一种杂原子微介孔复合材料及其合成方法与流程
本发明涉及一种合成杂原子微介孔复合材料的方法;更进一步说涉及一种含选自Fe、Sn、Zr、Ge、V、Cr、B、Mn的骨架杂原子和硅的分子筛。
背景技术:
杂原子硅分子筛(杂原子分子筛)的研究起源于上世纪七八十年代,目前已合成出多种杂原子分子筛,例如MFI型结构的TS-1、Fe-ZSM-5和Sn-ZSM-5分子筛等,MEL型结构的TS-2、FeS-2、CrS-2分子筛,MWW型结构的Ti-MCM-22、B-MWW,具有较大孔结构的TS-48分子筛。
1983年Taramasso在专利US 4410501中首次报道了水热晶化法合成钛硅分子筛的方法。该法是合成TS-1的经典方法,主要分制胶和晶化两步进行,合成过程如下:将正硅酸乙酯(TEOS)放入氮气保护无CO2的容器中,缓慢加入TPAOH(模板剂),然后慢慢滴加钛酸四乙酯(TEOT),搅拌lh,制得一种含有硅源、钛源和有机碱的反应混合物,加热,除醇,补水,175℃在自生压力釜下搅拌下,晶化10天,然后分离、洗涤、干燥、焙烧而得TS-1分子筛。然而该工艺中钛插入骨架过程影响因素众多,水解和成核的条件均不易控制,因此该法合成的TS-1分子筛存在催化活性低、稳定性差、难于合成和重现等弊端。
CN102757066A公开了一种合成B-β杂原子分子筛的方法,该方法按比例将硼源、模板剂、碱源、氟化物和水在室温下机械搅拌,待固体全溶,加入晶种和硅源形成初始凝胶,搅拌一定时间后装釜密封,在120℃~170℃下晶化2~5天,水热合成具有BEA结构的B-β杂原子分子筛。
中国专利CN98101357.0(CN1260241A)公开了钛硅分子筛重排技术,合成了具有独特空心结构的新型钛硅分子筛,不仅使合成TS-1的重现性大大增强,还增加了分子筛孔体积,大大提高了反应物分子在分子筛孔道中的传质扩散速率,催化性能增加。该专利公开的方法 将钛的水解溶液与已经合成出的TS-1分子筛按照分子筛(克):Ti(摩尔)=200~1500:1的比例混合均匀,将所得混合物在反应釜中与120~200℃下反应1~8天,过滤、洗涤并干燥。目前,HTS分子筛应用于催化氧化苯酚羟基化、环己酮氨肟化等过程已经实现工业化,具有反应条件温和、原子利用率高、工艺过程简单和副产物为水清洁高效等优点。
以上现有方法合成的杂原子硅分子筛主要以微孔为主,介孔体积不高。
技术实现要素:
本发明要解决的技术问题是提供一种杂原子微介孔复合材料(也称为杂原子硅微介孔复合分子筛、杂原子硅微介孔分子筛、杂原子硅微介孔复合材料或杂原子硅微介孔材料),该杂原子微介孔复合材料具有微孔和介孔复合结构,本发明要解决的另外技术问题是提供一种所述杂原子微介孔复合材料的合成方法。
本发明提供一种杂原子微介孔复合材料的合成方法,包括如下步骤:
(1)将杂原子源、模板剂、有机硅源、水和任选的无机铵源混合,水解赶醇;所述的模板剂包括有机季铵化合物和长链烷基铵化合物以及任选的有机胺;所述杂原子记为M,选自Fe、Co、Cu、Sn、Zr、B、Ge、V、Cr、Mn中的一种或多种;
(2)将步骤(1)所得产物于室温~50℃下老化;
(3)将步骤(2)所得到的老化产物与固体硅源混合均匀,然后在密闭反应釜中晶化,回收杂原子微介孔复合材料。
本发明提供的杂原子微介孔复合材料合成方法,优选包括以下步骤:
(1)将模板剂、杂原子源、有机硅源、任选的无机铵源和水混合,水解赶醇;所述水解赶醇通常在0~150℃例如0~100℃优选50~95℃下将所得到的混合物搅拌至少10分钟;所述搅拌的搅拌时间例如搅拌10分钟~50小时;其中,无机铵源(以NH4+计):杂原子源(以杂原子计)的摩尔比为0~5:1;
(2)将步骤(1)所得产物老化,所述老化为将步骤(1)所得产物于室温~50℃下静置1~60小时例如2~50小时或3~30小时,进一步例如3~15小时;
(3)将步骤(2)所得到的老化产物与固体硅源按照1:0.1~10的重量比混合均匀,然后在密闭反应釜中晶化,回收杂原子微介孔复合材料;其中所述的重量比例中,步骤(2)所得到的老化产物和固体硅源均以SiO2计;
其中,杂原子源与总硅源的摩尔比为0.005~0.05:1、水与总硅源的摩尔比为5~100:1;模板剂与总硅源的摩尔比为0.08~0.6:1例如为0.1~0.3:1;其中,所述的摩尔比中,总硅源以SiO2计,所述总硅源为以SiO2计的有机硅源和以SiO2计的固体硅源的总和,无机铵源以NH4+计,杂原子源以MO2计,水以H2O计;所述的无机铵源为无机铵盐和/或氨水。
本发明提供的杂原子微介孔复合材料合成方法,杂原子源与总硅源的摩尔比优选为0.01~0.05:1。步骤(3)中所述步骤(2)所得到的老化产物与固体硅源的摩尔比为1:0.1~10;其中所述的摩尔比中,所述步骤(2)所得到的老化产物以SiO2计,固体硅源以SiO2计。所述的模板剂为有机季铵化合物、长链烷基铵化合物和任选的有机胺,有机季铵化合物与总硅源的摩尔比为0.04~0.45:1,长链烷基铵化合物与总硅源的摩尔比为0.04~0.45:1,有机胺与总硅源的摩尔比为0~0.40:1。
本发明还提供一种杂原子微介孔复合材料,该杂原子微介孔复合材料具有以下特征:所述杂原子微介孔复合材料晶粒表面富硅,晶粒表面硅杂原子摩尔比与体相硅杂原子摩尔比的比值大于1.1,例如为1.1~5。其表面硅杂原子比与体相硅杂原子比的比值例如为1.2~4:1。
其中,表面硅杂原子比可采用TEM-EDX或离子激发腐蚀XPS方法测定得到,为距离晶粒表面不超过5nm例如1~5nm的原子层的硅杂原子比,体相硅杂原子比可通过化学分析的方法得到,或通过TEM-EDX在晶粒的中心区域例如距离晶粒表面距离大于20nm的区域测量得到,或通过XRF方法测量得到。
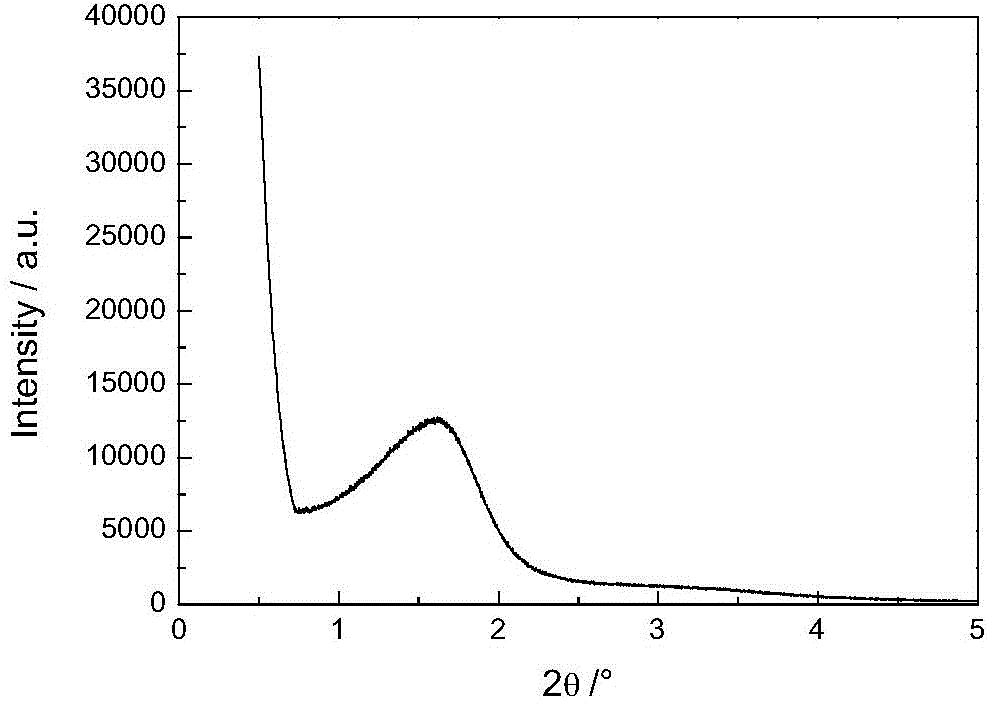
所述杂原子微介孔复合材料(也称杂原子微介孔分子筛),具有 微孔结构,还具有介孔结构,所述微孔的孔径小于1nm,所述介孔的孔径(直径)在2-8nm之间。所述杂原子微介孔复合分子筛XRD谱图中2θ角为0~3°和5~35°具有衍射峰。分子筛的XRD谱图在2θ角为5~35度具有衍射峰,表明分子筛中存在微孔结构;分子筛的2θ角为0~3°具有衍射峰,表明分子筛中存在介孔结构。
本发明所述杂原子微介孔复合材料,孔径小于1nm的微孔的体积即微孔体积为0.12~0.19mL/g,孔径为2-8nm介孔的体积即介孔体积为0.3~0.8mL/g。
本发明提供的杂原子微介孔复合材料合成方法,所制备的杂原子微介孔复合材料晶粒表面富硅,晶粒表面硅杂原子比明显高于体相硅杂原子比。此外,本发明提供的杂原子微介孔复合材料合成方法,使用相对廉价易得的固体硅源例如高纯度硅胶或/和白炭黑,部分代替价格昂贵有机硅源,能够降低分子筛生产过程的废物排放和节约原料成本的同时得到高性能的杂原子微介孔复合材料,所制备的分子筛具有更高的氧化活性。本发明提供的杂原子微介孔复合材料合成方法,可以在较低的模板剂用量和较低的水硅比情况下合成杂原子微介孔复合材料,可以降低杂原子微介孔复合材料的合成成本,提高合成分子筛晶化产物的固含量,提高单釜分子筛产量。
本发明提供的杂原子微介孔复合材料(也称杂原子硅微介孔复合分子筛),具有更高的表面硅与杂原子比和体相硅与杂原子比之比,具有更高的活性,用于双氧水参与的氧化反应,可以减少表面层中杂原子对双氧水的分解,有利于降低双氧水的无效分解副反应的活性,提高原料利用率。
附图说明
图1为实施例4的步骤(3)制备的含锡杂原子微介孔复合材料的XRD谱图(5-35°)。
图2为实施例4的步骤(3)制备的含锡杂原子微介孔复合材料的XRD谱图(0-5°)。
图3为实施例4的步骤(4)制备的含锡杂原子微介孔复合材料的TEM照片。
图4为利用TEM-EDX测量体相硅杂原子比和表面硅杂原子比的示意图,其中方框1示意测量颗粒边缘区域的硅杂原子比,方框2示意测量颗粒中心区域的硅杂原子比。由于颗粒边缘区域单位体积具有更高的外表面积,而中心区域的单位体积内对应外表面积较低,因此方框1和方框2内EDX测量结果可以反映体相与表面的硅杂原子比差异。
图5为含锡杂原子微介孔复合材料的低温N2吸附-脱附测得的孔分布曲线,表明所述材料中有介孔存在。
具体实施方式
本发明提供的杂原子微介孔复合材料的合成方法,可以在较低的模版剂用量情况下合成杂原子微介孔复合材料,例如模版剂与总硅源的摩尔比为0.1~0.3:1,进一步为0.1~0.25:1;本发明提供的方法中,可以在高固含量下合成杂原子微介孔复合材料,从可而减少水的使用量,提高单釜产量即在同样的合成反应器体积下合成更多的分子筛,因此所述的水与总硅源(以二氧化硅计)的摩尔比可以为5~80:1或5~50:1或5~30:1或6~20或6~15:1。
本发明提供的杂原子微介孔复合材料的合成方法,所述的杂原子源与总硅源的摩尔比为0.005~0.05:1例如为0.01~0.03:1进一步例如为0.01~0.025:1。
本发明提供的杂原子微介孔复合材料的合成方法,无机铵源与杂原子源的摩尔比为0~5:1例如为0.01~4:1优选为0.01~0.5:1。加入无机铵源,可以提高所合成分子筛的氧化活性,可以提高杂原子源的利用率(可以在同样的杂原子源使用量情况下具有更高的骨架杂原子硅比),降低杂原子源的使用量。
本发明提供的杂原子微介孔复合材料的合成方法,所述的模板剂与所述的总硅源的摩尔比不低于0.08:1,例如为0.08~0.6:1优选为0.1~0.3:1,例如为0.1~0.2:1。
本发明提供的杂原子微介孔复合材料的合成方法,所述有机硅源和固体硅源的摩尔比为1:0.1~10优选为1:1~9例如为1:2~8或者为1:3~7。以SiO2计的步骤(2)所得到的老化产物与固体硅源之比即等 于有机硅源和固体硅源的摩尔比。所述的固体硅源为无机硅源。
本发明提供的杂原子微介孔复合材料的合成方法,步骤(1)所述的模版剂包括有机季铵化合物和长链烷基铵化合物,可以任选还含有有机胺化合物,有机季铵化合物与总硅源的摩尔比为0.04~0.45:1,长链烷基铵化合物与总硅源的摩尔比为0.04~0.45:1,有机胺与总硅源的摩尔比为0~0.4:1。所述的有机季铵化合物例如为有机季胺碱和/或有机季铵盐。所述的有机胺为脂肪胺、芳香胺和醇胺中的一种或多种,所述的脂肪胺(也称脂肪胺类化合物),其通式为R3(NH2)n,其中R3为具有1~4个碳原子的烷基或者亚烷基,n=1或2;所述的醇胺(本发明也称醇胺类化合物)其通式为(HOR4)mNH(3-m),其中R4为具有1~4个碳原子的烷基,m=1、2或3。所述的脂肪胺例如乙胺、正丁胺、丁二胺或己二胺中的一种或多种;所述的芳香胺是指具有一个芳香性取代基的胺,例如苯胺、甲苯胺、对苯二胺中的一种或多种;所述的醇胺例如单乙醇胺、二乙醇胺或三乙醇胺中的一种或多种。所述的有机季铵碱例如四丙基氢氧化铵、四丁基氢氧化铵或四乙基氢氧化铵中的一种或多种;所述的有机季铵盐例如四丙基溴化铵、四丁基溴化铵、四乙基溴化铵、四丙基氯化铵、四丁基氯化铵或四乙基氯化铵中的一种或多种。
本发明提供的杂原子微介孔复合材料的合成方法,所述长链烷基铵化合物其通式为R5NH3X或者R5N(R6)3X,其中R5为碳原子数在12~18之间的烷基,R6为碳原子数在1~4之间的烷基;X为一价阴离子例如为OH-、Cl-、Br-;当X为OH-时,本发明称为碱式长链烷基铵化合物;所述的有机硅源为有机硅酯,所述的有机硅酯,其通式为Si(OR1)4,R1选自具有1~6个碳原子的烷基,所述的烷基是支链或直链烷基。所述的有机季铵碱例如四丙基氢氧化铵、四丁基氢氧化铵或四乙基氢氧化铵中的一种或多种;所述的有机季铵盐例如四丙基溴化铵、四丁基溴化铵、四乙基溴化铵、四丙基氯化铵、四丁基氯化铵或四乙基氯化铵中的一种或多种;所述的长链烷基铵化合物例如十六烷基三甲基溴化铵、十六烷基氯化铵、十六烷基三甲基氢氧化铵、长链烷基铵化合物为十四烷基三甲基溴化铵、十四烷基氯化铵、十四 烷基三甲基氢氧化铵、CTMAB(十六烷基三甲基溴化铵)、十二烷基三甲基溴化铵、十二烷基氯化铵、十二烷基三甲基氢氧化铵、十八烷基三甲基溴化铵、十八烷基氯化铵、十八烷基三甲基氢氧化铵中的一种或多种。
本发明提供的杂原子微介孔复合材料的合成方法,一种具体实施方式,所述的杂原子微介孔复合材料具有MFI结构,所述的有机季铵化合物包括四丙基氢氧化铵、四丙基氯化铵、四丙基溴化铵中的一种或多种。四丙基氢氧化铵、四丙基氯化铵、四丙基溴化铵中的一种或多种与总硅源的摩尔比不低于0.01:1,优选为0.02~0.45:1。
本发明提供的杂原子微介孔复合材料的合成方法,一种实施方式所述的杂原子微介孔复合材料具有MEL结构,所述的有机季铵化合物包括四丁基氢氧化铵、四丁基溴化铵或四丁基氯化铵中的一种或多种。四丁基氢氧化铵、四丁基溴化铵或四丁基氯化铵中的一种或多种的总和与与总硅源的摩尔比不低于0.01:1,优选为0.02~0.45:1。
本发明提供的杂原子微介孔复合材料的合成方法,一种实施方式,所述的杂原子微介孔复合材料具有BEA结构,所述的有机季铵化合物包括四乙基氢氧化铵、四乙基溴化铵、四乙基氯化铵中的一种或多种。所述的有机季铵化合物包括四乙基氢氧化铵、四乙基溴化铵、四乙基氯化铵中的一种或多种的总和与总硅源的摩尔比不低于0.01:1,优选为0.02~0.45:1。
本发明提供的杂原子微介孔复合分子筛的合成方法,有机季铵化合物与总硅源的摩尔比为0.05~0.45:1,长链有机胺化合物与总硅源的摩尔比为0.05~0.45:1。优选的情况下,所述的模板剂至少包括一种有机碱,所述的有机碱为有机季铵碱、有机胺、碱式长链烷基铵化合物中一种或多种,所述的模版剂中的有机碱与总硅源的摩尔比为0.04~0.5:1,例如0.05~0.45:1。
本发明提供的杂原子微介孔复合材料合成方法,步骤(1)中所述的有机硅源为有机硅酯,所述的有机硅酯,其通式为Si(OR1)4,R1选自具有1~6个碳原子的烷基例如R1为C1-C4的烷基,所述的烷基可以是支链烷基或直链烷基。所述的有机硅酯例如硅酸四甲脂、硅酸四 乙酯、硅酸四丁酯、二甲基二乙基硅酯中的一种或多种;其中优选硅酸四甲酯、硅酸四乙酯、二甲基二乙基硅酯中的一种或多种。本发明所说的固体硅源为高纯度的二氧化硅固体或者粉末,例如可以是白炭黑和/或者高纯度硅胶。优选情况下,以干基重量为基准所述固体硅源中SiO2含量不低于99.99重量%,且Fe、Al和Na杂质的总质量含量小于10ppm;例如SiO2含量为99.99~100重量%,通常为大于99.99且小于100重量%。所述的固体硅源可以是高纯度硅胶和/或白炭黑,优选白炭黑,其中所述硅胶中SiO2含量优选大于等于99.99重量%例如为大于99.99重量%且小于100重量%,且Fe、Al和Na杂质的质量含量小于10ppm。所述白炭黑的比表面积优选介于40~1000m2/g例如50-400m2/g之间,以白炭黑的干基重量为基准,所述白炭黑中SiO2含量优选大于等于99.99重量%例如为99.99~100重量%例如为大于99.99重量%且小于100重量%,所述白炭黑中Fe、Al和Na杂质的总质量含量小于10ppm。所述白炭黑可以商购,或者按照现有方法制备,例如按照专利CN200910227646.2提供的方法制备,一种制备方法是将四氯化硅与氢气和氧气发生燃烧反应得到。
所述的杂原子源为杂原子的有机化合物或杂原子的无机化合物,例如杂原子为金属时,所说杂原子源可以为无机金属盐或者有机金属酸酯。优选的,所述杂原子源为能够溶于水或与水反应发生水解反应产生杂原子的氧化物的含所述杂原子的化合物。所述的有机化合物例如杂原子的有机酸盐、醇盐、烷基化合物、苯基化合物、苄基化合物、羰基化合物、羧基化合物、醇化合物、烯基化合物、烷氧基化合物、有机卤化物、酰基化合物、四烷氧基杂原子酸酯(如M(alkoxy)4其中M代表所述的杂原子)中的一种或多种;杂原子的无机化合物可以是含有所述杂原子的无机盐或酸,例如杂原子的氯化物、杂原子的硫酸盐、杂原子的硝酸盐、杂原子的氢氧化物、杂原子的氧氯化物、杂原子的氧化物、杂原子的酸中的一种或多种,优选的,所述杂原子的无机盐或酸能够溶于水或与水反应形成水解产物。优选的,所述的杂原子源为四烷氧基杂原子酸酯M(alkoxy)4、杂原子的烷基化合物、四烷基杂原子化合物、杂原子的醇化合物、杂原子的羧基化合物、杂原子 的氯化物、杂原子的硫酸盐、杂原子的硝酸盐、杂原子的醋酸盐、杂原子的酸以及它们的水解产物中的一种或多种,其中四烷氧基杂原子酸酯分子中的烷氧基的碳原子数为1~6,例如碳原子数为1、2、3、4、5或6个。。杂原子源与总硅源(简称硅源)的摩尔比例如为0.008~0.035:1例如为0.01~0.03:1或0.01~0.025:1或0.015~0.025:1。本发明提供的方法可以使用无机杂原子源,与使用有机杂原子源相比,可以降低合成成本。所述杂原子M为Fe、Co、Cu、Sn、Zr、B、Ge、V、Cr、Mn中的一种或多种。
铁的有机化合物例如:醋酸亚铁、丙烯酸铁、乙醇铁、异丙氧铁、草酸高铁铵等中的一种或多种,铁的无机化合物例如氯化铁、氯化亚铁、氧化铁、氧化亚铁、五水硫酸铁等中的一种或多种。
锡的有机化合物例如:三丁基氯化锡、双乙酰丙酮基二丁基锡、三辛基氯化锡、三丁基乙烯基锡、三正丁基氢锡、四乙烯基锡、丙烯基三苯基锡、三正丁基溴化锡、二丁基二氯化锡、烯丙基三丁基锡、三甲基氯化锡、二甲基二氯化锡、四苯基锡、四甲基锡、三正丁基甲氧基锡、丁基三氯化锡、四烯丙基锡、三苯基氯化锡、四异丙基锡、甲基丙烯酸三丁基锡、四丁基锡、四乙基锡、乙醇锡、乙酸亚锡、二苯基二氯化锡、异丙氧基锡中的一种或多种;锡的无机化合物例如SnCl4、SnCl4·5H2O、SnCl2、SnCl2·2H2O、SnSO4中的一种或多种。
锆的有机化合物例如:异丙醇锆、四苄基锆、锆酸四丁酯、乙酰丙酮锆、四甲基丙稀酸锆、正丙醇锆、醋酸锆、叔丁醇锆、四乙氧基锆中的一种或多种;锆的无机化合物例如:氧氯化锆、氢氧化锆、硝酸锆、四氯化锆、Zr(SO4)2·4H2O中的一种或多种。
锗的有机化合物例如:苄基三氯化锗、乙基三氯化锗、三氯甲基锗、四正丁氧基锗烷、三丁基氯化锗、四乙基锗、三甲基氯化锗、三丁基乙烯锗、四正丁基锗、四甲基锗、三甲基氯化锗、三乙基氯化锗、四乙氧基锗、二氯二甲基锗、甲氧基锗、异丙醇锗、异丁基锗烷中的一种或多种;锗的无机化合物例如:四氯化锗、二氧化锗中的一种或多种。
钒的有机化合物例如:氧化二乙酰丙酮合钒、乙酰丙酮钒、三异 丙氧基氧化钒、三丙醇氧化钒、氧化三乙基钒中的一种或多种;钒的无机化合物例如:四氯化钒、三氯氧钒、硫酸氧钒、草酸氧钒、正钒酸钠、偏钒酸钠、偏钒酸铵、偏钒酸钾中的一种或多种。
铬的有机化合物例如:乙酰丙酮铬、苯甲酰丙酮铬、异丙醇铬中的一种或多种;铬的无机化合物例如六水氯化铬、Cr(NO3)3·9H2O中的一种或多种。
硼的有机化合物例如:三烷基硼化合物如三甲基硼、三乙基硼、三丙基硼、三丁基硼中的一种或多种;硼的无机化合物例如硼酸、偏硼酸、三氧化二硼中的一种或多种。
锰的有机化合物例如:乙酰丙酮锰、乙酸锰、二水合乙酸锰、四水合乙酸锰中的一种或多种;锰的无机化合物例如MnSO4·7H2O、MnCl2·4H2O、Mn(NO3)2·6H2O,Mn(C104)2·6H2O中的一种或多种。
本发明提供的杂原子微介孔复合材料合成方法,步骤(1)中所述的无机铵源为无机铵盐和/或氨水,所述的无机铵盐例如氯化铵、硝酸铵、硫酸铵中得到一种或多种。所述的无机铵源优选为氨水,以NH4+计的氨水与以杂原子(M)计的杂原子源的摩尔比为0~5:1,例如为0.01~4:1,例如为0.01~0.5:1。加入所述无机季铵盐,可以提高所合成分子筛的骨架杂原子的含量,提高分子筛的活性。
本发明提供的杂原子微介孔复合材料合成方法中,步骤(1)中将杂原子源、模板剂、有机硅源、无机铵源和水按混合,进行水解赶醇。所述水解赶醇,为在0~150℃优选0~100℃例如50~95℃搅拌至少10分钟,以使有机硅源和杂原子源水解,并降低所得混合物中有机硅源和有机杂原子源水解产生的醇(通常为一元醇)含量,即进行水解赶醇。通常搅拌时间为10~3000分钟,例如为2~30小时。通过水解赶醇,得到澄清透明的有机硅源和杂原子源水解液。优选情况下,步骤(1)得到的产物中有机硅源和有机杂原子源水解产生的一元醇的质量含量不超过10ppm,优选步骤(1)得到的混合物中一元醇的含量不高于10ppm(质量)。
本发明提供的杂原子微介孔复合材料合成方法,步骤(2)中,将步骤(1)所得产物老化,所述老化为在室温至50℃下将步骤(1) 所得产物静置1~60小时。所述室温为15~40℃;老化时间为1~60小时例如为2~50小时,优选3~30小时,例如3~15小时,老化过程中不进行搅拌,将所述物料即步骤(1)所得产物静置。
本发明提供的杂原子微介孔复合材料合成方法,步骤(3)中将步骤(2)得到的老化产物与固体硅源混合,以SiO2计,步骤(2)得到的产物与固体硅源的摩尔比为1:0.1~10(即所述有机硅源和固体硅源的摩尔比为1:0.1~10,例如可以是1:1~9、1:2~8、1:1~7或1:3~6。本发明提供的方法,可以使用较高比例的固体硅源,可以提高合成产物的固含量,从而在合成反应釜不变的情况下提高单次合成的产量。
本发明提供的杂原子微介孔复合材料合成方法,步骤(3)所述晶化,晶化的温度为110~200℃,晶化压力为自生压力,晶化的时间为2小时~20天,通常所述晶化的时间为0.5~20天,例如晶化时间为0.5~10天,进一步步骤(3)所述的晶化的温度为140~180℃例如为160~180℃,晶化时间优选为0.5~10天例如1~6天,进一步例如为1~3天。晶化压力为自生压力。所述晶化可以在不锈钢搅拌釜中进行。晶化升温可以一段升温也可以多段升温方式。升温速率可按照现有晶化升温方法进行,例如为0.5-1℃/min。所述晶化可以在不锈钢搅拌釜中进行。一种实施方式,所述晶化的晶化温度为160~180℃,晶化时间为0.5~6天例如1~3天,晶化压力为自生压力。一种实施方式,步骤(3)所述的晶化为:在100~130℃例如110~130℃下晶化0.5~1.5天,然后在160~180℃下晶化1~3天,晶化压力为自生压力。
本发明提供的杂原子微介孔复合材料合成方法,步骤(3)中所述回收杂原子微介孔复合材料为现有方法,包括将晶化产物过滤、洗涤和焙烧或者将晶化产物过滤、洗涤、干燥然后焙烧。过滤的目的为将晶化得到的杂原子微介孔复合材料与晶化母液分离,洗涤的目的是洗去吸附在分子筛颗粒表面的含硅的模板剂,例如可以在温度为室温~50℃,分子筛与水的重量比1:1~20例如1:(1-15)下进行混合洗涤然后过滤或用水淋洗。干燥的目的是除去分子筛中的大部分水分,以降低焙烧时候的水分蒸发量,干燥的温度可以为100~200℃。焙烧的 目的是除去分子筛中的模板剂,例如所述的焙烧的温度为350~650℃,焙烧时间为2-10小时。通过回收得到本发明所提供的杂原子微介孔复合材料产品。
本发明提供的杂原子微介孔复合材料合成方法中,步骤(3)回收得到的杂原子微介孔复合材料还可经过进一步处理,即本发明提供的杂原子微介孔复合材料合成方法,还可以包括步骤(4):
(4)将步骤(3)得到的杂原子微介孔复合材料在有机碱溶液中晶化处理,然后回收杂原子微介孔复合材料。该过程所得到的杂原子微介孔复合材料具有空心结构,本发明称之为杂原子微介孔复合材料重排(也称为分子筛重排)。其中杂原子微介孔复合材料(以SiO2计)与有机碱的摩尔比例为1:0.02~0.5例如为1:0.02~0.2;以SiO2计的杂原子微介孔复合材料与水的摩尔比为1:2~50例如为1:2~30或者为1:2~20,或者为1:5~10;晶化温度为120~200℃,时间为0.5~10天例如0.5~8天;晶化压力为自生压力,其中所述的有机碱优选有机季铵碱。优选,步骤(4)所述的晶化温度为150-200℃,晶化时间为0.5~10天或为1~6天,杂原子微介孔复合材料与水的摩尔比为1:2~30。回收方法为现有方法,通常包括将晶化产物过滤、洗涤、干燥然和焙烧,可参照步骤(3)所述的回收方法。所述的有机碱为有机胺和/或有机季铵碱;所述的有机胺为脂肪胺、芳香胺和醇胺中的一种或多种,所述的脂肪胺(也称脂肪胺类化合物),其通式为R3(NH2)n,其中R3为具有1~4个碳原子的烷基或者亚烷基,n=1或2;所述的醇胺(本发明也称醇胺类化合物)其通式为(HOR4)mNH(3-m),其中R4为具有1~4个碳原子的烷基,m=1、2或3。所述的脂肪胺例如乙胺、正丁胺、丁二胺或己二胺中的一种或多种;所述的芳香胺是指具有一个芳香性取代基的胺,例如苯胺、甲苯胺、对苯二胺中的一种或多种;所述的醇胺例如单乙醇胺、二乙醇胺或三乙醇胺中的一种或多种。所述的有机季铵碱例如四丙基氢氧化铵、四丁基氢氧化铵或四乙基氢氧化铵中的一种或多种。一种实施方式,步骤(4),所述的杂原子微介孔复合材料具有MFI结构,所述的有机季铵碱为四丙基氢氧化铵。一种实施方式,所述的杂原子微介孔复合材料具有MEL结构,步骤(4)所述的 有机季铵碱为四丁基氢氧化铵。一种实施方式,所述的杂原子微介孔复合材料具有BEA结构,步骤(4)所述的有机季铵碱为四乙基氢氧化铵。
步骤(4)本发明称之为杂原子微介孔复合材料重排,此过程可以进行一次,也可以重复一次或多次,所述重复,即将所述处理得到的杂原子微介孔复合材料代替步骤(3)得到的杂原子微介孔复合材料进行步骤(4)的处理。通过重排处理,可以得到具有二次孔结构的杂原子微介孔复合材料,所得杂原子微介孔复合材料具有空心结构即所述杂原子微介孔复合材料的晶粒为空心结构,该空心晶粒的空腔部分的径向长度为5~300nm,在25℃,P/P0=0.10,吸附时间1小时的条件下测得的苯吸附量为至少70毫克/克,该分子筛的低温氮吸附的吸附等温线和脱附等温线之间存在滞后环。重排后杂原子微介孔复合材料具有更大的孔体积和比表面积。
下面的实施例将对本发明作进一步的说明,但并不因此限制本发明。
实施例中的晶粒大小和表面硅杂原子比和体相硅杂原子比的测量方法采用TEM-EDX,TEM电镜实验在FEI公司Tecnai F20 G2S-TWIN型透射电子显微镜上进行,配有Gatan公司的能量过滤系统GIF2001,附件配备X射线能谱仪。电镜样品采用悬浮分散的方法制备在直径3mm的微栅上。实施例中每个样品随机选取20个颗粒测量其表面硅杂原子比和体相硅杂原子比,计算表面硅杂原子比和体相硅杂原子比的比值,然后取所述20个颗粒的平均值作为所述样品表面硅杂原子比和体相硅杂原子比的比值。
XRD测量方法:在Siemens D5005型X-射线衍射仪上进行样品的X-射线衍射(XRD)晶相图测定,射线源为CuKα管电压40kV,管电流40mA,扫描速度0.5°/min,扫描范围2θ=4°~40°。
BET比表面积和孔体积的测试方法采用氮吸附容量法,按照BJH计算方法。(参见石油化工分析方法(RIPP试验方法),RIPP151-90,科学出版社,1990年出版)
实施例和对比例中所用原料性质如下:
四丙基氢氧化铵,广东大有化工厂。
硅酸四乙酯,分析纯,国药集团化学试剂有限公司。
氨水,分析纯,浓度20重量%。
白炭黑,浙江巨化集团产品,型号AS-150;固含量大于95重量%,干基中二氧化硅含量大于99.99重量%,铁、钠和Al的总含量小于10ppm,比表面积为195m2/g。
其它试剂未经进一步说明的,均为市售商品,分析纯。
实施例1
(1)将浓度为20重量%的四丙基氢氧化铵(TPAOH)水溶液、十六烷基三甲基溴化铵(CTMAB)、五水硫酸铁、硅酸四乙酯(TEOS)、浓度为20重量%的氨水和水依次加入到烧杯中,放入带有加热和搅拌功能的磁力搅拌器上混合均匀,并在60℃下搅拌3小时,随时补充蒸发的水分,得到无色透明水解液;
(2)将所得水解液在一定温度(老化温度)下静置12小时进行老化,得到老化产物;
(3)在步骤(2)得到的老化产物中,搅拌下加入白炭黑粉末,加完后搅拌1小时混合均匀,形成一种“粘稠体”,将其转移到不锈钢密闭反应釜中,于150℃恒温晶化36小时,晶化产物经过过滤、用去离子水洗涤10次,每次用10倍于分子筛重量的去离子水,将滤饼置于120℃下干燥24小时,再置于550℃焙烧6小时,即可得本发明所述的铁杂原子微介孔复合材料产品,记为Fe-MFI-1;
(4)将6g所述Fe-MFI-1样品与浓度为20重量%的TPAOH水溶液均匀混合,所述Fe-MFI-1与TPAOH水溶液的重量比为1:5,于密闭的反应釜中于150℃晶化3天,过滤、洗涤,120℃下干燥24小时,550℃焙烧6小时,即可得重排杂原子铁杂原子微介孔复合材料产品,记为Fe-MFI-1-C。实施例1中各原料的配比和合成条件如表1所示。实施例1步骤(3)和步骤(4)所得产物的表面硅铁原子比与体相硅铁原子比之比见表2。
实施例2~3
按照实施例1的方法制备含铁杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
对比例1
参考文献(Journal of Catalysis 2014,312,263–270)进行Fe-MFI分子筛合成。具体步骤如下:
(1)称取0.46g五水硫酸铁、5.5g硫酸(质量分数95%-98%)以及25g去离子水混合均匀置于烧杯I中。
(2)在烧杯II中,称取21.32g九水硅酸钠溶于40.85g去离子水中。在搅拌的作用下,缓慢将烧杯II的溶液加入烧杯I中。接下来,称取3.33gTPABr(质量分数98%)加入上述混合物中,最终得到淡黄色的混合物。
(3)将步骤(2)最后所得的混合物置于170℃带搅拌的晶化釜内晶化3天,所得的产物经过过滤,洗涤,在室温条件下干燥24小时,再置于马弗炉中以2K/min-1的速率升至480℃焙烧4小时,最终得分子筛样品。
实施例4~10
按照实施例1的方法制备含锡杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
对比例2
参考文献(Microporous Materials,1997,12,331-340)进行Sn-MFI分子筛合成。具体步骤如下:
(1)称取1.9gSnCl4·5H2O,溶于10gH2O中得到溶液A,再称取5.34gNH4F溶于25g水中得到溶液B,在搅拌的作用下将这两种溶液A、B混合均匀得到溶液C,再将将9.78gTPABr加入56g水中得到溶液D,再将溶液D加入溶液C并搅拌30分钟得到溶液E;
(2)称取8.64g白炭黑在搅拌的作用下逐步加入溶液E中,并搅拌4小时得到均一的溶胶;
(3)将步骤(2)所得的溶胶装釜置于200℃静态晶化6天, 所得的产物经过滤,洗涤,并在120℃干燥5小时,然后在550℃焙烧3小时,最终得分子筛样品。
比较例1~3
按照实施例4的方法制备含锡杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
实施例11~13
按照实施例1的方法制备含锆杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
对比例3
参考文献(Catalysis Letters,1997,45,41-50)进行Zr-MFI分子筛合成。具体步骤如下:
(1)称取0.16g ZrCl4溶于5g去离子水,将此溶液加入21.25gTEOS中,缓慢搅拌20分钟,
(2)在上述溶液中逐滴加入40.72gTPAOH水溶液,待搅拌1小时后,再加入8g去离子水,并搅拌30分钟,得到澄清透明的溶胶。
(3)将步骤(2)所得的溶胶装釜置于160℃静态晶化48小时,所得的产物经过滤,洗涤,并在110℃干燥,550℃焙烧共16小时,最终得分子筛样品。
实施例14
按照实施例1的方法制备含硼锗杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
对比例4
参考文献(Microporous and Mesoporous Materials,2013,170,131–140)进行B-Ge-MFI分子筛合成。具体步骤如下:
(1)称取0.1g硼酸溶于5g去离子水中,再加入12.2g白炭黑,并搅拌15分钟,再逐滴加入3.3g质量分数为20%的TPAOH溶液并搅拌15分钟,然后称取0.1699gGeO2加入所得的溶液中,并持续搅 拌20分钟;
(2)将3.75gTPABr溶于8.61g去离子水中,再将此溶液加入步骤(1)所得的产物中,并搅拌1.5小时;
(3)将步骤(2)所得的产物装釜,并在60转/分钟的搅拌速率下,在160℃条件下晶化6天,晶化产物经过过滤、水洗、110℃过夜干燥,550℃焙烧6小时后,最终得分子筛样品。
实施例15、16
按照实施例1的方法制备含锗杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
对比例5
参考文献(J.Phys.Chem.1993,97,5678-5684)进行Ge-MFI分子筛合成。具体步骤如下:
(1)称取TEOS16.67g、TPABr0.66g、甲胺12.43g、HF(质量分数40%的水溶液)2.5g、去离子水30g加入烧杯中混合均匀,并剧烈搅拌30分钟以得到均匀的溶胶;
(2)称取四氯化锗0.43g缓慢加入步骤(1)所得的溶胶中,并搅拌2小时;
(3)将步骤(2)所得的产物装釜,并置于170℃条件下晶化18小时,所得的产物经过滤,洗涤,并在110℃干燥24h,550℃焙烧5小时,最终得分子筛样品。
实施例17
按照实施例1的方法制备含钒杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
对比例6
参考文献(Thermochimica Acta,2004,420,145–154)进行V-MFI分子筛合成。具体步骤如下:
(1)称取22.22g硅酸钠溶液(质量组成:8%Na2O,27%SiO2,65%H2O)与0.10g质量分数为50%的NaOH水溶液混合,得到碱性 溶液A;
(2)称取2.44g氟化钠溶于24.57g去离子水中,然后再分别称取0.38g硫酸氧钒(VOSO4)、4.53gTPABr溶于其中得到酸性溶液B;
(3)将溶液B倒入溶液A得到连续搅拌5小时以得到均一的溶胶,将此溶胶装釜并置于190℃条件下晶化3天,所得的产物经过滤,洗涤,并在110℃干燥24h,550℃焙烧5小时,得分子筛样品。
实施例18
按照实施例1的方法制备含铬杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
对比例7
参考文献(Applied Catalysis A:General,1996,134,L197-L201)进行Cr-MFI分子筛合成。具体步骤如下:
(1)将20gTEOS缓慢加入到由26g去离子水和0.96gCr(NO3)3·9H2O组成的溶液中,并剧烈搅拌半小时,得到溶液A。再称取2.4g质量分数为40%的HF水溶液,35g去离子水,0.32g NaOH,48.6g质量分数为20%的TPAOH水溶液混合均匀,得到溶液B;(2)将步骤(1)中所得的混合物B缓慢滴加到A产物中,并维持步骤(1)中的产物温度为0℃,搅拌2小时后产物pH值为5.3;
(3)将步骤(3)中所得的产物装釜并于170℃条件下静态晶化4天,晶化完成后所得的产物经过水洗,过滤,滤饼置于120℃干燥6小时,再置于550℃空气气氛下干燥12小时,最终得到分子筛样品。
实施例19
按照实施例1的方法制备含锰杂原子微介孔复合材料,其配比和合成条件见表1,表征和评价结果见表2。表1中未涉及的合成条件参考实施例1。
对比例8
参考文献(J.Am.Chem.Soc.2013,135,8594-8605)进行Mn-MFI分子筛合成。具体步骤如下:
(1)称取25gTEOS滴加到25g去离子水中,并搅拌半小时。然后滴加24gTPAOH水溶液(质量分数为25%)作为结构导向剂,继续搅拌3小时。此混合物称为溶液A。
(2)称取0.44g乙酰丙酮锰(III)溶于20mL乙醇中,再将此乙醇溶液滴加到溶液A中,并搅拌3小时,得到澄清的酒红色溶液B。
(3)将溶液B装釜,在180℃条件下晶化48h,所得的产物经过过滤,洗涤,在120℃条件下过夜干燥,再置于马弗炉中550℃焙烧5小时,最终得到淡粉色分子筛样品。
实施例20
对实施例1~19、对比例1~8和比较例1~3制备的杂原子微介孔复合材料进行环己烯的环氧化氧化反应评价,所用氧化剂为双氧水或者叔丁基过氧化氢为氧化剂,评价条件如下:采用250ml带加热搅拌和冷凝回流的三口烧瓶为反应器,反应温度为80℃,H2O2(或者叔丁基过氧化氢)/环己烯=3:1(摩尔比),环己烯质量30g,溶剂为丙酮,质量为40g,催化剂(杂原子微介孔复合材料)用量1.5g,反应时间4h,产物经离心分离出分子筛后液相采用安捷伦GC6890N分析,环己烯转化率及环氧环己烷选择性计算公式如下所示,结果见表2。
环己烯转化率=(1-反应后环己烯的摩尔数/反应前环己烯的摩尔数)×100%
环氧环己烷选择性=生成的环氧环己烷摩尔数/(反应前环己烯的摩尔数-反应后环己烯的摩尔数)×100%
实施例21
分别对实施例1~19、对比例1~8和比较例1~3步骤(3)制备的杂原子微介孔复合材料进行双氧水(浓度,30重量%)分解试验,试验条件如下:双氧水15g,杂原子微介孔复合材料2g,反应温度80℃,反应时间1小时,采用滴定法分析残余双氧水浓度,结果如表2所示。
需要说明的是,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其应视为本发明所公开的内容。
- 还没有人留言评论。精彩留言会获得点赞!