一种基于生物蛋白质信息网络比对的同源蛋白质检测方法与流程
本发明属于图算法领域,尤其涉及跨物种生物蛋白质信息网络中的同源蛋白质检测方法。
背景技术:
图论在生物系统建模中的应用是一个广阔的研究领域,包括基因组学和蛋白质组学。其中,一个重要方向为利用蛋白质-蛋白质相互作用网络(ppi)对细胞中蛋白质之间的相互作用的集合进行建模,通过比较不同物种的ppi网络,以揭示潜在生物学过程之间的相似性,挖掘物种之间的直系同源蛋白质。直系同源蛋白(orthologs)是指来自于不同物种的由垂直家系(物种形成)进化而来的蛋白,并且通常具有相似的功能。有效挖掘发现不同物种之间的直系同源蛋白,可以帮助人类深刻理解蛋白质之间的同源关系,预测未知的蛋白质功能,对于基因学和医药学的发展具有重要的指导意义。
传统同源蛋白质预测方法往往是仅基于蛋白质的序列进行的,通常假定具有相似序列或相似结构的两种蛋白质具有相似的功能。但是,高序列相似性不一定表明功能保守。由于蛋白质的功能位点通常只是整个序列的一个或几个小部分,因此即使所有功能位点完全不同,两种蛋白质也可能具有非常高的整体序列相似性。仅基于序列的方法会导致预测的同源蛋白质存在很多假阳性。通过生物蛋白质信息网络比对方法,能利用网络结构信息补充仅基于序列的方法,以发现不同物种之间具有高相似度的蛋白质对,再根据生物化学方法验证它们是否是真正具有同源性,就比较有针对性和高效。ppi网络比对产生两个ppi网络节点之间的映射关系,实质上是图论中的子图同构问题,这是一个无法在多项式时间内解决的难题。随着生物蛋白质信息网络规模的扩大,匹配问题面临着更加艰巨的挑战。
蛋白质网络比对分为局部比对算法和全局比对算法两类。局部比对算法采用计算局部网络相似性来进行蛋白质网络匹配,例如pathblast、mawish、alignnemo等,但通常会导致模棱两可的重叠对齐区域,一个物种的蛋白质网络子结构可能会匹配到另一个物种的蛋白质网络中的多个子结构,这可能会误导同源蛋白质对的确认。因此,更多的研究着眼于全局比对算法,例如isorank、l-graal、ghost、netal、magna、spinal、hubalign等。全局比对算法产生一对一的对齐,使得两个ppi网络中所有蛋白质节点都有各自的对应关系。但是现存的全局比对算法共有一个弊端,即匹配结果往往具有较好的拓扑质量,而具有较差的生物功能质量,导致同源蛋白质的预测结果并不理想。
技术实现要素:
本发明针对现有技术的不足,提供一种基于生物蛋白质信息网络比对的同源蛋白质检测方法。
本发明的技术方案为一种基于生物蛋白质信息网络比对的同源蛋白质检测方法,包含以下步骤:
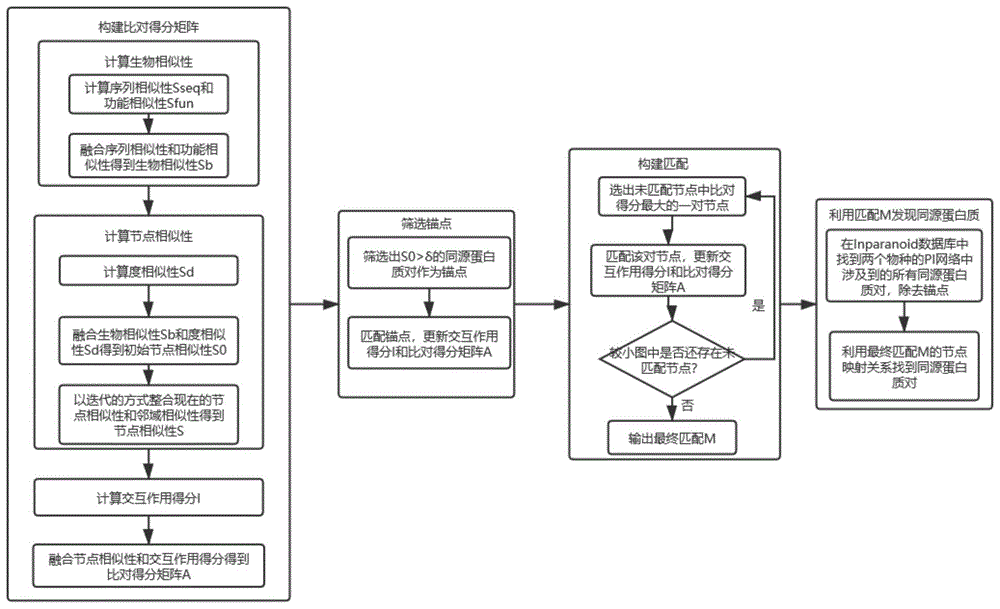
步骤1、构建得分矩阵(scorematrixconstruction)阶段,通过结合蛋白质交互(ppi)网络a和蛋白质交互(ppi)网络b各个节点对之间的生物相似性、拓扑相似性和交互作用信息构建一个初始的比对得分矩阵,具体是:首先结合蛋白质交互(ppi)网络a和蛋白质交互(ppi)网络b各个节点对之间的序列相似性和功能相似性得到生物相似性sb,然后整合生物相似性和蛋白质节点的度和邻域信息构建了节点相似性矩阵s。交互作用得分i由网络拓扑结构而得,反映了节点匹配对交互作用的保守性的影响。融合节点相似性s和交互作用得分i可构建比对得分矩阵a。
步骤2、锚点选取(anchorselection)阶段,通过选择并匹配锚点以提高比对得分矩阵a的置信度,具体是:首先从inparanoid数据库中下载比对物种之间的直系同源蛋白质对,并使用初始节点相似性过滤得到高质量的同源蛋白质对作为锚点并匹配,并将每一对锚点标记为已匹配节点,更新交互作用得分i和比对得分矩阵a,并删除它们在矩阵中的相关行及相关列,使得比对得分矩阵的置信度更高。
步骤3、比对构建(matchingconstruction)阶段,步骤2通过匹配锚点使得比对得分矩阵的置信度a比较高,此时再通过贪心搜索的策略去在两个ppi网络中寻找全局匹配是一种比较有效的做法,具体是:根据更新的比对得分矩阵a,找出两个ppi网络中还未标记为已匹配的节点中比对得分最高的节点对并匹配,也标记为已匹配节点,按照步骤2同样的方式更新i和a。重复上述操作,不断迭代,直到较小网络中的所有节点都在较大网络中找到了一一对应的匹配点。
步骤4、利用步骤3生成的ppi网络a和ppi网络b的匹配结果,结合生物数据库挖掘两个物种间的直系同源蛋白质,具体是:首先在inparanoid数据库中找到ppi网络a和ppi网络b中涉及到的所有同源蛋白质对,去除在步骤2结果中筛选作为锚点的蛋白质对,从步骤3生成的匹配结果可以挖掘到更多存在于inparanoid数据库中的同源蛋白质对。因此,这个一对一的映射关系可以对未来直系同源物的生物学研究提供指导作用。另外,最终的匹配结果同时具有较高的拓扑质量和生物功能质量,解决了现有生物蛋白质信息网络比对算法无法很好平衡这两者的问题。
在上述的基于生物蛋白质信息网络比对的同源蛋白质检测方法,步骤1中通过结合ppi网络a和ppi网络b各对节点之间的生物相似性,拓扑相似性以及交互作用信息来构造一个比对得分矩阵,具体是:
步骤1.1、计算生物相似性sb。为了计算序列相似性,首先通过blast计算出每一对ppi网络a中的节点u和ppi网络b中的节点v的序列分数bitscr(u,v)以及节点自身的序列分数bitscr(u,u)和bitscr(v,v),然后得到序列相似性
步骤1.2、整合生物相似性和蛋白质节点的度和邻域信息得到节点相似性s。假设n(u)和n(v)为u在g1中的邻居节点和v在g2中的邻居节点,则|n(u)|和|n(v)|为u和v的度,那么u和v之间的度相似性为
步骤1.3、计算交互作用得分i。u和v之间的交互作用得分i(u,v),表示两个节点匹配的情况下,与其相连的保守边的数目的估计值。每个节点i对它的任一邻居节点的依赖值都为该节点度的倒数
步骤1.4、融合节点相似性s和交互作用得分i,构建比对得分矩阵a。通过一个参数γ融合节点相似性s和交互作用得分i,构建初始比对得分矩阵a(u,v)=γ·s(u,v)+(1-γ)·i(u,v)。
在上述的基于生物蛋白质信息网络比对的同源蛋白质检测方法,步骤2中,筛选锚点,并通过匹配锚点提高比对得分的置信度,具体是:
步骤2.1、首先在inparanoid数据库中下载跨物种的直系同源蛋白质对,然后筛选出s0(u,v)>δ的同源蛋白质对作为锚点,其中,δ是一个阈值。
步骤2.2、然后匹配每一对锚点,并更新交互作用得分i和比对得分a。用交互作用得分增量矩阵id和交互作用得分减量矩阵ic来记录每匹配一对节点,交互作用得分i的更新。交互作用得分增量id(u,v)表示匹配节点u和v后,确定的保守交互作用数目。假设(i,j)是一对已经匹配的节点,在第k+1步,考虑匹配锚点u和v:若u是i的邻居节点,v是j的邻居节点,匹配u和v会增加一条确定的保守交互作用,第k+1步的交互作用得分增量idk+1(u,v)在第k步idk(u,v)的基础上加1,idk+1(u,v)=idk(u,v)+1;若u不是i的邻居节点,或者v不是j的邻居节点,确定的保守交互作用数目不变,idk+1(u,v)保持不变,idk+1(u,v)=idk(u,v)。但是如果将节点i和另一网络中的j匹配后,应该将i的依赖值从i的所有邻居节点的交互作用得分中减去,同理,节点j也应如此。交互作用得分减量矩阵ic1(u)和ic2(v)分别表示在ppi网络a中的节点u和ppi网络b中的节点v的交互作用得分中需要减去的依赖值。假设在k+1步,确定匹配的节点对为(i,j),则:若u是i的邻居节点,应该减去节点i的依赖值对其邻居u的影响,第k+1步的交互作用得分减量
在上述的基于生物蛋白质信息网络比对的同源蛋白质检测方法,步骤3中,基于比对得分矩阵,构建匹配结果,具体是:
步骤3.1、在步骤2中,已经匹配了所有的锚点对。对于剩余的ppi网络a和ppi网络b中未匹配的节点,采用贪心算法构建匹配。基于更新的比对得分矩阵a,每次选取比对得分最高的一对节点,标记这对节点为已匹配,根据这对匹配节点的信息,按照步骤2.2的描述更新交互作用得分i和比对得分a。
步骤3.2、重复步骤3.1的过程,不断迭代,直到较小网络中的所有节点都在较大网络中找到了一一对应的匹配点,得到了最终的比对结果。
在上述的基于生物蛋白质信息网络比对的同源蛋白质检测方法,步骤4中,利用生成的ppi网络a和ppi网络b的匹配结果,结合生物数据库挖掘两个物种间的直系同源蛋白质,具体是:
步骤4.1、在inparanoid数据库中检索ppi网络a和ppi网络b中涉及到的所有同源蛋白质对,去除在步骤2的结果中匹配的锚点对,用以验证最终匹配结果中蛋白质节点对之间的同源性。
步骤4.2、生成的匹配结果同时具有高拓扑质量和生物功能质量,最终匹配的节点对预测蛋白质之间的同源性具有指导作用,在匹配结果的基础上再进行生物化学验证更具有针对性和高效。
本发明具有以下优点:生物蛋白质信息网络比对很好地融合了生物学信息和网络拓扑信息,能够产生同时具有较高拓扑质量和生物功能质量的匹配结果,从而更有效地发现不同物种之间的直系同源蛋白质对。
附图说明
图1是本发明实施例的基于生物蛋白质信息网络比对的同源蛋白质检测方法流程图。
图2是本发明实施例的样本图数据。
图3是本发明实施例在样本图发现的被inparanoid证实的同源蛋白质对。
具体实施方式
本发明主要基于一种新提出的生物蛋白质信息网络比对算法,找到不同物种蛋白质交互网络之间的节点匹配关系,根据匹配结果在inparanoid数据库中进行查询,找到物种间的同源蛋白质对。通过本发明,我们提供了一种新的检测不同物种之间的同源蛋白质对的方法,比传统的方法更有效,且该算法与现有ppi网络比对算法相比,能够更好地平衡匹配结果的拓扑质量和生物功能质量。
本发明提供的方法能够用计算机软件技术实现流程。参见图1,实施例以酵母菌(yeast,生物代号scerevisiae)的蛋白质交互网络(ppi网络)和人类(human,生物代号hsapiens)的蛋白质交互网络(ppi网络)为例,样本图的属性信息参见图2,对本发明的流程进行一个具体的阐述,如下:
步骤1:首先我们通过计算融合序列相似性和功能相似性得到酵母菌和人类蛋白质节点的生物相似性,然后再整合生物相似性和蛋白质节点的度和邻域信息构建了酵母菌和人类的节点相似性矩阵。然后由网络拓扑信息得到两者初始的交互作用得分,融合节点相似性和交互作用得分产生酵母菌和人类的ppi网络中所有节点间的比对得分。
实施例中构建比对得分矩阵的具体实施过程如下:
首先,计算酵母菌蛋白质节点和人类蛋白质节点的序列相似性sseq和功能相似性sfun,并融合为生物相似性sb。计算序列相似性的方法为:通过blast计算出所有酵母菌ppi网络中的节点u和人类ppi网络中的节点v之间的序列分数bitscr(u,v),以及节点本身的序列分数bitscr(u,u)和bitscr(v,v),然后根据发明内容提到的
然后,整合生物相似性和蛋白质节点的度和邻域信息得到节点相似性s。计算度相似性的方法为:计算得到酵母菌ppi网络中的u和人类ppi网络中的v的邻居节点分别为n(u)和n(v),它们的度分别为|n(u)|和|n(v)|,然后由
其次,计算交互作用得分i。计算交互作用得分的方法为:计算得到酵母菌ppi网络中u的邻居节点u′的依赖值
最终,融合节点相似性s和交互作用得分i得到酵母菌ppi网络和人类ppi网络的比对得分矩阵a。融合节点相似性s和交互作用得分i构建比对得分矩阵的方法为:a(u,v)=γ·s(u,v)+(1-γ)·i(u,v),其中,γ的值在[0,1]之间可调节,这里选取为0.001。
步骤2:在直系同源蛋白质对中根据初始节点相似性筛选出锚点,匹配锚点对,更新交互作用得分i和比对得分矩阵a,提高比对得分矩阵的置信度。
实施例中筛选锚点,更新交互作用得分和比对得分的具体实施过程如下:
首先,筛选锚点。在inparanoid数据库中下载所有的酵母菌和人类的直系同源蛋白质对,然后筛选出s0(u,v)>δ的同源蛋白质对作为锚点,其中,δ是一个值在[0,1]之间的阈值,这里选取为0.7。
然后,根据匹配锚点更新交互作用得分i。更新交互作用得分的方法为:用交互作用得分增量矩阵id和交互作用得分减量矩阵ic来记录更新。假设(i,j)是一对已经匹配的节点,在第k+1步,考虑匹配锚点u和v:若u是i的邻居节点,v是j的邻居节点,idk+1(i,j)=idk(i,j)+1;若u不是i的邻居节点,或者v不是j的邻居节点,idk+1(i,j)=idk(i,j)。假设在第k+1步,确定匹配的节点对为(i,j),则:若u是i的邻居节点,
最终,更新比对得分矩阵a。更新比对得分的方法为:a(u,v)=γ·s(u,v)+(1-γ)·i(u,v),其中,γ的值在[0,1]之间可调节,这里选取为0.001。
步骤3:通过贪心搜索的策略构建全局匹配。根据比对得分矩阵a,找出酵母菌和人类ppi网络中未匹配节点中比对得分最高的节点对并匹配,按照步骤2的方式更新i和a。重复上述操作,不断迭代,直到酵母菌ppi网络中的所有节点都在人类ppi网络中找到了一一对应的匹配点。
步骤4:在inparanoid数据库中查询酵母菌ppi网络和人类ppi网络中涉及到的所有同源蛋白质对(orthologs),除去作为锚点的部分,然后根据最终匹配查询发现的同源蛋白质对。图3给出了匹配结果中发现的被inparanoid数据库证实的同源蛋白质对,该算法在酵母菌和人类ppi网络中找到14对同源蛋白质。该结果说明本发明提出的生物蛋白质信息网络比对算法可以有效地发现人类和酵母菌中的同源蛋白质对,该结果对预测未知功能蛋白具有指导意义。
本文中所描述的具体实施例仅仅是对本发明精神作举例说明。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或者超越所附权利要求书所定义的范围。
- 还没有人留言评论。精彩留言会获得点赞!