一种头孢米诺钠注射制剂的制备方法与流程
本发明属于生物医药技术领域,具体涉及一种头孢米诺钠注射制剂的制备方法。
背景技术:
头孢米诺是由日本明治制药开发的第四代头孢类抗生素(商品名为
本品对革兰阳性和革兰阴性菌具有广谱抗菌活性,尤其对链球菌(肠球菌除外)、大肠杆菌、肺炎克雷伯肝菌、变形杆菌属、流感嗜血杆菌、类杆菌属有很强的抗菌作用。本品对各种菌株产生的β内酰胺酶稳定,对脆弱类杆菌产生的β-内酰胺酶酶尤其稳定。
该药主要用于敏感菌引起的败血症、扁桃体周围脓肿、气管炎、细支气管炎、支气管扩张感染或慢性呼吸系统疾病的继发感染、肺炎、肺化脓性病变、肾孟肾炎、膀胱炎、胆囊炎、胆管炎、腹膜炎、盆腔腹膜炎、子宫附件炎、子宫内感染、盆腔类、子宫旁结缔组织炎等。在日本为期4年上市后的监测显示,其在包括老人和孕妇的人群中没有特殊的副作用发生,应用与其它头孢类药物相似。
目前,全世界预计到2018年医药市场销售额将达到5000亿~6060亿美元,其中抗感染用药占总份额的15%,约为600亿美元。头孢菌素类药物近10年来一直占据抗感染用药的第一位。根据国外报道,头孢菌素类药物的售额占全部抗感染用药的45%,年销售额约为230亿美元。
现在上述销售的头孢米诺钠制剂为粉针剂,但该粉针剂在临床使用时,临配之后,需要4小时内注射完毕,而很多医院的护士在临床上,临配的药物工作量以及给患者注射药物的工作量都非常大,因此,导致临配的头孢米诺钠不能在4小时内注射完成,给患者带来一定的风险,因此,研究新的头孢米诺钠及其制剂,具有重要意义。
技术实现要素:
基于上述原因,为了解决现有技术的不足,申请人经过多年创造性的试验,获得一种新的头孢米诺钠注射剂的制备方法,该制备方法得到的注射制剂与氯化钠注射液或葡萄糖注射液混合后,保持长期稳定性,不溶性微粒等几乎不增加。
本发明经过多次试验后,从众多药用辅料中,筛选得到枸橼酸钠,制备成粉针剂。
为实现上述目的,本发明的技术方案如下:
一种头孢米诺钠注射制剂的制备方法,头孢米诺钠原料与枸橼酸钠原料混合完全,灌装即得,其中头孢米诺钠原料与枸橼酸钠原料的重量比为1:0.04-0.06。所述的一种头孢米诺钠注射制剂的制备方法,其中头孢米诺钠原料由下述方法制备而成:取头孢米诺钠粗品,加入四氢呋喃溶解完全,加入丙酮后,得到结晶,获得头孢米诺钠原料药。
所述的一种头孢米诺钠注射制剂的制备方法其中该原料的制备方法中头孢米诺钠粗品的重量克数与四氢呋喃的体积毫升数比为1:3.5-5。
所述的一种头孢米诺钠注射制剂的制备方法,其中该原料的制备方法中头孢米诺钠粗品重量克数与丙酮体积毫升数比为1:4.5-6。
所述的一种头孢米诺钠注射制剂的制备方法,其中原料的制备方法为:
取头孢米诺钠粗品,加入四氢呋喃,升温至40-50℃,加入丙酮,搅拌均匀,5℃-10℃静置,析出结晶,过滤,滤饼用丙酮洗涤,干燥,得到头孢米诺钠原料。
所述的一种头孢米诺钠粉针剂,其中头孢米诺钠原料与枸橼酸钠原料的重量比为1:0.05。
本发明所述的头孢米诺钠粗品:由哈尔滨誉衡制药有限公司提供,购买日期2018年3月25日。
本发明所述的头孢米诺钠粗品中按无水物计算,含头孢米诺(c16h21n7o7s3)含量为85.9%。
本发明所述以及制备方法得到:头孢米诺钠原料为:为(+)-(6r,7s)-7-[(s)-2-(2-氨基-2-羧基乙硫基)乙酰氨基]-7-甲氧基-3-[[(l-甲基-1h-四氮唑-5-基)硫基]甲基]-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸钠七水合物,符合中国药典2015版关于头孢米诺钠项下各项标准。
本发明所述的四氢呋喃、丙酮等有机溶剂,均购自国药集团化学试剂北京有限公司,购买日期2018年1月11日。
本发明所述的枸橼酸钠等辅料,购自国药集团化学试剂北京有限公司,购买日期2018年1月11日。
本发具有下述有益的技术效果:
1、原料制备工艺简单,适合工业化生产。
2、本发明制备方法得到原料制备成粉针剂,复溶性好,杂质含量变化小,不溶性微粒增加量小。
3、本发明制剂中加入枸橼酸钠,有助于复溶,本发明制剂在氯化钠溶液和葡萄糖溶液中稳定性高。
附图说明
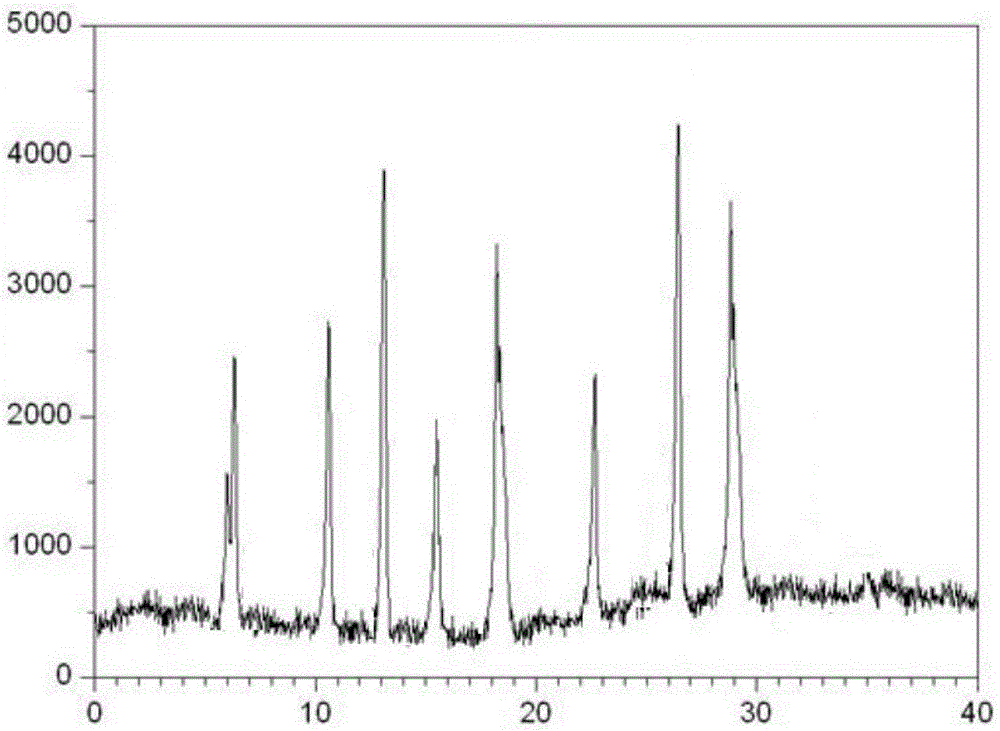
图1实施例3获得头孢米诺钠原料晶型图谱。由隆誉翼尧(北京)科技有限公司测定。
具体实施方式
以下通过具体实施方式的描述对本发明作进一步说明,但这并非是对本发明的限制,本领域技术人员根据本发明的基本思想,可以做出各种修改或改进,但是只要不脱离本发明的基本思想,均在本发明的范围之内。
本发明下述试验委托沈耀(香河)科技有限公司完成。
制备实施例
实施例1
取头孢米诺钠粗品100g,加入四氢呋喃350ml,升温至40℃,加入丙酮450ml,搅拌均匀,5℃-10℃静置,析出结晶,过滤,滤饼用丙酮洗涤,干燥,得到头孢米诺钠原料78.2g;经过检测,头孢米诺钠原料中残留溶剂的符合药典标准。经检测:为(+)-(6r,7s)-7-[(s)-2-(2-氨基-2-羧基乙硫基)乙酰氨基]-7-甲氧基-3-[[(l-甲基-1h-四氮唑-5-基)硫基]甲基]-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸钠七水合物。按无水物计算,含头孢米诺(c16h21n7o7s3)99.7%。有关物质i:单个最大杂质为0.014%,总杂质为0.078%;有关物质ⅱ:相对保留时间在0.82~0.10间的杂质为0.011%,相对保留时间小于0.82的杂质为未检出。
实施例2
取头孢米诺钠粗品100g,加入四氢呋喃500ml,升温至45℃,加入丙酮600ml,搅拌均匀,5℃-10℃静置,析出结晶,过滤,滤饼用丙酮洗涤,干燥,得到头孢米诺钠原料79.4g;经过检测,头孢米诺钠原料中残留溶剂的符合药典规定。经检测:为(+)-(6r,7s)-7-[(s)-2-(2-氨基-2-羧基乙硫基)乙酰氨基]-7-甲氧基-3-[[(l-甲基-1h-四氮唑-5-基)硫基]甲基]-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸钠七水合物。按无水物计算,含头孢米诺(c16h21n7o7s3)99.5%。有关物质i:单个最大杂质为0.016%,总杂质为0.124%;有关物质ⅱ:相对保留时间在0.82~0.10间的杂质为0.014%,相对保留时间小于0.82的杂质为未检出。
实施例3
取头孢米诺钠粗品100g,加入四氢呋喃450ml,升温至50℃,加入丙酮550ml,搅拌均匀,5℃-10℃静置,析出结晶,过滤,滤饼用丙酮洗涤,干燥,得到头孢米诺钠原料81.5g;经过检测,头孢米诺钠原料中残留溶剂的符合药典规定。经检测:为(+)-(6r,7s)-7-[(s)-2-(2-氨基-2-羧基乙硫基)乙酰氨基]-7-甲氧基-3-[[(l-甲基-1h-四氮唑-5-基)硫基]甲基]-8氧代-5-硫杂-1-氮杂双环[4.2.0]辛-2-烯-2-羧酸钠七水合物。按无水物计算,含头孢米诺(c16h21n7o7s3)99.8%。有关物质i:单个最大杂质为0.010%,总杂质为0.065%;有关物质ⅱ:相对保留时间在0.82~0.10间的杂质为0.011%,相对保留时间小于0.82的杂质为未检出。
【有关物质i】
取本品适量,加流动相溶解并稀释制成每lml中约含头孢米诺1.0mg的溶液,作为供试品溶液;精密量取适量,用流动相定量稀释制成每lml中约含头孢米诺10mg的溶液,作为对照溶液;精密量取对照溶液lml,用流动相定量稀释制成每lml中约含头孢米诺0.5mg的溶液,作为灵敏度溶液。照含量测定项下的色谱条件,取灵敏度溶液100μl液相色谱仪,主成分峰高的信噪比应大于10。精密量取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的3.5倍。供试品溶液色谱图中如有杂质峰,单个杂质峰面积不得大于对照溶液主峰面积的0.5倍(0.5%),各杂质峰面积的和不得大于对照溶液主峰面积的1.5倍(1.5%)。供试品溶液色谱图中小于灵敏度溶液主峰面积的峰忽略不计。
【有关物质ⅱ】
临用新制。取本品适量,精密称定,加水溶解并定量稀释制成每lml中约含头孢米诺1.0mg的溶液,作为供试品溶液;另取头孢米诺对照品适量,精密称定,加水溶解并定量稀释制成每lml中约含5μg的溶液,作为对照溶液;精密量取对照溶液1ml,用流动相定量稀释制成每lml中约含0.2μg的溶液,作为灵敏度溶液。照分子排阻色谱法(通则0514)测定。用球状亲水改性硅胶(分子量适用范围为聚合物500~15000)为填充剂(tsk-gelg2000swxl,7.8mm×30cm,5μm或效能相当的色谱柱以磷酸盐缓冲液(ph7.0)[0.005mol/l磷酸氢二钠溶液-0.005mol/l磷酸二氢钠溶液(61:39)]-乙腈(95:5)为流动相,流速为每分钟0.8ml,检测波长为254nm。取供试品溶液10ml,加0.lmol/l氢氧化钠溶液1ml,室温放置1分钟,再加0.lmol/l盐酸溶液1ml,摇匀,作为系统适用性溶液。取10μl注入液相色谱仪,记录色谱图;头孢米诺峰保留时间约为12分钟,头孢米诺峰与其前相邻降解杂质峰间的分离度应符合要求。取灵敏度溶液10μl注人液相色谱仪,记录色谱图,主成分峰高的信噪比应大于10。精密量取供试品溶液与对照溶液各10μl,分别注入液相色谱仪,记录色谱图;供试品溶液色谱图中如有杂质峰,相对保留时间在0.82~0.10间的杂质峰面积不得大于对照溶液主峰面积(0.5%),相对保留时间小于0.82的杂质峰面积的和不得大于对照溶液主峰面积的0.6倍(0.3%),供试品溶液色谱图中小于灵敏度溶液主峰面积的峰忽略不计。
按照“头孢米诺钠高分子聚合物的测定”【高丹玲.头孢米诺钠高分子聚合物的测定(j),海峡药学,2006,18(6):45-47】测定各样品中高分子聚合物的含量。
【含量测定】照高效液相色谱法(通则0512)测定。
色谱条件与系统适用性试验用十八烷基硅烷键合硅胶为填充剂;以醋酸溶液(1→100)-甲醇-四氢呋喃(990:5:5)为流动相,检测波长为254nm。取头孢米诺系统适用性对照品适量,加水溶解并稀释制成每lml中约含lmg的溶液,取10μl注入液相色谱仪,记录的色谱图应与标准图谱一致。
测定法取本品适量,精密称定,加水溶解并定量稀释制成每lml中约含头孢米诺1.0mg的溶液,作为供试品溶液,精密量取10μl注人液相色谱仪,记录色谱图;另取头孢米诺对照品适量,同法测定。按外标法以峰面积计算供试品中c16h21n7o7s3含量。
实施例4
制剂处方为:实施例2头孢米诺钠原料10g,枸橼酸钠0.4g;
制备方法为:取无菌头孢米诺钠与无菌枸橼酸钠混合完全,灌装,得到粉针剂100支。
实施例5
制剂处方为:实施例2头孢米诺钠原料10g,枸橼酸钠0.6g;
制备方法为:取无菌头孢米诺钠与无菌枸橼酸钠混合完全,灌装,得到粉针剂100支。
实施例6
制剂处方为:实施例2头孢米诺钠原料10g,枸橼酸钠0.5g;
制备方法为:取无菌头孢米诺钠与无菌枸橼酸钠混合完全,灌装,得到粉针剂100支。
本发明上述制剂符合中国药典(2015版)关于注射用头孢米诺钠项下各项要求,加速试验和稳定性试验也符合要求。
对比例1
市售头孢米诺钠原料(购自海南新世通制药有限公司,国药准字h20058078,购买日期2018年3月3日)
试验1
原料稳定性试验
对实施例1-3的头孢米诺钠原料和市售头孢米诺钠原料的化学稳定性进行了研究,考察条件为高温(60℃±2℃)、强光照射(4500lx±500lx),高湿(92.5%,rh)考察指标为外观,含量及有关物质。
试验结果见表1。
试验结论:上述试验表明:(1)本发明头孢米诺钠原料有关物质、高分子聚合物含量比对比例1的头孢米诺钠的原料含量低,具有更优秀的产品质量;
(2)经过稳定性试验表明,本发明有关物质以及高分子聚合物增加量小,而对比例1的头孢米诺钠原料有关物质、高分子聚合物含量变化大,充分说明本发明头孢米诺钠原料具有更好的稳定性。
试验2
制剂稳定性试验
对比例2:本发明实施例2头孢米诺钠原料10g,酒石酸钠0.5g;
制剂制备方法:取无菌头孢米诺钠原料和无菌酒石酸钠混合完全,灌装,得到粉针剂100支。
对比例3:本发明实施例2头孢米诺钠原料10g,碳酸钠0.5g;
制剂制备方法:取无菌头孢米诺钠原料和无菌碳酸钠混合完全,灌装,得到粉针剂100支。
对比例4:本发明实施例2头孢米诺钠原料10g。
制剂制备方法:取无菌头孢米诺钠原料,灌装,得到粉针剂100支。
试验方法:将实施例4-6和对比例2、对比例3、对比例4分别置于高温70℃,相对湿度95%和光照照6000lx条件下,于第10天取制剂,每支加氯化钠注射液250ml,加葡萄糖注射液250ml,常规(室温、市内常规光照)条件下容器密闭放置,不溶性微粒检查【按照中国药典附录不溶性微粒检查法光阻法,每毫升≥10μm微粒不能超过25个,每毫升≥25μm微粒不能超过5个】,试验结果见表2、表3。
表2制剂加入氯化钠注射液稳定性结果
表3制剂加入葡萄糖注射液稳定性结果
试验结论:经过常规放置,本发明的头孢米诺钠粉针剂4小时、6小时、8小时后,不溶性微粒依然符合质量标准要求,而加入酒石酸钠或碳酸钠或不加入辅料的头孢米诺钠粉针剂在4小时时,已经不符合质量要求,这充分说明,本发明头孢米诺钠粉针剂在临床配制后,可以经过长时间放置再使用。
- 还没有人留言评论。精彩留言会获得点赞!