一种高比表面积纳米晶氟化镁的制备方法与流程
本发明涉及一种高比表积氟化镁,具体涉及一种高温下比表面积大、纳米晶氟化镁的温和、简易及环境友好的制备方法。
背景技术:
氟化镁(MgF2)作为一种重要的化工、光学和催化材料,具有广泛的用途。它可用作电解铝的添加剂、冶炼金属镁的助熔剂、钛颜料的涂着剂、阴极射线屏的荧光材料。同时它也是一种无色透明的红外光学材料,具有较宽的透过范围和高的透过率,用于制作红外光学系统中的光学元件。此外,MgF2是一种可在腐蚀性HF、HCl气氛下长期稳定存在的催化材料,更重要的是,它的表面酸、碱性弱,与SiO2相似趋于化学惰性,很容易通过引入不同金属离子在较宽范围内精确调变MgF2催化剂表面酸碱性,因而可作为催化剂或载体用于腐蚀性气氛下的催化反应。例如,目前工业上生产氢氟烃(HFCs)、氢氟烯烃(HFOs)的过程中通常会涉及到气相氟化反应或脱卤化氢反应,常常用到MgF2负载型催化剂。因其具有较弱的酸碱性,在上述反应中副反应少、稳定性好,表现出较好的应用前景。
MgF2用作催化载体,必须要有大的比表面积,才能获得可观的催化活性。此外,目前大部分气相氟化反应或脱卤化氢反应是在300~500℃进行,因此MgF2用作催化载体时必须具有一定的高温热稳定性方可顺利使用。国内外相关制备大比表面积MgF2的文献、专利较少,特别是有关比表面积大于60m2/g且可在高温下稳定存在的高比表面积MgF2的制备。基于此,制备高比表面积、纳米晶MgF2对开发高活性催化剂或涂层材料意义非常重大。
仅有的报道中,德国化学家Kemnitz课题组,以金属醇盐,如甲醇镁为金属源,无水HF的醇、醚溶剂为氟源,通过无水溶胶-凝胶氟化法可制得比表面积大于100m2/g的高比表面积无定形MgF2(Erhard Kemnitz,Catal.Sci.Technol.,2015,5,786)。中国专利CN104437567公开了一种比表面积大于140m2/g的MgF2基催化剂的制备方法,是对不溶于水的金属镁盐在表面活性剂存在下进行回流反应,再经氟化处理获得。中国专利CN103482661公开了一种比表面积大于150m2/g的MgF2的制备方法,是在乙酸盐存在下将可溶性镁盐水溶液与氟化铵水溶液相混获得MgF2前驱体,再经焙烧制得纳米MgF2。中国专利CN104071814公开了一种比表面积大于70m2/g的MgF2的制备方法,是在碳源的存在下将可溶性镁盐水溶液与氟化铵水溶液相混获得MgF2前驱体,再经高温预碳化、高温碳化和高温除碳获得MgF2。
上述报道的MgF2制备方法,仍至少存有以下问题:(1)无水溶胶-凝胶氟化法是以价格昂贵的有机金属为金属源和无水HF的有机溶剂为氟源,且此法必须在无水条件下操作,过程复杂,难以适用工业生产,同时所得MgF2为无定形结构,高温下不稳定,易晶化为低比表面积氟化物;(2)碳化氟化法虽然原料易得,但工艺流程复杂,需反复进行高温碳化和除碳过程、能耗高,并且在温度高于350℃时,氟化镁比表面积迅速下降至30m2/g左右;(3)乙酸盐法虽然操作简便,但所需乙酸盐浓度高、用量大,成本较高,不适于工业生产。(4)回流反应采用的是不溶于水的镁源,产品流失严重,且回流反应能耗较高。
技术实现要素:
针对现有技术存在的缺陷与不足,本发明的目的是提供一种制备方法简单、操作温和、环境友好、生产成本低的纳米氟化镁的制备方法。本发明的这种氟化镁具有比表面积大、孔道结构可调、介孔孔道丰富、粒径在纳米尺度,易于对催化剂进行掺杂、改性,从而获得高活性、高稳定性的催化性能。
多元醇体系下的溶胶-凝胶法是一种温和条件下制备高比表面积、纳米尺度氟化镁的高效方法。在此温和氟化过程中,多元醇溶剂与镁源离子发生配位络合作用,抑制了所生成氟化物晶粒的增长、团聚,阻止了氟化物沉淀的生成,从而产生溶胶-凝胶过程,最终生成粒径在纳米范围以内氟化镁颗粒。此外,通过对多元醇的调变,可使镁源离子与之的配位能力发生改变,使生成的氟化镁按不同晶粒尺寸生长,从而获得具有不同比表面、孔道结构的氟化镁微晶。另外,在此溶胶-凝胶过程中不同镁源不会显著影响制备中的溶胶-凝胶化过程,具有较为宽泛的使用范围。
为了实现上述技术任务,本发明采用如下技术方案予以实现:
一种高比表面积纳米晶氟化镁的制备方法,包括以下步骤:
(1)将镁源、促凝剂和多元醇相混,20℃~80℃下回流6h以上,得反应液A;
所述镁源为硝酸镁、氯化镁、硫酸镁、甲醇镁、乙酸镁中的一种或任意几种的组合组成;
多元醇为乙二醇、二缩乙二醇、1,3-丙二醇、1,2-丙二醇、丙三醇中一种或任意几种的组合组成;
促凝剂为聚乙二醇、聚乙烯吡咯烷酮、平平加、柠檬酸、环糊精、聚乙烯醇、环氧乙烷中一种或任意几种的组合组成,镁源与促凝剂的质量比为1:0.5~10;
(2)在搅拌下,将氟化试剂加至反应液A中进行氟化处理,添加完毕后继续搅拌6h以上,得到液体溶胶;
所述氟化试剂为氟化氢、氟化铵水溶液中的一种,氟化试剂与镁源的摩尔比为3~6:1;
(3)将液体溶胶在100℃~160℃下静置老化24h以上,得到固体凝胶;
(4)将固体凝胶在140℃~200℃下干燥24h以上,最后在350℃~450℃下焙烧4h以上,制得高比表面积、纳米晶氟化镁。
进一步,步骤(2)中,所述氟化试剂的浓度为20wt.%~90wt.%;步骤(4)中,所述焙烧是在含氧气氛下进行;所述氟化镁具有晶型,焙烧后比表面积为60~200m2/g,孔径分布集中于15~30nm,粒径分布集中于27~68nm。
本发明的有益效果:本发明与现有技术相比,具有如下有益的技术效果:
①与无水溶胶-凝胶氟化法、碳化氟化法、乙酸盐氟化法和回流反应氟化法相比,本发明提供了一种原料易得、操作简单、制备条件温和、能耗低、环境友好的高比表面积、纳米晶氟化镁的制备方法;②本发明提供的制备方法可很容易实现对所制氟化镁比表面积、孔道、颗粒尺寸、晶型的调变,可制得比表面积大于60m2/g、晶粒尺寸在70nm以内的纳米晶氟化镁,而无水溶胶-凝胶氟化法所得氟化镁为无定形,高温焙烧比表面积会迅速下降,碳化氟化法需经多次高温焙烧,能耗高,而乙酸盐氟化法对乙酸盐用量大,成本高,回流反应法也存在产品流失严重、能耗高的难题;③本发明对所用镁源无特别要求,适用性广,成本低;④本发明所制氟化镁溶胶具有腐蚀性小的特点,易于进行材料表面涂覆;⑤采用高比表面积纳米晶氟化镁做催化载体,可实现反应温度大于350℃的催化反应。
附图说明
图1为高比表面积纳米晶氟化镁的X射线衍射图
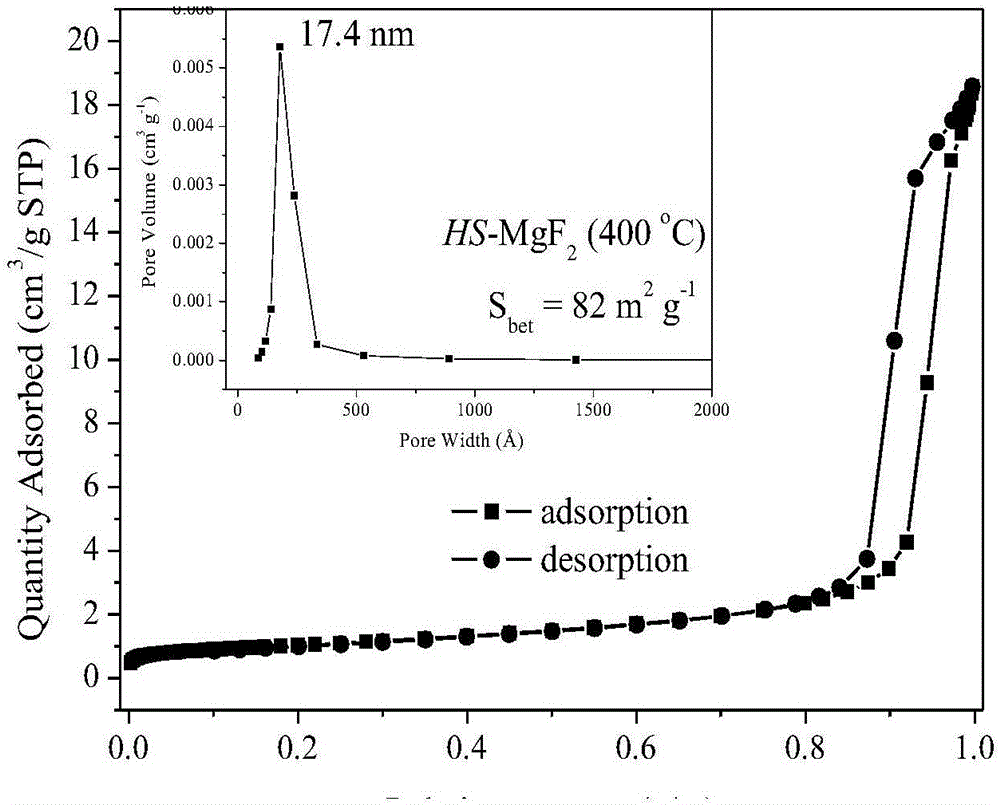
图2为高比表面积纳米晶氟化镁的等温吸附曲线图
具体实施方式
实施例1:制备高比表面积、纳米晶氟化镁
将1.0M镁源溶于50mL多元醇溶剂中,在20~80℃搅拌下回流处理6h,再将氟化试剂在搅拌下滴加到上述溶液中,滴加时间为30min,滴加完毕后再搅拌6h,得到液体溶胶;然后在100~160℃下静止老化24h以上,得到固体凝胶;再在140~200℃下干燥24h以上,最后在空气氛350~450℃下焙烧4h以上,制得高温下高比表面积纳米晶氟化镁。不同镁源、多元醇溶剂、促凝剂、氟化试剂、焙烧温度下制得的氟化镁织构性能见表1所示。
表1实施例1的氟化镁物化性质结果
实施例2:制备高比表面积纳米晶氟化镁
将1.0M硝酸镁和聚乙二醇溶于50mL乙二醇中,在30℃搅拌下回流处理6h,再将HF水溶液(40wt.%)在搅拌下滴加到上述溶液中,滴加时间为30min,滴加完毕后再搅拌6h,得到液体溶胶;然后在100℃下静止老化24h以上,得到固体凝胶;再在140℃下干燥24h以上,最后在400℃下焙烧4h以上,制得高温下高比表面积纳米晶氟化镁。不同聚乙二醇用量制得的氟化镁织构见表2所示。
表2实施例2的氟化镁物化性质结果
实施例3:制备高比表面积纳米晶氟化镁
将1.0M硝酸镁和聚乙二醇(Mg/胶凝剂质量比为1:3)溶于50mL乙二醇中,在30℃搅拌下回流处理6h,再将HF水溶液在搅拌下滴加到上述溶液中,滴加时间为30min,滴加完毕后再搅拌6h,得到液体溶胶;然后在100℃下静止老化24h以上,得到固体凝胶;再在140℃下干燥24h以上,最后在400℃下焙烧4h以上,制得高温下高比表面积纳米晶氟化镁。不同HF浓度制得的氟化镁织构见表3所示。
表3实施例3的氟化镁物化性质结果
实施例4:制备高比表面积纳米晶氟化镁
将1.0M硝酸镁和聚乙二醇(Mg/胶凝剂质量比为1:3)溶于50mL乙二醇中,在30℃搅拌下回流处理6h,再将HF水溶液(40wt.%)在搅拌下滴加到上述溶液中,滴加时间为30min,滴加完毕后再搅拌6h,得到液体溶胶;然后在100℃下静止老化24h以上,得到固体凝胶;再在140℃下干燥24h以上,最后在400℃下焙烧4h以上,制得高温下高比表面积纳米晶氟化镁。不同Mg/HF摩尔比制得的氟化镁织构见表4所示。
表4实施例4的氟化镁物化性质结果
实施例5:制备高比表面积纳米晶氟化镁
将1.0M硝酸镁和聚乙二醇(Mg/胶凝剂质量比为1:3)溶于50mL乙二醇中,在30℃搅拌下回流处理6h,再将HF水溶液(40wt.%)在搅拌下滴加到上述溶液中,滴加时间为30min,滴加完毕后再搅拌6h,得到液体溶胶;然后在100℃下静止老化24h以上,得到固体凝胶;再在140℃下干燥24h以上,最后在400℃下焙烧4h以上,制得高温下高比表面积纳米晶氟化镁。不同气氛下制得的氟化镁织构见表5所示。
表5实施例5的氟化镁物化性质结果
- 还没有人留言评论。精彩留言会获得点赞!