纳米载药释药系统及其应用与制备方法、2,6-二苯基吡啶衍生物的制备方法与流程
本发明涉及生物医药技术领域,特别涉及一种纳米载药释药系统及其应用与制备方法、2,6-二苯基吡啶衍生物的制备方法。
背景技术:
随着人口结构和癌症谱的变化,癌症已成为我国首要、全球第二大死亡原因和重大公共健康问题。据国际顶级期刊《柳叶刀》发表的《globalburdenofdisease》,过去十年癌症的发病率和死亡率分别增长30.5%、17.8%[1,2]。2015年全球共有1750万新增癌症患者,870万人死于癌症[3];其中我国有429.2例新发肿瘤病例和281.4万例死亡病例[4],这意味着我国平均每天1.2万人新患癌症、7500人死于癌症;2017年美国有168.9万例新发肿瘤病例,60.1万人死于癌症[5],相当于美国平均每天4600人新患癌症、1650人死于癌症。
2018年国际顶级期刊《柳叶刀》发表了2000-2014年持续性的全球71个国家超过3750万患者癌症生存率大数据[2],我国两大高发癌症——肝癌和肺癌的5年存活率仍然没有改善。2000-2014年我国肝癌的5年存活率从11.7%上升到14.1%,但仍然是我国台湾地区的一半;而肺癌的5年存活率从18.7%上升到19.8,仍处于10%-20%的平均范围之内;胃癌的5年生存率从30.2%提升到35.9%,但仍然只有韩国的一半。令人揪心的我国儿童癌症的5年生存率是许多国家的一半,比如芬兰、瑞典、和丹麦[6]。
2018年美国癌症年报指出:男性一生中被诊断为癌症的概率为39.7%,而女性的概率为37.6%,按照这个比例推算,一家3代中出现至少一位癌症患者的概率会超过95%。未来没有癌症患者的家庭几乎凤毛麟角[7]。因此,加强癌症防治药物及药物制剂迫在眉睫。
目前有4000多个候选药物进入美国fda临床试验,然而没有一种可以作为有效治疗癌症的标准,例如蒽环类抗癌药物阿霉素广泛用于乳腺癌、卵巢癌、骨癌及其他类型的癌症中的化疗,但是其水溶性有限和体内分布较差,fda警告其副作用:局部组织坏死(如不是静脉注射)、致命的充血性心力衰竭、急性骨髓性白血病、骨髓增生异常综合征、严重的骨髓抑制和白血球减少症减少等[8]。
长春新碱是who基本药物清单的抗癌药物,用于治疗淋巴瘤、非小细胞肺癌、膀胱癌、脑癌和睾丸癌,但是它会诱导周边神经病变、低钠血症、白血球和血小板降低,还会引起胃肠道不良反应、高血压、抑郁、疼痛、肌肉痉挛、眩晕、便秘和脱发等副作用[9]。此外长春新碱在生物体液中的溶解度较低,因此生物利用度较低[9]。喜树碱由于羧酸酯结构更倾向与血清白蛋白结合,导致较低的生物利用度和药效。化学修饰获得大量的水溶性喜树碱衍生物,然而只有伊立替康和拓扑利康被fda批准上市。虽然拓扑利康不良反应较少(10%-29%),但是其半衰期较短(2-3h),同时也会引起便秘、肥胖、发烧、腹痛、骨痛、口疮、食欲不振、皮疹、呼吸短促、咳嗽、头疼等不良反应[10]。伊立替康虽然半衰期适中(6-12h),但是腹泻和免疫抑制较严重[11]。新批准的抗癌药物darzalex、emplicity和farydak也饱受毒副作用质疑[8]。
目前,临床常用的抗癌药物,存在生物利用度低、易被快速清除等局限性,需要较大剂量和更高频率的给药;其非特异性分布导致较大的毒副作用,在杀死癌细胞的同时也会导致正常细胞死亡;肿瘤细胞对一种抗肿瘤药物产生耐药性的同时,对结构和作用机制不同的抗肿瘤药物产生交叉耐药性,即多要耐药性,这是导致肿瘤化疗失败的主要原因;尽管有些抗癌药物的疗效提高了,但是他们很难到达转移性肿瘤的位点,这些药物对转移瘤的作用还存疑[12];因此,常规的抗癌药物已逐渐不能满足临床癌症治疗用药的需求,而新兴的多学科交叉的纳米药物能够解决上述这些问题。
纳米药物进入体内,被内吞进入细胞,经由内体-溶酶体通路,到达溶酶体,而溶酶体内环境为ph5-6的酸性环境,且其中含有大量酶类,会导致对纳米药物的降解及破坏,从而导致所载药物失活而无法发挥药效。因此需要采取必要措施使纳米载体快速逃逸溶酶体,避免活性药物被降解,而细胞器靶向和受体靶向,提高药效、降低毒副作用。目前溶酶体逃逸的策略主要有胀破溶酶体法、溶酶体破坏剂等。前者主要是利用质子海绵效应,纳米药物可以吸收大量的质子,是的溶酶体质子泵持续开放,增加溶酶体渗透压,使溶酶体涨破,实现药物的溶酶体逃逸。因此,基于肿瘤微环境的ph响应纳米载体/药物,在正常组织中保持稳定,而通过epr效应富集到肿瘤部位后,在肿瘤组织或溶酶体的酸性环境中快速释放。溶酶体破坏剂主要以蜂毒肽、富含精氨酸的穿膜肽等为代表,能够破坏溶酶体体膜,但是这种破坏作用没有选择性,即对正常细胞膜也存在无法避免的破坏。常见的具有溶酶体逃逸功能的材料主要有聚乙烯亚胺及其衍生物、穿膜肽、融合肽、电荷转换脂质等。然而,聚乙烯亚胺及其衍生物存在细胞毒性和体内积蓄毒性,而穿膜肽存在非特异性细胞结合,融合肽和电荷转换脂质在聚乙二醇存在的时候往往无法实现溶酶体逃逸。因此,寻找新型具有溶酶体逃逸功能的纳米载体对避免药物被降解和靶向细胞器和靶组织具有重要的作用。
技术实现要素:
本发明的主要目的是提出一种纳米载药释药系统及其应用与制备方法、2,6-二苯基吡啶衍生物的制备方法,旨在解决传统纳米载药释药系统溶酶体逃逸功能弱的技术问题。
为实现上述目的,本发明提出了一种纳米载药释药系统,所述纳米载药释药系统包括:如结构式(i)的2,6-二苯基吡啶衍生物、药物及载体材料;
其中,结构式(i)中的n=8或n=9或n=10或n=11或n=12。
可选地,所述2,6-二苯基吡啶衍生物、所述药物及所述载体材料之间的摩尔浓度之比为:25~50:50~500:50~75。
可选地,所述纳米载药释药系统为纳米脂质体、胶束和纳米粒中的一种或其组合。
可选地,所述药物为小分子抗肿瘤药物或核酸药物;
其中,所述小分子抗肿瘤药物为盐酸阿霉素、盐酸表阿霉素、博来霉素、长春花碱、达卡巴嗪、紫杉醇、多西紫杉醇、喜树碱、灯盏花素、地西他滨、卡培他滨、索拉非尼、舒尼替尼、帕唑帕尼或硼替佐米中的一种或其组合;所述所述核酸药物为核酸适配体、抗体基因、核酶、反义核酸、mrna、sirna、shrna、mirna、circrna和lncrna中的一种或其组合。
本发明还提供了一种纳米载药释药系统在制备肿瘤细胞内溶酶体逃逸和细胞器靶向的抗肿瘤纳米药物中的应用,其中,所述纳米载药释药系统为如上所述的纳米载药释药系统。
本发明还提供了一种纳米载药释药系统在制备用于治疗胰腺癌、乳腺癌、肝癌、肺癌、结肠癌、前列腺癌、皮肤癌、膀胱癌、卵巢癌及子宫癌的药物上的应用,其中,所述纳米载药释药系统为如上所述的纳米载药释药系统。
本发明还提供了一种纳米载药释药系统的制备方法,所述纳米载药释药系统的制备方法包括超声法,所述超声法的具体制备步骤如下:
1)将结构式(i)的2,6-二苯基吡啶衍生物和载体材料溶解在有机溶剂中,减压旋转蒸发形成有机膜;
2)于步骤1)中形成的有机膜中加入药物的水溶液,水合、涡旋超声,得到如权利要求1-5任意一项所述的纳米载药释药系统。
本发明还提供了一种纳米载药释药系统的制备方法,所述纳米载药释药系统的制备方法还包括微流控法,所述微流控法的具体制备步骤如下:
1)将结构式(i)的2,6-二苯基吡啶衍生物和载体材料溶解在乙醇中,得到第一混液;
2)将药物溶解在3%~15%的葡萄糖溶液或者醋酸钠缓冲液中,得到第二混液;
3)将所述第一混液与所述第二混液按照3:1的比例、12ml/min的流速进行微流控混合,然后通过透析膜透析过滤,得到如权利要求1-5任意一项所述的纳米载药释药系统。
本发明还提供了一种如结构式(i)的2,6-二苯基吡啶衍生物的制备方法,所述2,6-二苯基吡啶衍生物的制备方法包括第一合成方法,所述第一合成方法包括如下步骤:
a、boc保护:将2,6-二溴吡啶-4-胺、二碳酸二叔丁酯和4-二甲基吡啶溶解于四氢呋喃溶液中,得反应体系;然后将所述反应体系升温至25~80℃,至完全反应;回收溶剂,并用有机相萃取残渣,得第一萃取相;用水相洗涤所述第一萃取相并干燥,得第二萃取相;硅胶柱分离纯化所述第二萃取相,得黄色固体叔丁基(2,6-二溴代吡啶-4-基)氨基甲酸酯;
b、甲基化:取氢化钠和四氢呋喃,混匀、反应,得第一溶液;取叔丁基(2,6-二溴代吡啶-4-基)氨基甲酸酯和四氢呋喃溶液,混匀、反应,得第二溶液;取碘甲烷和四氢呋喃溶液,混匀、反应,得第三溶液;将所述第二溶液缓慢加入所述第一溶液,混匀,然后于低温环境下,加入所述第三溶液,混匀得反应体系;至反应完全,将所述反应体系淬灭、稀释、萃取、洗涤、干燥;分离、浓缩、纯化,得黄色固体叔丁基(2,6-二溴代吡啶-4-基)n-甲基-氨基甲酸酯;
c、脱boc保护:在具塞反应器中,充入惰性气体,将所述叔丁基(2,6-二溴代吡啶-4-基)n-甲基-氨基甲酸酯溶解于hcl/ea溶液中,充分反应,至所述叔丁基(2,6-二溴代吡啶-4-基)n-甲基-氨基甲酸酯完全消耗;除去溶剂,得黄色固体2,6-二溴代-n-甲基吡啶-氨基盐酸盐;
d、亲核取代:取氢化钠和四氢呋喃,混匀、反应,得第一反应液;取2,6-二溴代-n-甲基吡啶-氨基盐酸盐和四氢呋喃溶液,混匀、反应,得第二反应液;将所述第二反应液缓慢加入所述第一反应液中,充分反应后,将反应体系温度降至-5~5℃,然后分批加入2-溴代-n,n-二甲基乙基-1-胺,并缓慢将反应体系的温度升至室温,充分反应后,进行淬灭反应,得淬灭液;将所述淬灭液固液分离,并萃取残渣,得萃取液;将所述萃取液分离纯化,得淡黄色油状化合物n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺;
e、suzuki反应:取所述n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺、5-溴-2-甲氧基苯基硼酸和四氢呋喃溶液,混匀、反应,得第一混液;加入碳酸钠溶液,混匀、反应,得第二混液;并用惰性气体置换空气,加入四(三苯基膦)钯,再次用惰性气体置换空气,60~100℃回流,至反应完全,然后进行淬灭反应、萃取,得萃取液,将所述萃取液干燥、过滤、固液分离,然后进行硅胶柱色谱纯化,得黄色固体n1-(2,6-二(5-溴代-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺;
f、sonogashira偶联:取所述n1-(2,6-二(5-溴代-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺、1-十二炔、三苯基膦和无水四丁基氟化铵,混匀、反应,得第一混合物;按体积比为2:1~1:2的比例分别量取二甲基甲酰胺和二异丙胺,混匀反应,得第二反应液;然后用惰性气体置换空气,并加入双(三苯基膦)二氯化钯,再次用惰性气体置换空气,密封,得第三反应混合物;将所述第三反应混合物于60~100℃下充分反应,稀释、萃取,得萃取液;将所述萃取液干燥、固液分离,然后进行硅胶色谱纯化,得褐油状化合物n1-(2,6-二(5-(十二-1-炔-1-基)-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺;
g、氢化还原:取所述n1-(2,6-二(5-(十二-1-炔-1-基)-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺和乙醇,混匀、反应,然后加入10%钯/碳催化剂进行催化,得催化反应物,然后将所述催化反应物体系于氢气环境中置换空气,并于室温、0.6~1.0mpa的氢气压力下搅拌所述催化反应物,至反应完全,过滤除去催化剂,然后洗涤、固液分离,然后进行硅胶柱色谱纯化分离,得如结构式(i)的2,6-二苯基吡啶衍生物,其中,n=12。
本发明还提供了一种如结构式(i)的2,6-二苯基吡啶衍生物的制备方法,所述2,6-二苯基吡啶衍生物的制备方法还包括第二合成方法,所述第二合成方法包括如下步骤:
a1、芳香硝基的取代反应:取2,6-二溴-4-硝基吡啶和n,n-二甲基甲酰胺溶液,混匀、反应,并于低温环境下加入氢化钠,然后于室温下充分反应,得第一反应液;加入n1,n2,n2-三甲基-1,2-乙二胺于所述第一反应液中,至反应完全后,进行淬灭反应、萃取,得萃取液;将所述萃取液进行洗涤、干燥、浓缩,然后硅胶柱层析纯化,得棕色油状化合物n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺;
b1、suzuki反应:取所述n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺、5-溴-2-甲氧基苯基硼酸、碳酸钠及四氢呋喃溶液/水,混匀、反应,加入四(三苯基膦),并于惰性气体环境,60℃~100℃下,过夜反应,至反应完全后,用饱和碳酸钠稀释,得稀释液;然后萃取所述稀释液,得萃取液;将所述萃取液进行洗涤、干燥、减压浓缩后,进行硅胶层析纯化,得棕色油状化合物n1-(2,6-双(3-溴代苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺;
c1、sonogashira偶联:取所述n1-(2,6-双(3-溴代苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺、1-十二炔、三苯基膦和n,n-二甲基甲酰胺/2,6-二异丙基苯胺溶液,混合,然后加入干燥的四丁基氟化铵,并于惰性气体环境中置换空气,得第一反应体系;于所述第一反应体系中加入二氯二(三苯基膦)合钯,再次于惰性气体环境中置换空气,密封,得第二反应体系;将所述第二反应体系于60℃~100℃温度下,过夜反应,至反应完全后,稀释,得稀释液;萃取所述稀释液,得萃取液;将所述萃取液进行洗涤、干燥、减压浓缩后,进行硅胶层析纯化,得棕色油状化合物n1-(2,6-双(3-(十二烷-1-炔-1-基)苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺;
d1、氢化还原:取所述n1-(2,6-双(3-(十二烷-1-炔-1-基)苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺和乙醇,混匀,然后加入10%钯/碳催化剂进行催化,得催化反应物,然后将所述催化反应物体系于氢气环境中置换空气,并于室温、0.6~1.0mpa的氢气压力下搅拌所述催化反应物,过夜,至反应完全,过滤除去催化剂,然后洗涤、减压蒸干后,进行硅胶柱色谱纯化分离,得如结构式(i)的2,6-二苯基吡啶衍生物,其中,n=12。
相较于现有技术,本发明取得了以下有益效果:
本发明技术方案中,通过一种新设计合成的2,6-二苯基吡啶衍生物、药物及载体材料构建了一种纳米载药释药系统,以实现快速打开肿瘤细胞内的溶酶体,使得药物能快速逃逸至细胞质中,从而使负载的药物/sirna等载体免受降解和破坏,进而提高细胞器靶向性,提高药效和降低毒副作用。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图示出的结构获得其他的附图。
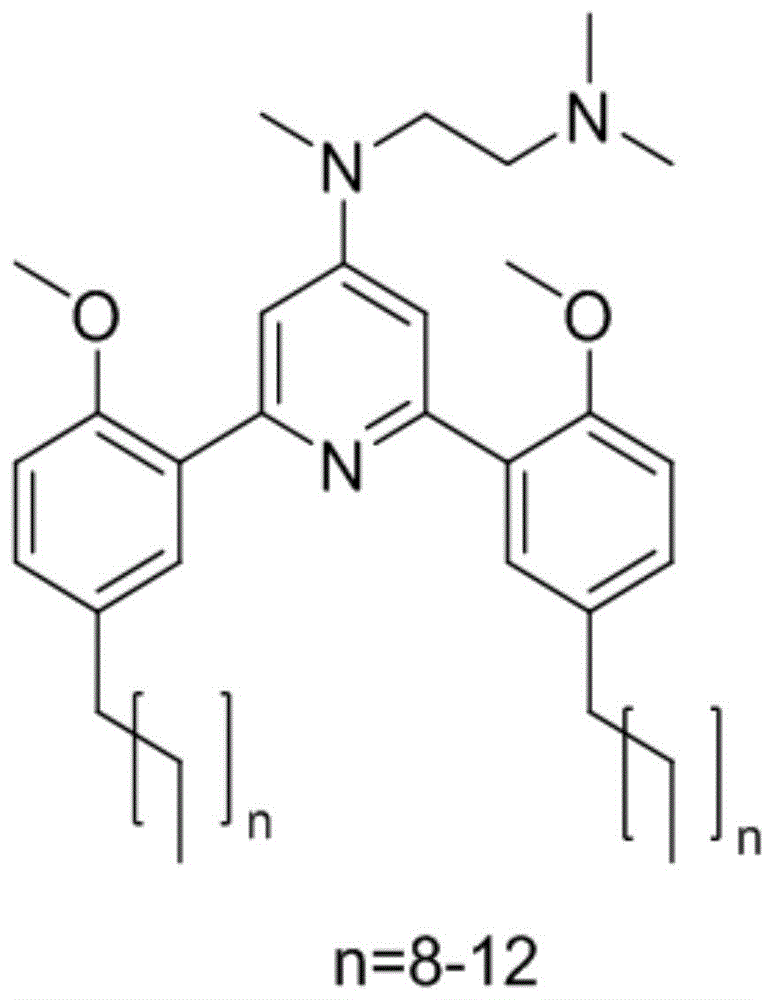
图1为本发明纳米载药释药系统实施例中2,6-二苯基吡啶衍生物的结构式示意图;
图2为本发明一实施例中2,6-二苯基吡啶衍生物的1h-nmr表征图谱;
图3为图2所示的2,6-二苯基吡啶衍生物的13c-nmr表征图谱;
图4为图2所示的2,6-二苯基吡啶衍生物的esi-ms表征图谱;
图5为本发明另一实施例中2,6-二苯基吡啶衍生物的1h-nmr表征图谱;
图6为图5所示的2,6-二苯基吡啶衍生物的13h-nmr表征图谱;
图7为图5所示的2,6-二苯基吡啶衍生物的esi-ms表征图谱;
图8为本发明一实施例中纳米载药释药系统负载磺基罗丹明在hela细胞内的转运激光共聚焦图像;
图9为本发明一实施例中纳米载药释药系统负载磺基罗丹明与hela细胞内溶酶体的manders共定位系数柱形图;
图10为本发明一实施例中纳米载药释药系统dox释放率统计图。
本发明目的的实现、功能特点及优点将结合实施例,参照附图做进一步说明。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
需要说明,若本发明实施例中有涉及方向性指示(诸如上、下、左、右、前、后……),则该方向性指示仅用于解释在某一特定姿态(如附图所示)下各部件之间的相对位置关系、运动情况等,如果该特定姿态发生改变时,则该方向性指示也相应地随之改变。
另外,若本发明实施例中有涉及“第一”、“第二”等的描述,则该“第一”、“第二”等的描述仅用于描述目的,而不能理解为指示或暗示其相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。另外,各个实施例之间的技术方案可以相互结合,但是必须是以本领域普通技术人员能够实现为基础,当技术方案的结合出现相互矛盾或无法实现时应当认为这种技术方案的结合不存在,也不在本发明要求的保护范围之内。
相关技术中,纳米药物进入体内后被内吞进细胞,然后经由内体-溶酶体通路,到达溶酶体。一般情况下,溶酶体内环境的ph为5~6的酸性环境,且其中含有大量酶类,会导致纳米药物的降解及破坏,从而导致所载药物失活而无法发挥药效。因此,需要采取必要措施使纳米载体快速逃逸溶酶体,以避免活性药物被降解,而提高细胞器靶向性和靶点靶向性,提高药效,并降低毒副作用。
基于此,本发明提出了一种纳米载药释药系统,所述纳米载药释药系统包括:如结构式(i)的2,6-二苯基吡啶衍生物、药物及载体材料;
其中,结构式(i)中的n=8或n=9或n=10或n=11或n=12。
由上,本发明中通过合成一种新的如结构式(i)的2,6-二苯基吡啶衍生物,并利用此2,6-二苯基吡啶衍生物与载体材料一起构建纳米药物载体以搭载药物,构成能将药物送入细胞靶向点的纳米材料系统,从而实现对病变细胞的治疗。需进一步说明的是,本发明构建的纳米材料系统为一种ph响应型的纳米载药释药系统,其能够响应不同ph条件,并通过内吞与溶酶体膜进行融合。具体地,在正常生理ph=7.4的条件下,该纳米载药释药系统不会发生质子化和构型翻转,此时,该纳米载药释药系统不会破坏生物膜,并在该纳米载药释药系统的作用下可将药物运输至靶向点;在ph=5.2的酸性条件下,该纳米载药释药系统会发生质子化,进一步地与其自身的甲氧基发生氢键作用,使得载体材料的烃链相对方向发生反转,从而使得与纳米药物融合的溶酶体外壳打开,药物快速释放,进而实现药物的溶酶体逃逸。
可选地,所述2,6-二苯基吡啶衍生物、所述药物及所述载体材料之间的摩尔浓度之比为:25~50:50~500:50~75。所述纳米载药释药系统为纳米脂质体、胶束和纳米粒中的一种或其组合。所述药物为小分子抗肿瘤药物或核酸药物;其中,所述小分子抗肿瘤药物为盐酸阿霉素、盐酸表阿霉素、博来霉素、长春花碱、达卡巴嗪、紫杉醇、多西紫杉醇、喜树碱、灯盏花素、地西他滨、卡培他滨、索拉非尼、舒尼替尼、帕唑帕尼、硼替佐米和核酸药物中一种或其组合;所述核酸药物为核酸适配体、抗体基因、核酶、反义核酸、mrna、sirna、shrna、mirna、circrna和lncrna中的一种或其组合。所述载体材料为磷脂、磷脂-聚乙二醇、胆固醇和壳聚糖中的一种或其组合。
需要解释的是,所述小分子抗肿瘤药物为分子量小于1000的有机化合物。
本发明还提供了一种纳米载药释药系统在制备肿瘤细胞内溶酶体逃逸和细胞器靶向的抗肿瘤纳米药物中的应用,其中,所述纳米载药释药系统为如上所述的纳米载药释药系统。
本发明还提供了一种纳米载药释药系统在制备用于治疗胰腺癌、乳腺癌、肝癌、肺癌、结肠癌、前列腺癌、皮肤癌、膀胱癌、卵巢癌及子宫癌的药物上的应用,其中,所述纳米载药释药系统为如上所述的纳米载药释药系统。
本发明还提供了一种纳米载药释药系统的制备方法,所述纳米载药释药系统的制备方法包括超声法,所述超声法的具体制备步骤如下:
1)将结构式(i)的2,6-二苯基吡啶衍生物和载体材料溶解在有机溶剂中,减压旋转蒸发形成有机膜;
2)于步骤1)中形成的有机膜中加入药物的水溶液,水合、涡旋超声,得到如权利要求1-5任意一项所述的纳米载药释药系统。
本发明还提供了一种纳米载药释药系统的制备方法,所述纳米载药释药系统的制备方法还包括微流控法,所述微流控法的具体制备步骤如下:
1)将结构式(i)的2,6-二苯基吡啶衍生物和载体材料溶解在乙醇中,得到第一混液;
2)将药物溶解在3%~15%的葡萄糖溶液或者醋酸钠缓冲液中,得到第二混液;
3)将所述第一混液与所述第二混液按照3:1的比例、12ml/min的流速进行微流控混合,然后通过透析膜透析过滤,得到如权利要求1-5任意一项所述的纳米载药释药系统。
具体地,为进一步说明本发明中的纳米载药释药系统,通过以下实验进行详细阐述。
一、2,6-二苯基吡啶衍生物的制备
1、2,6-二苯基吡啶衍生物的制备过程中遇到的难题。
在jeanneleblond团队在化学期刊《angewandtechemieinternationaledition》和《nanoscale》上报道的2,6-二苯基吡啶类化合物的合成方法中,由于单甲基化的产物和原料的亲和性至少相当,因此会进一步的甲基化生成叔胺基甚至季铵盐,如第一合成路径,难以生成本发明中的目标产物tm如结构(1)的2,6-二苯基吡啶衍生物。
第一合成路径
基于上述第一合成路径,还曾通过第二合成路径以进一步实现对目标产物tm的合成,具体地,包括:首先在boc保护氨基后进行甲基化,以避免季铵盐的形成。然后进一步采用suzuki反应,以获得2,6-二苯基吡啶类化合物6(具体地,所述2,6-二苯基吡啶类化合物的化学结构如第二合成路径中的化合物6所示),再经过sonogashira偶联、还原和脱boc保护,以获得化合物9(具体地,所述化合物9的化学结构如第二合成路径中的化合物9),但是,所述化合物9无论是经过sn2亲核取代反应还是经过还原酰胺法和tbs保护的溴乙醇反应,均无法获得目标产物tm。
第二合成路径
基于上述第二合成路径,本发明还拟通过改变合成路线的优先顺序,以合成目标产物tm,如第三合成路径。上述第三合成路径即是先将化合物6进行脱boc保护,然后再进行sn2亲核取代反应,但是在进行sn2亲核取代反应的时候未发生相关的反应,通过所述第三合成路径也无法获得目标产物tm。
第三合成路径
2、2,6-二苯基吡啶衍生物合成路径
基于上述第一合成路径、第二合成路径以及第三合成路径的探索,本发明探索获得至少两种可以获得目标产物tm的合成路径(第四合成路径和第五合成路径),具体如下:
第四合成路径
第五合成路径
3、第一实施例组。
为对本发明中的目标产物tm(即2,6-二苯基吡啶衍生物)的合成路径的进一步说明,通过以下第一实施例组来进行详细阐述。应当理解,下述实施例组中选用的试剂组分及其含量并不唯一,还包括其他作用类似或者含量相近的足以获得本发明中需要合成的2,6-二苯基吡啶衍生物的其他实施例组。
3.1目的产物tm的合成:
实施例a(即ph响应脂质的合成)
1)叔丁基(2,6-二溴代吡啶-4-基)氨基甲酸酯(即第四合成路径中的化合物2)的合成:于500ml的圆底烧瓶中,将2,6-二溴吡啶-4-胺(19.5g,77.3mmol,1eq)、boc2o(即二碳酸二叔丁酯)(18.5g,85.1mmol,1.1eq)和dmap(即4-二甲氨基吡啶)(2.5g,19.3mmol,0.25eq)溶解于250ml四氢呋喃溶液中。反应缓慢升至60c并反应16小时tlc和lc-ms检测反应完全后,减压回收thf(四氢呋喃),并用乙酸乙酯(250ml)溶解残渣,用饱和nh4cl溶液(200ml*3)和饱和食盐水(200ml)洗涤乙酸乙酯相,na2so4干燥,减压回收溶剂,硅胶柱色谱(石油醚:乙酸乙酯=100:0→0:100)分离纯化,获得25g黄色固体叔丁基(2,6-二溴代吡啶-4-基)氨基甲酸酯(其产率为:91.8%)。其中,esi-ms:m/z[m+h]+=352.9。
2)叔丁基(2,6-二溴代吡啶-4-基)n-甲基-氨基甲酸酯(即第四合成路径中的化合物3)的合成:于500ml的干燥圆底烧瓶中加入nah(混悬在矿物油中60%含量,11.3g,285mmol,4eq)缓慢加入200ml四氢呋喃,室温搅拌10分钟,然后将步骤1)中获得的叔丁基(2,6-二溴代吡啶-4-基)氨基甲酸酯(25g,71.0mmol,1eq)的thf溶液(100ml)缓慢加入到反应液中,室温下搅拌0.5小时,然后在0℃碘甲烷(15.1g,106.5mmol,1.5eq)的thf溶液(20ml),反应液在室温下搅拌3小时。tlc和lc-ms检测提示所述叔丁基(2,6-二溴代吡啶-4-基)氨基甲酸酯反应完全后,将反应降至0℃,加入水并用乙酸乙酯(300ml)稀释,有机相用200ml饱和食盐水洗涤,无水na2so4干燥,减压浓缩,获得黄色固体叔丁基(2,6-二溴代吡啶-4-基)n-甲基-氨基甲酸酯(其产率为:98.2%)。其中,1hnmr(400mhz,cdcl3)δ(ppm)1.53(s,9h),3.28(s,3h),7.52(s,2h);esi-ms:m/z[m+h]+=367.0。
3)2,6-二溴代-n-甲基吡啶-氨基盐酸盐(即第四合成路径中的化合物14)的合成:在具塞和充满氩气的100ml圆底烧瓶中,将步骤2)中获得的叔丁基(2,6-二溴代吡啶-4-基)n-甲基-氨基甲酸酯(25g,68.3mmol,1eq)溶解于hcl/ea溶液,搅拌3小时,待tlc和lc-ms检测所述叔丁基(2,6-二溴代吡啶-4-基)n-甲基-氨基甲酸酯消耗完全后,减压蒸除溶剂,获得黄色固体2,6-二溴代-n-甲基吡啶-氨基盐酸盐(20g,66.2mmol,其产率为:96.9%)。需要说明的是,获得的所述2,6-二溴代-n-甲基吡啶-氨基盐酸盐在此不进行其他处理,直接用作下一步反应的反应原料。其中,esi-ms:m/z[m+h]+=266.9。
4)n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺(即第四合成路径中的化合物15)的合成:于500ml的圆底烧瓶中加入nah(混悬在矿物油中60%含量,21.2g,530mmol,8eq),缓慢加入200ml四氢呋喃,室温搅拌10分钟,缓慢加入步骤3)中获得的所述2,6-二溴代-n-甲基吡啶-氨基盐酸盐(20g,66.2mmol,1eq)的thf(100ml)溶液,室温搅拌30分钟后,将反应降至0℃,并于0oc下,分批加入2-溴代-n,n-二甲基乙基-1-胺(溴酸盐,18.5g,79.5mmol,1.2eq),反应缓慢升温至室温,并搅拌16个小时。tlc和lc-ms检测所述2,6-二溴代-n-甲基吡啶-氨基盐酸盐消耗完全后,用甲醇淬灭反应,减压回收溶剂后,干法上样进行硅胶柱色谱分离(dcm:meoh=80:1),获得淡黄色油状的n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺(14.3g,42.4mmol,其产率为:64%)。其中,1hnmr(400mhz,cd3od)δ(ppm)2.23(s,6h),2.43(t,j=7.4hz,2h),2.93(s,3h),3.44(t,j=7.4hz,2h),6.74(s,2h);esi-ms:m/z[m+h]+=338.0。
5)n1-(2,6-二(5-溴代-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺(即第四合成路径中的化合物16)的合成:于500ml的圆底烧瓶中,将步骤4)中获得的所述n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺(14.3g,42.4mmol,1eq)和5-溴-2-甲氧基苯基硼酸(24.5g,106mmol,2.5eq)溶解于250ml的四氢呋喃,加入na2co3(22.5g,212mmol,5eq,120ml蒸馏水)。在氩气置换空气3次,加入pd(pph3)4(即四(三苯基膦)钯)(4.3g,3.7mmol,0.8eq),再次用氩气置换空气3次。80℃回流48小时,tlc和lc-ms检测所述n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺反应完全后,饱和碳酸钠溶液淬灭反应,乙酸乙酯萃取3次,每次100ml,合并的有机相经na2so4干燥、过滤、减压回收溶剂后,进行硅胶柱色谱分离(dcm:meoh=80:1),获得14g黄色固体n1-(2,6-二(5-溴代-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺(25.5mmol,其产率为:60%)。其中,1hnmr(400mhz,cd3od)δ(ppm)2.23(s,6h),2.43(t,j=7.4hz,2h),2.93(s,3h),3.44(t,j=7.4hz,2h),6.74(s,2h);esi-ms:m/z[m+h]+=338.0。
6)n1-(2,6-二(5-(十二-1-炔-1-基)-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(即第四合成路径中的化合物17)的合成:将步骤5)中获得的所述n1-(2,6-二(5-溴代-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙基二胺(14g,25.5mmol,1eq)置于500ml圆底烧瓶中。加入1-十二炔(14.9g,89.25mmol,3.5eq)和pph3(即三苯基膦)(2.0g,7.65mmol,0.3eq)。加入无水四丁基氟化铵(tbaf,77.1g,296.8mmol,7eq),然后加入150ml二甲基甲酰胺和150ml二异丙胺(dipa)。氩气置换空气3次。加入pdcl2(pph3)2(即双(三苯基膦)二氯化钯)(1.75g,2.5mmol,0.1eq),氩气置换空气3次,密封。混合物在80℃下搅拌16h,然后用饱和的na2co3稀释,乙酸乙酯萃取3次。有机层经na2so4干燥,减压回收溶剂。粗品经硅胶柱色谱(dcm:meoh=80:1)纯化,得到褐油状的n1-(2,6-二(5-(十二-1-炔-1-基)-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(12g,16.7mmol,其产率为:65%)。其中,1hnmr(400mhz,cdcl3)δ(ppm)0.85(t,j=6.6hz,6h),1.25-1.37(m,28h),1.43(quint,j=7.4hz,4h),1.56(quint,j=7.4hz,4h),2.30(s,6h),2.37(t,j=7.2hz,2h),2.53(t,j=7.6hz,4h),3.04(s,3h),3.51(t,j=7.6hz,2h),3.85(s,6h),6.88(d,j=6.8hz,2h),6.90(s,2h),7.36(dd,j1=2.0hz,j2=2.0hz,2h),7.81(d,j=2.4hz,2h)。
7)n1-(2,6-二(5-十二炔-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(即第四合成路径中的目的产物tm1)的合成:将步骤6)中获得的所述n1-(2,6-二(5-(十二-1-炔-1-基)-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(12g,16.7mmol,1eq)置于250ml圆底瓶,溶于120ml乙醇。加入10%的pd/c(1.5g),用氢气置换瓶内空气3次。在室温、1个大气压的氢气压力下搅拌4小时。tlc或lc-ms检测所述n1-(2,6-二(5-(十二-1-炔-1-基)-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺反应完全后,过滤除去催化剂,并用乙醇洗涤三次,合并滤液减压蒸干。粗产物经硅胶柱色谱(pe:ea=100:0→0:100,dcm:meoh=30:1)纯化,得到微黄色的n1-(2,6-二(5-十二炔-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(7.2g,9.88mmol,其产率为:60%)。其中,1hnmr(400mhz,cdcl3)δ(ppm)0.87(t,j=6.6hz,6h),1.24-1.37(m,36h),1.58(quint,j=7.4hz,4h),2.31(s,6h),2.54(t,j=7.6hz,2h),2.59(t,j=7.8hz,4h),3.04(s,3h),3.52(t,j=7.8hz,2h),3.83(s,6h),6.89(d,j=8.4hz,2h),6.99(s,2h),7.12(dd,j1=2.4hz,j2=2.4hz,2h),7.64(d,j=2.0hz,2h)。13cnmr(400mhz,cdcl3)δ(ppm)14.15,22.72,29.47-29.72,31.66,31.96,35.16,38.06,45.91,50.10,55.98,56.03,106.47,111.50,128.82,130.55,131.46,135.28,153.17,155.02,155.94。esi-ms:m/z[m+h]+=364.9。
实施例b(即非ph响应脂质tm2的合成)
1)n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(即第五合成路径中的化合物19)的合成:往2,6-二溴-4-硝基吡啶(即第五合成路径中的化合物18)(50.0g,177.3mmol)的dmf(即n,n-二甲基甲酰胺)(300ml)溶液中,0℃条件下加入nah(10.6g,266.0mmol),室温搅拌1h。然后加入n1,n1,n2-三甲基-1,2-乙二胺(18.1g,177.3mmol),在室温下搅拌过夜。tlc或lc-ms检测反应完全后,加水淬火,用乙酸乙酯(200ml×2)提取。合并的有机层用100ml×3的盐水洗涤,na2so4干燥,浓缩,粗产物经硅胶柱层析(dcm:ea=2:1)纯化,得到棕色油状的n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(47.5g,其产率为:79.4%)。
2)n1-(2,6-双(3-溴代苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(即第五合成路径中的化合物20)的合成:将步骤1)中获得的所述n1-(2,6-二溴代吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(27.5g,81.6mmol)、5-溴-2-甲氧基苯基硼酸(32.8g,163.2mmol)和na2co3(17.3g,163.2mmol)混合于四氢呋喃/水(300ml,2:1)溶液,加入pd(pph3)4(4.7g,4.08mmol),在氮气保护下,80℃搅拌过夜。tlc和lc-ms监测反应,待原料完全消耗,混合物用饱和的na2co3稀释,然后用乙酸乙酯(200ml×2)萃取。合并有机层,用200ml饱和食盐水洗涤,na2so4干燥,减压浓缩,粗产物用硅胶柱层析(dcm:ea=2:1)纯化,得到棕色油状的n1-(2,6-双(3-溴代苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(32.1g,其产率为:80.4%)。
3)n1-(2,6-双(3-(十二烷-1-炔-1-基)苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(即第五合成路径中的化合物21)的合成:将步骤2)中获得的所述n1-(2,6-双(3-溴代苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(21.2g,43.3mmol)、1-十二炔(14.5g,86.6mmol)和pph3(11.3g,13.0mmol)混合于dmf/dipa(即n,n-二甲基甲酰胺/2,6-二异丙基苯胺)(240ml,1:1)溶液中。然后加入干燥的四丁基氟化铵(135.8g,519.6mmol)(预先从1.0m的tbaf四氢呋喃溶液中高真空干燥所得),用氩气置换反应瓶内空气3次。加入pd(pph3)2cl2(3.0g,4.33mmol),氩气置换3次,密封。混合物在80℃下搅拌过夜。tlc和lc-ms监测反应完全后,用饱和na2co3溶液稀释,用乙酸乙酯(200ml×2)萃取。合并有机层,用200ml饱和食盐水洗涤,na2so4干燥,浓缩,粗产物用硅胶柱层析(dcm:etoac=2:1)纯化,得到棕色油状的n1-(2,6-双(3-(十二烷-1-炔-1-基)苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(21.5g,其产率为:75%)。
4)n1-(2,6-双(3-十二烷基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(即第五合成路径中的目的产物tm2)的合成:在步骤3)中获得的所述n1-(2,6-双(3-(十二烷-1-炔-1-基)苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(17.2g,26.0mmol)的乙醇(200ml)溶液中,加入10%的pd/c(8.0g),用氢气置换瓶内空气3次。在室温、0.6mpa的氢气压力下搅拌过夜。tlc或lc-ms检测反应完全后,过滤除去催化剂,并用乙醇洗涤三次,合并滤液减压蒸干。粗产物经硅胶柱色谱(pe:ea=100:0→0:100,dcm:meoh=30:1)纯化,得到棕色的蜡状物n1-(2,6-双(3-十二烷基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺(10.5g,其产率为:60.3%)。其中,1hnmr(400mhz,cdcl3)δ7.88(s,2h),7.84(d,j=7.8hz,2h),7.37(t,j=7.6hz,2h),7.24(d,j=8.6hz,1h),7.21(s,1h),6.90(s,2h),3.78–3.64(m,2h),3.13(s,3h),2.76(t,j=7.3hz,2h),2.73–2.66(m,4h),2.50(s,6h),1.71–1.60(m,4h),1.36–1.19(m,36h),0.87(t,j=6.8hz,6h)。esi-ms:m/z[m+h]+=668.3。
并且基于上述实施例a和实施例b的合成路线和方案,得到如图2至图7所示的结果数据图。
3.2结果数据分析。具体地,如图2至图7所示,其中,图2为目的产物tm1(即n1-(2,6-二(5-十二炔-2-甲氧基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺)的核磁共振氢谱图,图3为tm1的核磁共振碳谱图,图4为tm1的质谱图;图5为目的产物tm2(即n1-(2,6-双(3-十二烷基苯基)吡啶-4-基)-n1,n2,n2-三甲基-1,2-乙二胺)的核磁共振氢谱图,图6为tm2的核磁共振碳谱图,图7为tm2的质谱图。由图2至图4可知,在1hnmr(400mhz,cdcl3)中δ(ppm)=0.87(t,j=6.6hz,6h),1.24-1.37(m,36h),δ(ppm)=1.58(quint,j=7.4hz,4h),2.31(s,6h),δ(ppm)=2.54(t,j=7.6hz,2h),δ(ppm)2.59(t,j=7.8hz,4h),3.04(s,3h),δ(ppm)=3.52(t,j=7.8hz,2h),3.83(s,6h),δ(ppm)=6.89(d,j=8.4hz,2h),6.99(s,2h),δ(ppm)=7.12(dd,j1=2.4hz,j2=2.4hz,2h),δ(ppm)=7.64(d,j=2.0hz,2h);13cnmr(400mhz,cdcl3)中δ(ppm)=14.15、22.72、29.47-29.72、31.66、31.96、35.16、38.06、45.91、50.10、55.98、56.03、106.47、111.50、128.82、130.55、131.46、135.28、153.17、155.02、155.94;esi-ms:m/z[m+h]+=364.9;可知,tm1的化学式为:
由图5至图7可知,在1hnmr(400mhz,cdcl3)中δ(ppm)=0.87(t,j=6.6hz,6h),1.24-1.37(m,36h),δ(ppm)=1.58(quint,j=7.4hz,4h),2.31(s,6h),δ(ppm)=2.54(t,j=7.6hz,2h),δ(ppm)=2.59(t,j=7.8hz,4h),3.04(s,3h),δ(ppm)=3.52(t,j=7.8hz,2h),3.83(s,6h),δ(ppm)=6.89(d,j=8.4hz,2h),6.99(s,2h),δ(ppm)=7.12(dd,j1=2.4hz,j2=2.4hz,2h),δ(ppm)=7.64(d,j=2.0hz,2h);在13cnmr(400mhz,cdcl3)中δ(ppm)=14.15、22.72、29.47-29.72、31.66、31.96、35.16、38.06、45.91、50.10、55.98、56.03、106.47、111.50、128.82、130.55、131.46、135.28、153.17、155.02、155.94;esi-ms:m/z[m+h]+=364.9;可知,tm2的化学式为:
二、纳米载药释药系统的制备。
为对本发明中的纳米载药释药系统做进一步的说明,通过以下实验来进行详细阐述。应当理解,本发明中的纳米载药释药系统并不限于下述实验中涉及的具体数据,还包括能本发明提及的其他数据范围。
1、纳米载体的制备。
1.1制备方法。
将实验一中制备的目标产物tm1以及dspc(即二硬脂酰基磷脂酰胆碱)和dspe-peg2000(即二硬脂酰基磷脂酰乙醇胺-聚乙二醇2000)均分别先用氯仿制备成20mg/ml的储备液,然后分别按照下述表1所示的摩尔比例混合于5ml圆底烧瓶中,减压旋蒸去氯仿,室温真空烘干12小时,然后取1ml的30mm磺基罗丹明溶液进行水合反应,充分涡旋后使用超声破壁机处理样本,并得到如表1所示的数据结果。
表1
1.2结果分析。由上表1可知,在上述目标产物tm1以及dspc和dspe-peg2000的配比下,纳米载体均表现出低于200nm的流体动力学粒径,但是在hplc检测下,随着目标产物tm1的摩尔浓度的提高,其在载体材料中的融合更低。说明,由于目标产物tm1的脂溶性过高,在所述目标产物tm1在纳米载药释药系统中的比例超过50%时,该目标产物tm1会出现与载体材料的不完全融合,导致目标产物tm1的不必要消耗浪费。
2、纳米载药释药系统在细胞内的转运。
2.1实验操作。
取40mg/ml的脂质tm1乙醇溶液,与20mg/ml的dspc和dspe-peg2000氯仿氯仿溶液,按照40:55:5的摩尔比混合于5ml圆底烧瓶,减压旋蒸除去氯仿,室温真空烘干12h,取30mm磺基罗丹明溶液按照上述摩尔比的50当量水合,充分涡旋后使用超声破壁机处理样品,制成脂质体。
将hela细胞接种至24孔板中,每孔30000个细胞,孵育24小时,然后用mem培养基洗涤2次,分别用实验一中获得的能实现ph响应的目标产物tm1孵育1小时和12小时,和不能实现ph响应的目标产物tm2孵12小时,脂质体包裹的磺基罗丹明和游离的罗丹明(阴性对照)浓度均为900nm。孵育后用mem培养基冲洗3次,并在30℃下用100nmlyso
2.2结果分析。
由图8可知,其中a为未经纳米载体包裹的磺基罗丹明b在hela细胞中的转运,b为经纳米载体(含有能实现ph响应的目标产物tm1)(即ph响应脂质体)包裹的磺基罗丹明b在hela细胞中的转运,c为经纳米载体(含不能实现ph响应的目标产物tm2)(即非ph响应脂质体)包裹的磺基罗丹明b在hela细胞中的转运。可知,在a中,向培养的hela细胞中加入未经脂质体包裹的、游离的红色荧光染料磺基罗丹明b,并孵育12小时后,无论是溶酶体内还是细胞质中,都观测不到磺基罗丹明b的红色荧光,表明游离的磺基罗丹明b自身无法穿越细胞质膜,进入不了细胞;在b中,向培养的hela细胞中加入包裹了磺基罗丹明b的且能实现ph响应的纳米载体,孵育1小时后,磺基罗丹明b被迅速内吞进入细胞,经由经内体-溶酶体通路,到达溶酶体。荧光显微镜图片显示磺基罗丹明b与溶酶体共定位效率mcc值为0.80。而孵育12小时后,磺基罗丹明b与溶酶体共定位效率mcc值降低为0.29,大量磺基罗丹明b从溶酶体中逃逸至细胞质,并经细胞内转运,弥散分布于细胞质大部分区域;在c中,向培养的hela细胞中加入包裹了磺基罗丹明b的且不能实现ph响应的纳米载体,孵育12小时后,磺基罗丹明b始终滞留于溶酶体,其共定位效率mcc值为0.87,进一步证实,ph不敏感的纳米载体包裹的磺基罗丹明b无法实现有效的溶酶体逃逸。
由图9可知,在图9中每个视野中至少含有4个细胞,**代表学生t检测中p≤0.01。12小时后,在含有ph响应脂质体的细胞中检测到mcc的含量在0.3左右,而在含有非ph响应脂质体的细胞中检测到mcc的含量为0.9左右,可知含有目标产物tm1的ph响应脂质体和溶酶体膜的融合性更高,且其与peg的兼容性更高,有利于纳米载药释药系统在溶酶体内的逃逸。
3、纳米载药释药系统的制备及药物释放。
3.1纳米载药释药系统的制备及阿霉素(dox)的释放率测定。
按照50:10:37.5:2.5的摩尔比例分别量取40mg/ml阳离子脂质的乙醇溶液、dspc、胆固醇和dspc-peg2000,混合于5ml的圆底烧瓶中,减压旋蒸除去乙醇,室温真空烘干12小时,取上述摩尔比的200当量dox的5%葡萄糖溶液,充分涡旋后使用超声波破壁机处理样品。取100ul上述脂质体(即纳米载药释药系统),在2个小棕瓶中分别加入4mlpbs7.4、pbs5.2释放液,并各设置一组平行组,然后将上述加入pbs释放液的脂质体放入装有样品的透析袋中,间隔一定时间取释放液检测dox的含量,并得到如图10所示的结果数据图。
3.2结果分析。由图10可知,脂质体在ph5.2的pbs液中释放dox的速率远远大于脂质体在ph7.4的pbs液中释放dox的速率。并且,在释放至96小时时,脂质体在ph5.2的pbs液中释放了近54%的dox,而在ph7.4的pbs液中释放了近19%的dox,可见,含有ph响应脂质体的纳米载药释药系统比含有非ph响应脂质体的纳米载药系能释放更多的药物分子,大大提高了药物分子的溶酶体逃逸和细胞器靶向性,对临床医学有很大的促进意义。
以上所述仅为本发明的可选实施例,并非因此限制本发明的专利范围,凡是在本发明的发明构思下,利用本发明说明书及附图内容所作的等效结构变换,或直接/间接运用在其他相关的技术领域均包括在本发明的专利保护范围内。
参考文献:
1.global,regional,andnationalage-sexspecificmortalityfor264causesofdeath,1980–2016:asystematicanalysisfortheglobalburdenofdiseasestudy2016.thelancet,2017.390(10100):p.1151-1210.
2.globalburdenofdiseasecancercollaboration,fitzmauricec,allenc,barberrm,barregardl,bhuttaza,etal.global,regional,andnationalcancerincidence,mortality,yearsoflifelost,yearslivedwithdisability,anddisability-adjustedlife-yearsfor32cancergroups,1990to2015:asystematicanalysisfortheglobalburdenofdiseasestudy.jamaoncol.2017apr1;3(4):524-548.
3.globalburdenofdiseasecancer,c.,global,regional,andnationalcancerincidence,mortality,yearsoflifelost,yearslivedwithdisability,anddisability-adjustedlife-yearsfor32cancergroups,1990to2015:asystematicanalysisfortheglobalburdenofdiseasestudy.jamaoncology,2017.3(4):p.524-548.
4.chen,w.,etal.,cancerstatisticsinchina,2015.ca:acancerjournalforclinicians,2016.66(2):p.115-132.
5.siegel,r.l.,k.d.miller,anda.jemal,cancerstatistics,2017.ca:acancerjournalforclinicians,2017.67(1):p.7-30.
6.allemani,c.,etal.,globalsurveillanceoftrendsincancersurvival2000–14(concord-3):analysisofindividualrecordsfor37513025patientsdiagnosedwithoneof18cancersfrom322population-basedregistriesin71countries.thelancet,2018.391(10125):p.1023-1075.
7.siegel,r.l.,k.d.miller,anda.jemal,cancerstatistics,2018.ca:acancerjournalforclinicians,2018.68(1):p.7-30.
8.pantapasis,k.,etal.,chapter2-bioengineerednanomaterialsforchemotherapy,innanostructuresforcancertherapy.2017,elsevier.p.23-49.
9.narayan,v.andd.vaughn,pharmacokineticandtoxicityconsiderationsintheuseofneoadjuvantchemotherapyforbladdercancer.expertopinionondrugmetabolism&toxicology,2015.11(5):p.731-742.
10.mould,d.r.,etal.,populationpharmacokineticandadverseeventanalysisoftopotecaninpatientswithsolidtumors.clinicalpharmacology&therapeutics,2002.71(5):p.334-348.
11.minev,b.,cancermanagementinman:chemotherapy,biologicaltherapy,hyperthermiaandsupportingmeasures.kindleed.2011,netherlands:springer.
12.schroeder,a.,etal.,treatingmetastaticcancerwithnanotechnology.naturereviewscancer,2011.12:p.39.
13.shi,j.,etal.,cancernanomedicine:progress,challengesandopportunities.naturereviewscancer,2017.17:p.20.
14.matsumoto,y.,etal.,vascularburstsenhancepermeabilityoftumourbloodvesselsandimprovenanoparticledelivery.naturenanotechnology,2016.11:p.533.
15.curry,f.-r.e.,drugdelivery:redefiningtumourvascularbarriers.naturenanotechnology,2016.11:p.494.
16.gottesman,m.m.,t.fojo,ands.e.bates,multidrugresistanceincancer:roleofatp–dependenttransporters.naturereviewscancer,2002.2:p.48.
17.pasut,g.,a.guiotto,andf.m.veronese,protein,peptideandnon-peptidedrugpegylationfortherapeuticapplication.expertopinionontherapeuticpatents,2004.14(6):p.859-894.
18.ferrari,m.,nanogeometry:beyonddrugdelivery.naturenanotechnology,2008.3:p.131.
19.srinivasarao,m.,c.v.galliford,andp.s.low,principlesinthedesignofligand-targetedcancertherapeuticsandimagingagents.naturereviewsdrugdiscovery,2015.14:p.203.
20.peer,d.,etal.,nanocarriersasanemergingplatformforcancertherapy.naturenanotechnology,2007.2:p.751.
21.jiang,w.,etal.,nanoparticle-mediatedcellularresponseissize-dependent.naturenanotechnology,2008.3:p.145.
22.pastan,i.,etal.,immunotoxintherapyofcancer.naturereviewscancer,2006.6:p.559.
23.zhou,j.andj.rossi,aptamersastargetedtherapeutics:currentpotentialandchallenges.naturereviewsdrugdiscovery,2016.16:p.181.
24.kedmi,r.,etal.,amodularplatformfortargetedrnaitherapeutics.naturenanotechnology,2018.13(3):p.214-219.
25.rosenblum,d.,etal.,progressandchallengestowardstargeteddeliveryofcancertherapeutics.naturecommunications,2018.9(1):p.1410.
- 还没有人留言评论。精彩留言会获得点赞!