一种沸石分子筛的合成方法与流程
本发明涉及一种沸石的合成方法,属于分子筛催化材料合成领域。
背景技术:
沸石分子筛是化学工业中重要的吸附和催化材料。人类在十九世纪就发现和认识到天然沸石独特性质和重要性。随后人们尝试模仿天然沸石的地质生成条件,采用高温水热合成技术(温度高于200℃和高压大于100个大气压)进行合成人工沸石。1850年代开发出低温水热合成技术(反应温度25-150℃,反应压力为一至几十个大气压),合成出a、x、l、y、丝光沸石等低硅铝比的沸石。大大降低的合成难度和生产成本。随着石油工业的发展,沸石的合成技术也取得的巨大的进步,不仅开发出上百种类型的新结构沸石,还在开发出溶剂热合成、微波合成、气相合成法、极浓体系合成法、太空合成等不同的合成方式。
水热合成是最常用的沸石合成方式,一般是将硅源、铝源、碱、模板剂和水制成凝胶,然后在高温密闭的环境中,压力在一至几十个大气压的自生压条件下合成沸石晶体。
专利cn101096274a公开了一种富铝beta沸石的制备方法,其过程是先将硅源和铝源在水解剂存在下制备硅铝共凝胶,经过老化和焙烧后粉碎作为硅铝源;然后加入由四乙基铵阳离子、铵根离子、氟离子和水组成的溶液中,置于密闭反应器中,水热晶化晶化得到沸石产物。
专利us6827924公开了一种纳米eu-1沸石的合成方法,其合成方法是将模板剂四乙基氢氧化铵,添加剂甲醇、甲苯等有机物与硅铝源混合,然后置于密闭反应器中,最后采用水热晶化方式合成出纳米eu-1沸石。
专利cn101254929a公开了一种高硅铝比nay分子筛的制备方法,该方法是不使用机模板剂,合成方式是水热晶化,晶化反应分为两步进行,在第一步晶化反应结束后向反应体系再添加硅铝凝胶,最终水热晶化出nay分子筛。
微波合成法与水热合成方法类似,不同之处在于加热方式采用微波加热方式。如cn104556129a公开了一种超细t型分子筛沸石膜的微波合成法,是先将载体管打磨、超声清洗干燥,放入t型分子筛溶液中浸渍干燥;然后放入晶化母液中,采用微波加热合成出t型分子筛沸石膜。
专利cn105967203a公开了一种粉煤灰mfi沸石分子筛的制备方法,是先活化粉煤灰,再与十六烷基三甲基溴化铵,正硅酸乙酯,去离子水混合,最后置入反应釜微波加热晶化,合成出mfi沸石分子筛。
气相合成法是一种新型的分子筛制备方法,这种方法是将合成原料先制备成干胶,再将干胶置于反应器上部,水或模板剂的混合溶液置于反应器下部,干胶与液体不接触,然后在高温自生压力的条件下进行晶化反应。如专利cn1583561a公开了一种蒸汽相中自转晶制备镁碱沸石的方法,是将碱金属、三价元素的氧化物、四价元素的氧化物、水组成的混合物制成凝胶,经脱水得到干胶,在四氢呋喃/水的蒸汽相中晶化合成出镁碱沸石。
专利cn101962195a一种多级孔道钛硅沸石ts-1的制备方法,采用气相法合成出ts-1沸石。该专利是用含糖的反应物料制备干胶,糖在制造干胶过程中炭化,最后也是在干胶与溶液不接触的状态下合成出ts-1沸石。
极浓体系合成法,非常类似于水热晶化法,但二者有本质的区别。极浓体系合成法中水的用量极低,合成原料在晶化过程中近似于固态,无法像在溶液体系中那样进行物质传输。如专利cn101402049a一种甲醇制丙烯催化剂的制备方法,是先将硅源、铝源、有机模板剂、碱、水混合;再高温蒸发浓缩使原料形成湿润的胶状形态;最后置于晶化釜中晶化出zsm-5沸石。
专利cn102060309a一种丝光沸石及其制备方法丝光沸石及其制备方法,是先制备成初始凝胶混合物,然后在60℃-120℃下进行第一阶段的晶化反应,形成沸石微晶;最后升温到120℃-200℃进行第二阶段的晶化反应合成出丝光沸石。晶化体系中水/二氧化硅的摩尔比为1-7,水含量极低,也属于极浓体系合成。
离子液体合成是一种特殊的沸石分子筛合成技术。离子液体是一种由有机阳离子与有机或无机阴离子组成的一类有机盐,高温下呈液体状态。在离子液体中合成分子筛可以在常压下进行,因而可以降低反应压力,减少安全风险。但是需要使用昂贵的离子液体参与反应,大规模工业推广应用难度很大。
目前,大多数的沸石分子筛的合成具有一定的难度,合成条件一般比较苛刻,多是采用高温高压合成,安全隐患多。离子液体合成法虽然可在常压下合成沸石,但离子液体的使用却会大大提升了合成成本。因此降低反应温度与合成压力,使用廉价的合成原料,是降低生产成本和提升生产安全性的主要研究方向之一。
技术实现要素:
针对现有技术的不足,本发明提供了一种沸石分子筛的合成方法。本发明提供的合成方法属于常压合成,生产成本低廉,所制备的沸石分子筛的性质可以满足催化工业的需要。
本发明提供一种沸石分子筛的合成方法,所述合成方法包括如下内容:
(1)将无机碱、铝源、硅源、水混合,混合均匀后得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,当反应器温度大于100℃后,向反应器中通入100℃的水蒸气进行晶化反应,反应温度为100~240℃,反应时间为10~150h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到沸石。
本发明方法中,步骤(1)中所述无机碱可以是naoh、koh、lioh中的一种或多种;铝源可以是铝酸钠、硫酸铝、氯化铝、硝酸铝中的一种或多种;硅源可以是白碳黑、硅胶、硅溶胶或水玻璃中的一种或多种。
本发明方法中,步骤(3)中所述的反应器设置有进气管线和出气管线,其中进气管线与水蒸气源连通,出气管线与外界大气连通,用于保持反应器内的压力为常压。当反应器温度大于100℃后,向反应器中通入100℃的水蒸气进行晶化反应。
本发明方法中,步骤(4)中所述洗涤为用蒸馏水洗涤;所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
本发明方法中,步骤(1)在合成凝胶时,还可以加入模板剂,所述模板剂的种类与所要制备的分子筛种类有关。
与现有技术相比,本发明沸石合成方法具有以下优点:现有沸石分子筛的合成方法需要在高压条件下进行,生产过程中存在高压爆炸危险,而且生产设备也需要满足高温耐压要求,生产成本高。而本发明提供的沸石分子筛的合成方法是在常压条件下进行的,不但避免了高压爆炸的危险,而且生产设备使用的是非高温耐压的材质,大大节省了生产成本。
附图说明
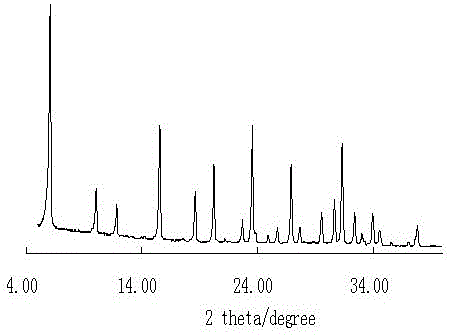
图1为实施例1得到样品的xrd谱图。
图2为实施例4得到样品的xrd谱图。
图3为实施例7得到样品的xrd谱图。
图4为实施例10得到样品的xrd谱图。
图5为实施例13得到样品的xrd谱图。
图6为实施例16得到样品的xrd谱图。
图7为实施例19得到样品的xrd谱图。
图8为实施例22得到样品的xrd谱图。
具体实施方式
下面通过具体实施例对本发明氧化铝的制备方法予以详细的描述,但并不局限于实施例。
本发明提供一种沸石的合成方法,所述合成方法包括如下内容:
(1)将无机碱、硅源、铝源、水混合,混合均匀后得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为100~240℃,反应时间为10~150h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到沸石。
本发明方法中,步骤(1)中所述无机碱可以是naoh、koh、lioh中的一种或多种;硅源可以是白碳黑、硅胶、硅溶胶或水玻璃中的一种或多种;铝源可以是铝酸钠、硫酸铝、氯化铝、硝酸铝中的一种或多种。
本发明方法中,步骤(2)中所述的反应器设置有进气管线和出气管线,其中进气管线与水蒸气源连通,出气管线与外界大气连通,用于保持反应器内的压力为常压。
本发明方法中,步骤(3)中所述洗涤为用蒸馏水洗涤;所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
本发明方法可以用于合成不同类型的沸石,如现有常见的x沸石、y沸石、beta沸石、zsm-5沸石、zsm-12沸石、zsm-35沸石、eu-1沸石、丝光沸石等。
第一方面,当用本发明方法合成x沸石时,所述合成方法包括如下内容:
(1)将无机碱、硅源、铝源、水按照摩尔比1~12na2o:1~10sio2:a12o3:80~700h2o混合,混合均匀后得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为100~140℃,反应时间为10~40h,优选反应温度为105~135℃,反应时间为15~35h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到沸石。
上述方法中,步骤(1)将无机碱、硅源、铝源、水按照摩尔比1.5~11na2o:1.5~9sio2:a12o3:85~650h2o混合。
上述方法中,步骤(4)所述洗涤为用蒸馏水洗涤;步骤(4)所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
第二方面,当用本发明方法合成y沸石时,所述合成方法包括如下内容:
(1)按照摩尔比10~16na2o:al2o3:10~23sio2:260~400h2o,将氢氧化钠、水、铝酸钠和水玻璃混合均匀,于15~40℃老化,得到y结构导向剂;
(2)将无机碱、硅源、铝源、水按照摩尔比为4~15na2o:al2o3:4~25sio2:150~1200h2o混合均匀,然后加入步骤(1)中制备的y结构导向剂混合后得到凝胶;
(3)将步骤(2)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(4)将步骤(3)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为100~160℃,反应时间为10~60h;
(5)将步骤(4)得到的固体产物洗涤干燥,得到沸石。
上述方法中,步骤(2)所述y结构导向剂的加入量以体积计算为整个凝胶的1~20%,优选2~15%。
上述方法中,步骤(2)将无机碱、硅源、铝源、水按照摩尔比为5~12na2o:al2o3:5~20sio2:200~1000h2o混合均匀。
上述方法中,步骤(4)所述洗涤为用蒸馏水洗涤;步骤(4)所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
第三方面,当用本发明方法合成beta沸石时,所述合成方法包括如下内容:
(1)将无机碱、硅源、铝源、水按照摩尔比为3~12na2o:20~150sio2:a12o3:500~3000h2o混合,混合均匀后得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为100~180℃,反应时间为10~96h,优选反应温度为105~160℃,反应时间为20~90h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到beta沸石。
上述方法中,步骤(1)将无机碱、硅源、铝源、水按照摩尔比4~10na2o:25~120sio2:a12o3:600~2500h2o混合。
上述方法中,步骤(1)中加入模板剂,所述模板剂为四乙基氢氧化铵和/或四乙基溴化铵。当加入模板剂时,步骤(1)无机碱、硅源、铝源、水、模板剂的摩尔比为3~12na2o:20~150sio2:a12o3:500~3000h2o:0.5~30模板剂;优选为4~10na2o:25~120sio2:a12o3:600~2500h2o:1~25模板剂。
上述方法中,步骤(4)所述洗涤为用蒸馏水洗涤;步骤(4)所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
第四方面,当用本发明方法合成zsm-5沸石时,所述合成方法包括如下内容:
(1)将无机碱、硅源、铝源、水按照摩尔比为0.45~3.5na2o:30~120sio2:a12o3:800~2000h2o混合,混合均匀后得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为140~220℃,反应时间为20~70h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到沸石。
上述方法中,步骤(1)将无机碱、硅源、铝源、水按照摩尔比为0.5~3na2o:35~100sio2:a12o3:1000~1800h2o混合。
上述方法中,其中,步骤(1)中加入模板剂,所述模板剂为四丙基氢氧化铵、正丁胺、乙二胺、己二胺中的一种或几种。当加入模板剂时,步骤(1)中无机碱、硅源、铝源、水、模板剂的摩尔比为0.45~3.5na2o:30~120sio2:a12o3:800~2000h2o:5~20模板剂,优选为0.5~3na2o:35~100sio2:a12o3:1000~1800h2o:7~18模板剂。
上述方法中,步骤(3)中晶化反应温度为150~210℃,反应时间为20~60h。
上述方法中,步骤(4)所述洗涤为用蒸馏水洗涤;步骤(4)所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
第五方面,当用本发明方法合成zsm-12沸石时,所述合成方法包括如下内容:
(1)将无机碱、硅源、铝源、水按照摩尔比为1~10na2o:25~120sio2:a12o3:600~2000h2o混合,混合均匀后得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为140~220℃,反应时间为20~70h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到沸石。
上述方法中,步骤(1)将无机碱、硅源、铝源、水按照摩尔比1.5~8na2o:30~100sio2:a12o3:800~1800h2o混合。
上述方法中,步骤(3)中晶化反应温度为150~210℃,反应时间为30~60h。
上述方法中,其中,步骤(1)中加入模板剂,所述模板剂为甲基三乙基氯化铵。当加入模板剂时,步骤(1)中无机碱、硅源、铝源、水、模板剂的摩尔比为1~10na2o:25~120sio2:a12o3:600~2000h2o:2~25模板剂,优选为1.5~8na2o:30~100sio2:a12o3:800~1800h2o:4~20模板剂。
上述方法中,步骤(4)所述洗涤为用蒸馏水洗涤;步骤(4)所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
第六方面,当用本发明方法合成丝光沸石时,所述合成方法包括如下内容:
(1)将无机碱、硅源、铝源、水按照摩尔比为6~35na2o:10~120sio2:a12o3:700~3500h2o混合均匀后,在加入晶种,得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为140~220℃,反应时间为10~50h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到沸石。
上述方法中,步骤(1)中所述的晶种为丝光沸石,步骤(1)中晶种加入量以丝光沸石计与所加入的硅源以sio2计的质量比为0.005~0.06:1,优选为0.01~0.04:1。
上述方法中,步骤(1)中将无机碱、硅源、铝源、水按照摩尔比为8~30na2o:15~100sio2:a12o3:800~3000h2o混合均匀后,在加入晶种,得到凝胶。
上述方法中,其中,步骤(3)中晶化反应温度为150~210℃,反应时间为15~45h。
上述方法中,步骤(4)所述洗涤为用蒸馏水洗涤;步骤(4)所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
第七方面,当用本发明方法合成eu-1沸石时,所述合成方法包括如下内容:
(1)将无机碱、硅源、铝源、水按照摩尔比为0.3~7na2o:25~110sio2:a12o3:450~1600h2o,优选0.5~6na2o:30~100sio2:a12o3:500~1500h2o混合均匀后得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为170~240℃,反应时间为20~110h,优选反应温度为180~230℃,反应时间为25~100h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到沸石。
上述方法中,其中,步骤(1)中加入模板剂,所述模板剂为溴化六甲双铵,当加入模板剂时,步骤(1)中无机碱、硅源、铝源、水、模板剂的摩尔比为0.3~7na2o:25~110sio2:a12o3:450~1600h2o:1~6模板剂,优选为0.5~6na2o:30~100sio2:a12o3:500~1500h2o:2~5模板剂。
上述方法中,其中,步骤(1)中将无机碱、硅源、铝源、水按照摩尔比0.5~6na2o:30~100sio2:a12o3:500~1500h2o混合均匀后得到凝胶。
上述方法中,步骤(4)所述洗涤为用蒸馏水洗涤;步骤(4)所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
第八方面,当用本发明方法合成方沸石时,所述合成方法包括如下内容:
(1)将无机碱、硅源、铝源、水按照摩尔比为:6~30na2o:5~30sio2:a12o3:500~2000h2o混合,混合均匀后得到凝胶;
(2)将步骤(1)得到的凝胶于80~160℃条件下干燥,直至水分完全蒸发;
(3)将步骤(2)得到的混合物装入反应器,向反应器中通入水蒸气进行晶化反应,反应温度为100~170℃,反应时间为20~110h;
(4)将步骤(3)得到的固体产物洗涤干燥,得到沸石。
上述方法中,其中,步骤(1)将无机碱、硅源、铝源、水按照摩尔比7~25na2o:8~25sio2:a12o3:600~1800h2o混合。
上述方法中,其中,步骤(3)中晶化反应温度为110~160℃,反应时间为25~100h。
上述方法中,步骤(4)所述洗涤为用蒸馏水洗涤;步骤(4)所述的干燥条件是100~150℃条件下处理5~15h,优选110~140℃条件下处理6~12h。
实施例1
首先取4g氢氧化钠、3.7g铝酸钠置于50ml蒸馏水中,搅拌直至全部溶解。再缓慢添加3.5g白炭黑,搅拌30min。装入反应器中110℃晶化3h。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至110℃时,开始通入100℃的水蒸气,在此条件下晶化20h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl1,所得样品xrd谱图如图1所示,为高结晶度的x沸石。
实施例2
首先取2g氢氧化钠、3g铝酸钠置于30ml蒸馏水中,搅拌直至全部溶解。再缓慢添加2g白炭黑,搅拌30min。装入反应器中80℃晶化3.5h。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至105℃时,开始通入100℃的水蒸气,在此条件下晶化10h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl2,所得样品为高结晶度的x沸石。
实施例3
首先取5g氢氧化钠、3g铝酸钠置于60ml蒸馏水中,搅拌直至全部溶解。再缓慢添加4.5g白炭黑,搅拌30min。装入反应器中100℃晶化4h。然后在100℃条件下6燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至135℃时,开始通入100℃的水蒸气,在此条件下晶化40h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl3,所得样品为高结晶度的x沸石。
实施例4
(1)将3.56g氢氧化钠和1.33g铝酸钠溶于24ml蒸馏水中,待溶解完全后向其中加入11ml水玻璃,然后搅拌至均匀,在35℃下静置24h,得到y结构导向剂。
(2)将1.5g铝酸钠和3g氢氧化钠溶于50ml蒸馏水中至溶解完全,然后在搅拌条件下,向其中加入4g白炭黑,搅拌25min;然后加入步骤(1)得到的y结构导向剂2ml,搅拌30min。
(3)然后在100℃条件下干燥,直至水分完全蒸发。
(4)然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至110℃时,开始通入100℃的水蒸气,在此条件下晶化30h。
(5)最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl4,所得样品xrd谱图如图2所示,为高结晶度的y沸石。
实施例5
(1)将3.56g氢氧化钠和1.33g铝酸钠溶于24ml蒸馏水中,待溶解完全后向其中加入11ml水玻璃,然后搅拌至均匀,在35℃下静置24h,得到y结构导向剂。
(2)将0.62g铝酸钠和3.5g氢氧化钠溶于80ml蒸馏水中至溶解完全,然后在搅拌条件下,向其中加入8g白炭黑,搅拌25min;然后加入步骤(1)得到的y结构导向剂9ml,搅拌30min。
(3)然后在100℃条件下干燥,直至水分完全蒸发。
(4)然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至100℃时,开始通入100℃的水蒸气,在此条件下晶化60h。
(5)最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl5,所得为高结晶度的y沸石。
实施例6
(1)将3.56g氢氧化钠和1.33g铝酸钠溶于24ml蒸馏水中,待溶解完全后向其中加入11ml水玻璃,然后搅拌至均匀,在35℃下静置24h,得到y结构导向剂。
(2)将3.2g铝酸钠和5g氢氧化钠溶于25ml蒸馏水中至溶解完全,然后在搅拌条件下,向其中加入3g白炭黑,搅拌25min;然后加入步骤(1)得到的y结构导向剂1.6ml,搅拌30min。
(3)然后在100℃条件下干燥,直至水分完全蒸发。
(4)然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至160℃时,开始通入100℃的水蒸气,在此条件下晶化15h。
(5)最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl6,所得为高结晶度的y沸石。
实施例7
取0.75g氢氧化钠溶解于50ml蒸馏水,待溶解完全后加入5mlteaoh(25wt%),搅拌30min。接着加入0.45g铝酸钠,搅拌直至全部溶解。再缓慢添加5g白炭黑,搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至140℃时,开始通入100℃的水蒸气,在此条件下晶化60h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl7,所得样品xrd谱图如图3所示,为高结晶度的beta沸石。
实施例8
取0.25g氢氧化钠溶解于90ml蒸馏水,待溶解完全后加入5mlteaoh(25wt%),搅拌30min。接着加入1g硝酸铝,搅拌直至全部溶解。再缓慢添加3.5g硅胶,搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至105℃时,开始通入100℃的水蒸气,在此条件下晶化90h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl8,为高结晶度的beta沸石。
实施例9
取1.5g氢氧化钠溶解于30ml蒸馏水,待溶解完全后加入8mlteaoh(25wt%),搅拌30min。接着加入0.3g铝酸钠,搅拌直至全部溶解。再缓慢添加16g白炭黑,搅拌30min。然后在120℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至160℃时,开始通入100℃的水蒸气,在此条件下晶化20h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl9,为高结晶度的beta沸石。
实施例10
取0.12g氢氧化钠溶解于50ml蒸馏水中,再加入2.5ml25%的四丙基氢氧化铵中,搅拌30min。接着加入0.5g铝酸钠,搅拌30min。再缓慢添加7g白炭黑,搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至185℃时,开始通入100℃的水蒸气,在此条件下晶化30h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl10,所得样品xrd谱图如图4所示,为高结晶度zsm-5沸石。
实施例11
取0.5g氢氧化钠溶解于30ml蒸馏水中,再加入2ml25%的四丙基氢氧化铵中,搅拌30min。接着加入0.5g铝酸钠,搅拌30min。再缓慢添加4g白炭黑,搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至150℃时,开始通入100℃的水蒸气,在此条件下晶化60h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl11,所得样品为高结晶度zsm-5沸石。
实施例12
取2.5g氢氧化钠溶解于80ml蒸馏水中,再加入3ml25%的四丙基氢氧化铵中,搅拌30min。接着加入0.35g铝酸钠,搅拌30min。再缓慢添加16.5g白炭黑,搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至210℃时,开始通入100℃的水蒸气,在此条件下晶化20h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl12,所得样品为高结晶度zsm-5沸石。
实施例13
取0.12g氢氧化钠溶解于50ml蒸馏水中,再加入3g甲基三乙基氯化铵中,搅拌30min。接着加入0.5g铝酸钠,搅拌30min。再缓慢添加7g白炭黑,搅拌30min。再装入密闭反应器中185℃预晶化5h。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至185℃时,开始通入100℃的水蒸气,在此条件下晶化50h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl13,所得样品xrd谱图如图5所示,为高结晶度zsm-12沸石。
实施例14
取0.2g氢氧化钠溶解于30ml蒸馏水中,再加入0.5g甲基三乙基氯化铵中,搅拌30min。接着加入0.5g铝酸钠,搅拌30min。再缓慢添加7g白炭黑,搅拌30min。再装入密闭反应器中180℃预晶化4h。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至210℃时,开始通入100℃的水蒸气,在此条件下晶化60h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl14,所得样品为高结晶度zsm-12沸石。
实施例15
取2.5g氢氧化钠溶解于80ml蒸馏水中,再加入5甲基三乙基氯化铵中,搅拌30min。接着加入0.35g铝酸钠,搅拌30min。再缓慢添加6.5g白炭黑,搅拌30min。再装入密闭反应器中175℃预晶化5h。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至150℃时,开始通入100℃的水蒸气,在此条件下晶化30h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl15,所得样品为高结晶度zsm-12沸石。
实施例16
取1.4g氢氧化钠、1g铝酸钠置于50ml蒸馏水中,搅拌直至全部溶解。再缓慢添加7.2g白炭黑,剧烈搅拌30min。然后加入0.25g丝光沸石晶种,剧烈搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至170℃时,开始通入100℃的水蒸气,在此条件下晶化30h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl16,所得样品xrd谱图如图6所示,为高结晶度的丝光沸石。
实施例17
取0.3g氢氧化钠、1.3g铝酸钠置于30ml蒸馏水中,搅拌直至全部溶解。再缓慢添加18g白炭黑,剧烈搅拌30min。然后加入0.2g丝光沸石晶种,剧烈搅拌25min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至210℃时,开始通入100℃的水蒸气,在此条件下晶化45h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl17,所得样品为高结晶度丝光沸石。
实施例18
取2.5g氢氧化钠、1g铝酸钠置于80ml蒸馏水中,搅拌直至全部溶解。再缓慢添加4g白炭黑,剧烈搅拌30min。然后加入0.4g丝光沸石晶种,然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至150℃时,开始通入100℃的水蒸气,在此条件下晶化15h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl18,所得样品为高结晶度丝光沸石。
实施例19
取0.85g氢氧化钠溶解于50ml蒸馏水,搅拌30min。接着加入0.6g铝酸钠,搅拌30min。再加入1g溴化六甲双铵,搅拌30min。再缓慢添加6.5g白炭黑,搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至185℃时,开始通入100℃的水蒸气,在此条件下晶化50h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl19,所得样品xrd谱图如图7所示,为高结晶度eu-1沸石。
实施例20
取0.35g氢氧化钠溶解于30ml蒸馏水,搅拌30min。接着加入0.6g铝酸钠,搅拌30min。再加入0,5g溴化六甲双铵,搅拌30min。再缓慢添加16g白炭黑,搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至180℃时,开始通入100℃的水蒸气,在此条件下晶化10h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl20,所得样品为高结晶度eu-1沸石。
实施例21
取3.5g氢氧化钠溶解于90ml蒸馏水,搅拌30min。接着加入0.6g铝酸钠,搅拌30min。再加入2.5g溴化六甲双铵,搅拌30min。再缓慢添加4.5g白炭黑,搅拌30min。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至230℃时,开始通入100℃的水蒸气,在此条件下晶化25h。最后将固体产物洗涤至中性,在120℃干燥12h,所得样品编号为cl21,所得样品为高结晶度eu-1沸石。
实施例22
取.0.5g氢氧化钠、1g铝酸钠、置于30ml蒸馏水中,搅拌直至全部溶解。再添加16g白炭黑。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至160℃时,开始通入100℃的水蒸气,在此条件下晶化100h。将所得到的产物用蒸馏水洗涤4次至中性,120℃干燥12h,所得样品编号cl22,所得样品如图8所示,为纯净的方沸石。
实施例23
取5g氢氧化钠、1g铝酸钠、置于95ml蒸馏水中,搅拌直至全部溶解。再添加3g白炭黑。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至110℃时,开始通入100℃的水蒸气,在此条件下晶化25h。将所得到的产物用蒸馏水洗涤4次至中性,120℃干燥12h,所得样品为方沸石,编号为cl23。
实施例24
取1.5g氢氧化钠、1.5g铝酸钠、置于70ml蒸馏水中,搅拌直至全部溶解。再添加9g白炭黑。然后在100℃条件下干燥,直至水分完全蒸发。然后将所得混合物置于反应器中(反应器的进气管线接通水蒸气源,出气管线联通外界大气),待反应器温度升温至120℃时,开始通入100℃的水蒸气,在此条件下晶化50h。将所得到的产物用蒸馏水洗涤4次至中性,120℃干燥12h,所得样品为方沸石,编号为cl24。
- 还没有人留言评论。精彩留言会获得点赞!