节疼痛”、“疲劳感”)获得改善的时间)通过被验者本人以4阶段[0:无,1:轻症,2:中程度,3:重症]评估,评估临床试验用药对于安慰剂的有效性。另外,作为有效性的次要评估项目,通过使用鼻腔或咽头的分泌液的流行性感冒病毒效价,对于临床试验用药的有效性或副作用亦进行评估。对象为选择全部符合下述基准的患者。(a)12岁以上、低于65岁的男性或女性患者。(b)亦符合以下的任一项,诊断为流行性感冒病毒感染症的患者。·有38℃以上的发热(腋温)·因流行性感冒病毒感染症引起的以下的全身症状及呼吸器官症状中各个有1项以上为中等程度以上的症状·全身症状(头痛,轻微发烧或恶寒,肌肉或关节疼痛,疲劳感)·呼吸器官症状(咳嗽,喉咙痛,鼻塞)(c)发病起至48小时的患者(登记时)。然而,发病的定义为以下中的任何一种。·体温开始上升的时点(正常体温至少上升1℃以上)·具有1项以上全身症状及呼吸器官症状中的任何一种症状的时点临床试验用药的给予方法(i)被验药化合物ii-6的20mg片。(ii)安慰剂或对照药化合物ii-6的20mg片的安慰剂。奥司他韦75mg胶囊奥司他韦75mg胶囊的安慰剂:与奥司他韦75mg胶囊不能区分的胶囊。给予量及给予方法将判断为合格的20至64岁的患者以2:2:1的比率,随机分配给化合物ii-6的单次给予(对应体重为40或80mg)群、奥司他韦75mg1日2次且给予5日的给予组或安慰剂组中的任何一组。将判断为合格的12至19岁的患者以2:1的比率,随机分配给化合物ii-6的单次给予(对应体重为40或80mg)群或安慰剂给予组中的任何一组。化合物ii-6的给予量于体重低于80kg被验者给予40mg,80kg以上的被验者给予80mg。每个给予组的临床试验用药[化合物ii-6组]第1天:口服给予化合物ii-6的20mg片(对应体重为2片或4片)。口服给予奥司他韦的安慰剂胶囊,1日2次(早、晚),每次1胶囊。第2天至第5天:口服给予奥司他韦的安慰剂胶囊,1日2次(早、晚),每次1胶囊。[奥司他韦组]第1天:口服给予化合物ii-6的安慰剂片(对应体重为2片或4片)。口服给予奥司他韦的75mg胶囊,1日2次(早、晚),每次1胶囊。第2天至第5天:口服给予奥司他韦的75mg胶囊,1日2次(早、晚),每次1胶囊。[安慰剂组]第1天:口服给予化合物ii-6的安慰剂片(对应体重为2片或4片)。口服给予奥司他韦的安慰剂胶囊,1日2次(早、晚),每次1胶囊。第2天至第5天:口服给予奥司他韦的安慰剂胶囊,1日2次(早、晚),每次1胶囊。另外,“第1天”表示给予初日,“第2天至第5天”表示从给予初日算起的第2天至第5天。有效性的主要评估项目有效性的主要评估项目为至流行性感冒症状消失为止的时间(流行性感冒患病期间)。设成从开始给予时点至流行性感冒症状消失为止的时间。流行性感冒症状的消失指记录被验者的患者日记中流行性感冒七种症状(咳嗽、喉咙痛、头痛、鼻塞、轻微发烧或恶寒、肌肉或关节疼痛、疲劳感)全都成为“0:无”或“1:轻症”的时点,该状态至少持续21.5小时(24小时-10%)。有效性的次要评估项目有效性的次要评估项目如以下所述。(1)于各点的流行性感冒病毒效价阳性患者的比例(2)于各点从病毒效价的基线的变化量(3)以病毒效价为基础,至病毒排出停止的时间(4)副作用的表现频率病毒的效价以下述的步骤测定。(1)将接种于平底96孔微量盘的mdck-siat1细胞于37±1℃、5%co2的恒温箱内培养1日。(2)将标准株(流行性感冒病毒ah3n2、a/victoria/361/2011、保存条件:-80℃、源自:国家感染疾病研究所(nationalinstituteofinfectiousdiseases)、检体(将于化合物ii-6的第三期临床试验中从患者收集者保管于超低温冷冻库)及细胞对照用的培养基以10倍连续稀释法稀释至101-107倍。(3)于倒立显微镜下确认以片状存在的细胞后,除去培养基,以100μl/孔加入新的培养基。(4)除去培养基。(5)将于上述(2)调整的各个检体(100-107),以每1检体使用4孔,以100μl/孔接种。(6)于室温进行1000rpm、30分钟的离心吸附。(7)离心后,除去培养基,以新的培养基将细胞洗净1次。(8)将新的培养基以100μl/孔添加的。(9)于5%co2恒温箱内以33±1℃保温3日。(10)保温后于倒立显微镜下评估细胞变性作用(cpe)。病毒效价阳性的判定方法通过根据上述病毒效价的测定方法测定,超过检测限界的情况判断为阳性。主要评估项目的分析对于为主要评估项目的流行性感冒患病期间记载主要分析及次要分析。主要分析于itti集团实施。(1)主要分析将12至64岁的患者作为对象,将给予前的流行性感冒七种症状的合计评分(11点以下、12点以上)及地区(日本/亚洲,其他地区)作为分层因子,以分层一般化wilcoxon检测比较安慰剂组与临床试验用药的给予组。另外,对各组绘制kaplan-meier生存曲线,算出流行性感冒患病期间的中值及其95%置信区间、流行性感冒患病期间的组间差及其95%置信区间。(2)次要分析将20至64岁的患者作为对象,通过与主要分析相同的方法比较化合物ii-6组与奥司他韦组的流行性感冒患病期间。次要评估项目的分析于化合物ii-6组与安慰剂组之间、化合物ii-6组与奥司他韦组之间(20至64岁的年龄层)比较以下的有效性的次要评估项目。(1)于各时点中的流行性感冒病毒效价阳性患者的比例只将于visit1开始给予前的时点病毒效价为定量下限以上的患者包含于分析中。应用将每个visit给予前流行性感冒七种症状的合计评分及地区作为分层因子的mantel-haenszel检测,于2组间比较病毒效价阳性的患者的比例。(2)于各时点中从病毒效价的基线的变化量只将于visit1开始给予前的时点病毒效价为阳性的患者包含于分析中。应用将每个visit给予前的流行性感冒七种症状的合计评分及地区作为分层因子的vanelteren检测,于2组中比较从流行性感冒病毒效价的基线的变化量。(3)以病毒效价为基础至病毒停止排出的时间只将于visit1开始给予前的时点病毒效价为定量下限以上的患者包含于分析中。应用将给予前的流行性感冒七种症状的合计评分及地区作为分层因子的分层一般化wilcoxon检测。(4)副作用的表现频率计算每个给予组的副作用发现件数及例数。(1)对于主要评估项目的结果(流行性感冒的患病期间)于随机挑选的1436人的患者中有1366人(化合物ii-640mg或80mg给予组578人、奥司他韦给予组498人及安慰剂组290人)完成试验。对于主要评估项目,itti病例(遵守gcp、给予临床试验用药且确认为流行性感冒病毒感染的病例)以1064人的患者构成。符合计划集病例以990人(化合物ii-640mg或80mg给予组427人、奥司他韦给予组351人、及安慰剂组212人)构成。以下的表表示分析的结果。[表23]a对安慰剂或对奥司他韦。b自助重抽(bootstrap)推定。c将地区及给予前流行性感冒七种症状的合计评分作为分层因子,症状未消失的患者于最终评估时点中止。于itti集团的流行性感冒患病期间(中值)(95%ci)对于化合物ii-6组53.7小时(95%ci:49.5,58.5),安慰剂组为80.2小时(95%ci:72.6,87.1),化合物ii-6组与安慰剂组之差为-26.5小时。流行性感冒患病期间于使用分层一般化wilcoxon检测的主要分析与安慰剂组比较,化合物ii-6组显著的缩短(p<.0001)。于20岁以上、低于65岁的部分集团的流行性感冒患病期间,对于化合物ii-6组53.5小时(95%ci:48.0,58.5),奥司他韦组为53.8小时(95%ci:50.2,56.4),化合物ii-6组与奥司他韦组之差为-0.3小时。流行性感冒患病期间于分层一般化wilcoxon检测,化合物ii-6组与奥司他韦组之间无显著差异。次要评估项目的分析(1)于各点的流行性感冒病毒效价阳性患者的比例以下的表表示分析的结果。[表24]第2天表示从给予第一天算起24小时后,第3天表示48小时后、第4天表示72小时后、第5天表示96小时后、第6天表示120小时后、第9天表示192小时后。a对安慰剂或对奥司他韦。mantel-haenszel检测。地区及给予前流行性感冒七种症状的合计评分作为分层因子,将给予前病毒效价为阳性的集团作为对象。病毒效价阳性患者的比例于第2天,与安慰剂组比较,于化合物ii-6组为显著的低(mantel-haenszel检测:p<.0001)、于第3天亦相同,与安慰剂组比较,于化合物ii-6组为显著的低(p<.0001)。于20岁以上、低于65岁的部分集团的病毒效价阳性患者的比例于第2天及第3天,与奥司他韦组比较,于化合物ii-6组为显著的低(p<.0001)。(2)于各点中从病毒效价的基线的变化量以下的表表示分析结果。[表25]单位:log10[tcid50/ml]。第2天表示从给予第一天算起24小时后,第3天表示48小时后、第4天表示72小时后、第5天表示96小时后、第6天表示120小时后、第9天表示192小时后。a对安慰剂或对奥司他韦。vanelteren检测。将地区及给予前流行性感冒七种症状的合计评分作为分层因子,将给予前病毒效价为阳性的集团作为对象。病毒效价于第2天与安慰剂组比较,于化合物ii-6组显著的减少,于第3天亦相同,与安慰剂组比较,显著的减少(vanelteren检测:p<.0001)。于20岁以上、低于65岁的部分集团的病毒效价于第2天及第3天,与奥司他韦组比较,于化合物ii-6组为显著的减少(p<.0001)。(3)以病毒效价为基准,至病毒停止排出的时间以下的表表示分析结果。[表26]a对安慰剂或对奥司他韦。b将地区及给予前流行性感冒七种症状的合计评分作为分层因子。病毒效价未消失的患者于最终评估时点中止。将于第1天病毒效价为阳性且至病毒停止排出的时间无缺失的患者作为分析对象。以病毒效价为基础至病毒停止排出的时间(中值)对于化合物ii-6组24.0小时,安慰剂组为96.0小时,与安慰剂组比较,化合物ii-6组为显著的缩短(分层一般化wilcoxon检测:p<.0001)。于20岁以上、低于65岁的部分集团的至病毒停止排出的时间,化合物ii-6组为24.0小时,奥司他韦组为72.0小时,与奥司他韦组比较,于化合物ii-6组显著的缩短(p<.0001)。(4)不良事件的表现频率无不能否定因果关系的严重不良事件的报告。不能否定因果关系的不良事件于化合物ii-6组为610例中表现27例(4.4%、37件),于安慰剂组为309例中表现12例(3.9%、19件),于奥司他韦组为513例中表现43例(8.4%、53件)。化合物ii-6组与安慰剂组之间的表现率在统计学上未看到显著差异(费雪的正确性检测(fisher'sexact检测))、两侧p值:0.8627)。然而,化合物ii-6组的表现率比奥司他韦组更显著的低(费雪的正确性检测、两侧p值:0.0088)。试验例17临床试验(ph3:小儿)实施流行性感冒病毒感染患者单次口服给予临床试验用药(有效成分(化合物ii-6):5mg、10mg、20mg、40mg)的有效性及安全性的评估。主要评估项目为于流行性感冒患病期间(从开始给予临床试验用药的流行性感冒症状(“咳嗽”、“流鼻涕/鼻塞”及“发热”)至获得改善的时间),保护者或被验者本人通过评估/测定,评估临床试验用药的有效性。另外,对于“咳嗽”及“流鼻涕/鼻塞”以4阶段[0:无,1:轻症,2:中程度,3:重症]进行评估。对象为选择全部符合下述基准的患者。(a)6个月以上、低于12岁的男性或女性患者。(b)亦符合以下中任一项,诊断为流行性感冒病毒感染症的患者。·通过鼻腔或咽头的分泌液的流行性感冒快速诊断[rapidantigentest(rat)]为阳性·有38℃以上的发热(腋温)·于7岁以上的患者,因流行性感冒病毒感染症引起的呼吸器官症状(咳、流鼻涕/鼻塞)中,有1个项目以上为中程度以上的症状。(c)发病起至48小时的患者(登记时)。然而,发病的定义为将发热超过37.5℃作为最初确认的时点。(d)体重为5kg以上的患者。临床试验用药的给予方法(i)被验药化合物ii-6的5mg片:化合物ii-6的10mg片的半片。化合物ii-6的10mg片。化合物ii-6的20mg片。给予量及给予方法于患者,将以体重为基础换算过的用量于第1天单次口服给予(参照下述表)。[表27]有效性的主要评估项目有效性的主要评估项目为至流行性感冒症状消失的时间(流行性感冒患病期间)。设成从开始给予时点至流行性感冒症状消失的时间。流行性感冒症状的消失为从开始给予的时点满足下述的a及b的时点,其临床状态至少持续21.5小时(24小时-10%)。a.根据患者的日记,“咳嗽”及“流鼻涕/鼻塞”双方都为“0:无”或“1:轻症”b.体温(腋温)低于37.5℃主要评估项目的分析对于为主要评估项目的流行性感冒患病期间,记载主要分析。主要分析于itti集团实施。(1)主要分析描绘至流行性感冒症状(“咳嗽”、“流鼻涕/鼻塞”及“发热”)消失的时间(流行性感冒患病期间)的kaplan-meier曲线,算出至流行性感冒症状完全消失的时间的中值及其95%置信区间。观察期间中流行性感冒症状未完全消失的患者作为中止例处理。(1)对于主要评估项目的结果(流行性感冒的患病期间)对于主要评估项目,以103人的患者构成。于itti集团的流行性感冒患病期间(中值)为44.6小时(95%ci:38.9,62.5)。[产业上的可利用性]对于“通过对于含有式(i)表示的化合物的制剂照射光,制剂的色差会变高”这一问题发现了的新的见解。对此,通过在制剂的表面通过包覆光稳定化物质及聚合物,可减低制剂的色差。通过此,含有式(i)表示的化合物的制剂即使在光照射下亦可稳定地保存。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1