一种艰难梭菌毒素A/B的融合蛋白及其编码基因与应用的制作方法与工艺
本发明属于生物免疫学领域,尤其涉及一种艰难梭菌毒素A/B的融合蛋白及其编码基因与应用。
背景技术:
艰难梭菌相关性腹泻(CDAD)的发生率近年来不断升高,成为医院内腹泻的主要原因。目前医院内抗生素相关性腹泻的治疗方法是使用万古霉素或甲硝唑等抗生素,通常情况下抗生素可以有效治疗CDAD,但约有20~30%的复发患者因菌株的抗药性而无法得到治疗。随着菌株的毒力不断增强和对抗生素的耐药性,需要发展新的治疗方法,而疫苗和可以中和毒素与干扰艰难梭菌发病机制的抗体药物是CDAD预防和治疗的主要发展方向。艰难梭菌产生的毒素A与毒素B是其主要致病因素,采用纯化的毒素A和毒素B作为预防CD感染疫苗的研究已经进入临床II期实验;而用于治疗复发性CDAD的针对毒素A和毒素B的全人单抗药物也已经进入临床研究。毒素A被认为在CDAD的发病过程中起到主要的作用,研究表明艰难梭菌毒素A羧基末端受体结合区是毒素A与肠壁细胞结合的关键蛋白区域。毒素B可能在CDAD的发病过程中仅起到辅助作用。但是近年报道的艰难梭菌临床感染病例,有些菌株只有毒素B的表达,没有毒素A的表达,而病人也表现出腹泻、巨结肠症等症状。而且在CDAD抗体治疗动物实验中,同时使用毒素A和毒素B的抗体可以大大提高对动物的保护效率。在疫苗的制备过程中也同时使用毒素A和毒素B作为抗原。利用重组表达的毒素A和毒素B作为预防CDAD疫苗的研究以经有报道,均是单独表达毒素A或毒素B。这样在制备预防CDAD疫苗的过程中,就需要两套表达和纯化系统,大大增加了疫苗研发过程中的难度和工作量,在以后的生产过程中也会对场地、设备和质控等提出更高的要求。此外,毒素A包含2700多个氨基酸残基,毒素B包含近2600多个氨基酸残基,如果表达完整的毒素A和毒素B的融合蛋白,则其至少含4600个氨基酸残基,分子量接近500KD,非常困难。
技术实现要素:
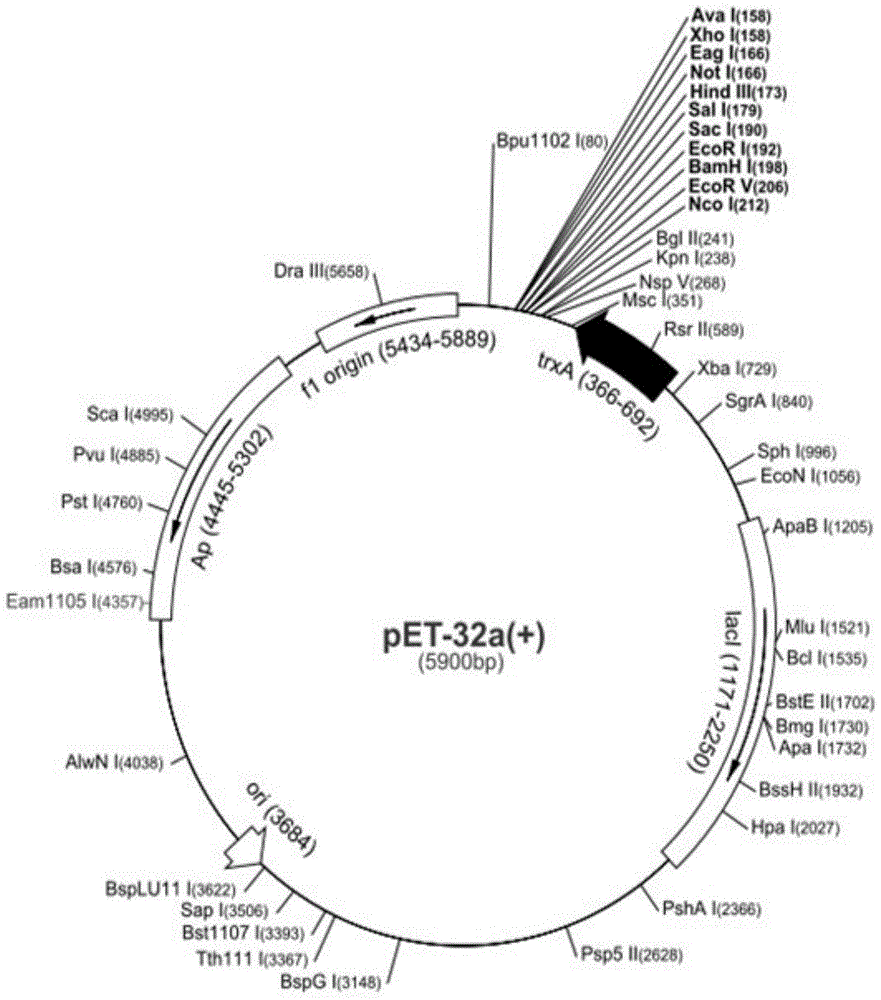
本发明的目的在于提供一种艰难梭菌毒素A/B的融合蛋白,旨在解决分别表达和纯化艰难梭菌毒素A和艰难梭菌毒素B所存在的工作量大、研发难度高的问题。本发明的再一目的在于提供上述艰难梭菌毒素A/B的融合蛋白的编码基因。本发明的再一目的在于提供上述艰难梭菌毒素A/B的融合蛋白的应用。本发明是这样实现的,一种艰难梭菌毒素A/B的融合蛋白,其氨基酸序列如SEQIDNO:2所示。本发明进一步提供了上述艰难梭菌毒素A/B的融合蛋白的编码基因,其核苷酸序列如SEQIDNO:1所示。本发明进一步提供了上述艰难梭菌毒素A/B的融合蛋白的纯化方法,将所述艰难梭菌毒素A/B的融合蛋白用Q树脂纯化,洗脱的盐浓度为在350~550mM。本发明进一步提供了上述艰难梭菌毒素A/B的融合蛋白的纯化方法,将所述艰难梭菌毒素A/B的融合蛋白与可亲和层析的标签融合表达,利用亲和层析树脂进行第一步纯化,洗脱下的艰难梭菌毒素A/B的融合蛋白切除标签后,再经过第二步亲和层析去除标签。本发明进一步提供了一种表达载体,由上述的艰难梭菌毒素A/B的融合蛋白的编码基因插入到原核表达载体的多克隆位点构建而成。优选地,所述原核表达载体为pET32b。本发明进一步提供了一种大肠杆菌宿主细胞,包含上述表达载体,该表达载体由上述的艰难梭菌毒素A/B的融合蛋白的编码基因插入到原核表达载体的多克隆位点构建而成。本发明进一步提供了上述艰难梭菌毒素A/B的融合蛋白在制备单克隆抗体、Elisa诊断检测试剂盒和胶体金检测试剂盒上的应用。本发明进一步提供了上述艰难梭菌毒素A/B的融合蛋白在制备预防艰难梭菌引起的腹泻及其它相关疾病疫苗上的应用。本发明克服现有技术的不足,提供一种艰难梭菌毒素A/B的融合蛋白,其氨基酸序列如SEQIDNO:2所示,该融合蛋白的编码基因序列为SEQIDNO:1,SEQIDNO:1序列包括艰难梭菌毒素A的羧基端结构域、艰难梭菌毒素B的羧基端结构域和编码柔性多肽的核苷酸基因序列,其中,艰难梭菌毒素A羧基端结构域的核苷酸序列如SEQIDNO:3所示,艰难梭菌毒素B羧基端结构域的核苷酸序列如SEQIDNO:4所示序列,在序列SEQIDNO:1中,艰难梭菌毒素A羧基端结构域序列和艰难梭菌毒素B羧基端结构域序列通过柔性多肽相连,柔性多肽基因片段的核苷酸序列如SEQIDNO:5所示,编码为甘氨酸-甘氨酸-甘氨酸-丝氨酸(GGGS)四肽序列的重复,重复次数可以是0次到多次,优选为4次。在本发明中,所选取的艰难梭菌毒素A羧基端和艰难梭菌毒素B羧基端核苷酸序列即符合大肠杆菌密码子偏爱性,同时通过同义密码子替换改善了原始序列多重复区对化学基因合成的影响;并且优化是在保证艰难梭菌外毒素A羧基端和艰难梭菌毒素B羧基端氨基酸序列不变的情况下,通过密码子改变对羧基端基因重复序列进行调整,充分考虑大肠杆菌密码子利用偏好进行基因序列优化,艰难梭菌外毒素A羧基端基因片段和艰难梭菌外毒素B羧基端基因片段可分别在大肠杆菌原核系统中高效表达。本发明通过构建艰难梭菌毒素A和艰难梭菌毒素B的融合基因,可以获得同时带有毒素A和毒素B免疫原性的融合蛋白,减少CDAD疫苗研发和生产的难度以及成本。附图说明图1是本发明实施例1中艰难梭菌毒素A羧基端和艰难梭菌外毒素B羧基端重组基因序列构建示意图;图2是本发明实施例2中原核表达载体pET32b的质粒图谱;图3是本发明实施例2中表达载体转化子质粒pET32b(+)-toxA-toxB的菌落PCR电泳结果图;图4是本发明实施例2中表达载体转化子质粒pET32b(+)-toxA-toxB被XhoI与XbaI双酶切后的电泳结果图;图5是本发明实施例3中艰难梭菌毒素A/B的融合蛋白诱导表达后的SDS-PAGE电泳结果;图6是本发明实施例4中艰难梭菌毒素A/B的融合蛋白的金属螯合层析纯化结果图;图7是本发明实施例4中艰难梭菌毒素A/B的融合蛋白在凝血酶酶切后电泳结果图;图8是本发明实施例5中原核表达载体pET-N-toxA-toxB的菌落PCR电泳结果图;图9是本发明实施例6中艰难梭菌毒素A/B的非融合蛋白诱导表达后的SDS-PAGE电泳结果图;图10是本发明实施例7中艰难梭菌毒素A/B的非融合蛋白经Q树脂纯化后的SDS-PAGE电泳结果图;图11是本发明实施例8中艰难梭菌毒素A/B的融合蛋白的凝血活性测定结果图。具体实施方式本发明的技术方案在于:通过PCR扩增分段获得经密码子改造后的艰难梭菌毒素A羧基端基因与原核表达载体pET32b(+)组成的载体片段、艰难梭菌毒素B羧基端结构域与柔性链的融合基因片段,获得的融合片段及获得的载体分别酶切回收后,在DNALigase作用下进行连接,获得含艰难梭菌毒素A羧基端结构域和艰难梭菌毒素B羧基端结构域的融合基因的重组表达质粒pET32b(+)-ToxA-ToxB。重组质粒转化表达宿主BL21(DE3)菌株后可被IPTG诱导表达,融合蛋白经相关鉴定为目的蛋白。另经凝血酶酶切后,可以释放完整的艰难梭菌外毒素A羧基端蛋白和毒素B羧基端的融合蛋白,凝集试验表明该方法表达的毒素A羧基端和毒素B羧基端的融合蛋白有凝集活性。为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。下列实施例中未注明具体条件的实验方法,通常按照常规条件,或按照制造厂商所建议的条件。实施例中所述的“室温”是指进行试验的操作间的温度,一般为25℃。实施例1密码子优化的编码基因序列为SEQIDNO:1的构建如图1所示,toxinA羧基端受体结合区域的792bp和toxinB羧基端的1770bp通过18个氨基酸的柔性链连接形成C-AB的融合蛋白序列,包括以下具体步骤:选取艰难梭菌外毒素A(ToxinA)基因羧基端972个碱基序列(共编码324个氨基酸),艰难梭菌外毒素B(ToxinB)基因羧基端1770个碱基序列(共编码590个氨基酸),分析该编码序列,找出影响基因合成的重复序列,如SEQIDNO:6所示。同时分析该序列与大肠杆菌密码子使用偏好不同的位点,用高频密码子替换低频密码子;另外,在兼顾大肠杆菌密码子偏好的基础上,用高频或中频密码子对重复区内的序列进行同义密码子替换,获得新的优化后的艰难梭菌外毒素A基因羧基端核苷酸序列SEQIDNO:3以及艰难梭菌外毒素B基因羧基端核苷酸序列SEQIDNO:4。为了明确显示密码子改造的位点,现将艰难梭菌外毒素A羧基端原始序列与改造后序列进行比对,比对结果如以下所示(R2-3表示重复序列2到3,R4-8表示重复序列4到8,R9-13表示重复序列9到13,R14-17表示重复序列14到17,其它以此类推):R2-3氨基酸ThrAsnGlyLysTyrPheLysTyr优化前ACTAATGGTAAATATTTTAAATAT优化后ACAAACGGCAAGTACTTCAAGTACR4-8氨基酸GlnSerLysLeuThrLeuAsnLysLys优化前CAAACTAAATTAACTTTGAATAAAAAA优化后CAGAGCAAGCTGACGCTGAACAAGAAG氨基酸TyrPheAspAsnAsnSerLysAlaLys优化前TATTTTGATAATAACTCAAAAGCAAAA优化后TACTTCGACAACAATAGCAAGGCCAAG氨基酸TyrTyrPheAsnLeuGlnIleTyrAsn优化前TATTACTTTAATTTGGAAATTTATAAT优化后TACTATTTCAACCTGCAGATCTACAACR9-13氨基酸LeuLeu优化前TTGTTG优化后CTGCTGR14-17氨基酸LeuGlnPheLeuThrLeuAsnGlyLys优化前CTTCAATTCTTAACTTTGAATGGTAAA优化后CTGCAGTTTCTGACACTGAACGGAAAGR18-22氨基酸LysLeuThrLeuAsnGlyLysLysGlyLys优化前AAATTAACTTTGAATGGCAAAAAAGGTAAA优化后AAGCTGACGCTTAACGGTAAGAAGGGCAAG氨基酸TyrPheLeuThrIleGlyLysTyrLeuAsn优化前TACTTTCTTACTATTGGTAAATATCTTAAC优化后TATTTCCTGACGATCGGAAAGTACCTGAAT氨基酸ThrAlaThrGlyThrIleLysSerThrGly优化前ACTGCTACTGGAACTATTAAATCAACTGGT优化后ACAGCAACAGGCACAATCAAGTCTACAGGAR23-26氨基酸LeuLeuArg优化前CTTCTTCGA优化后CTGCTGCGTR27-29氨基酸GlnAsnArgPheLeuLeuIleGlyAsnAsn优化前CAAAATAGATTCCTACTAATAGGTAATAAT优化后CAGAACCGTTTTCTGCTGATTGGCAACAAC氨基酸SerThrThrGlyTyrProAla优化前TCAACTACTGGTTATCCTGCT优化后TCTACAACCGGCTACCCGGCAR30-32氨基酸LeuLeuLeu优化前CTACTTCTT优化后CTGCTGCTG根据上述优化原则,参考大肠杆菌密码子偏爱性设计出相应的核苷酸序列,通过18个氨基酸的柔性多肽链的核苷酸序列SEQIDNO:5(C)连接艰难梭菌外毒素A核苷酸序列SEQIDNO:3羧基端和艰难梭菌外毒素核苷酸序列SEQIDNO:4羧基端,得到融合蛋白的编码基因序列为SEQIDNO:1(C-AB),编码的氨基酸序列如SEQIDNO:2所示。对艰难梭菌毒素A/B的融合蛋白的核苷酸序列SEQIDNO:1进行全基因人工合成并测序正确。实施例2原核表达载体构建一、原核表达载体pET32b(+)-toxA-toxB的构建与鉴定1、目的基因片段获得:设计一套PCR引物扩增合成的pUCE-toxA-toxB的艰难梭菌外毒素核苷酸序列SEQIDNO:3和SEQIDNO:4,其中5’端为平末端,3’端引入NruI酶切位点。PCR产物纯化试剂盒回收PCR反应后的产物,然后经NruI酶切后释放3’端含有粘性末端的艰难梭菌外毒素A羧基端和外毒素B羧基端基因片段。扩增艰难梭菌外毒素A羧基端和外毒素B羧基端基因片段用引物序列如下,反向引物引入NruI酶切位点“TCGCGA”:正向引物1:GCCTCTACAGGATATACAAGTA反向引物1:GTGTCGCGACTCGAGTCATTATTC2、pET32b(+)载体(如图2所示)片段:设计一套PCR引物扩增pET32b(+)的部分序列,其中5’端引入NruI酶切位点,3’端引入平末端。PCR产物纯化试剂盒回收PCR反应后的产物,然后经NruI酶切后释放5’端含有粘性末端的pET-32b载体片段。扩增pET-32b载体片段用引物序列如下:正向引物2:GAGTCGCGACACCACCACCACCACCACTGAGAT,反向引物2:GATCATACCAGAACCGCGTGGCAC;3、连接反应:上述步骤获得的载体和两段目的基因在T4DNALigase作用下,设计连接体系,其中体系中片段:载体的质量浓度比设定为7:1。温度设定为16℃,过夜连接,获得含有目的基因的表达载体;4、转化及鉴定:氯化钙法将获得含有目的基因的表达载体转化大肠杆菌DH5α,均匀涂布于含Amp抗性的LB琼脂平板,37℃培养过夜。次日挑取24个单菌落进行试管培养,并提取质粒。对转化子质粒进行二种鉴定,分别为菌落PCR鉴定,结果如图3所示,菌落PCR结果应为3394bp条带,图中的1-4样品均为阳性,因此挑取阳性样品进行酶切实验。XhoI单酶切鉴定、XhoI和XbaI双酶切鉴定,结果如图4所示,此二酶双酶切后片段大小应分别为5335bp与3246bp,由图可知1、2、3、4、号质粒可能为阳性转化子,选择鉴定均显阳性的转化子1号送出测序,测序正确后正式命名为pET32b(+)-toxA-toxB。实施例3艰难梭菌毒素A/B的融合蛋白的表达1、诱导表达及鉴定:将鉴定后的pET32b(+)-toxA-toxB质粒用氯化钙法转化大肠杆菌宿主菌BL21(DE3),从中随机挑选单菌落进行表达。即用无菌枪头挑取单菌落于含有100μg/mlAmp的LB培养基中37℃过夜培养,转速设定为220rpm。次日清晨按1:10接种于新鲜的LB培养基中,37℃,220rpm培养至OD值约为0.6时加入IPTG至终浓度为0.4mM,诱导在22℃条件下进行,诱导时间分别设定为3h和8h,分别取样进行8%的SDS-PAGE电泳检测,结果如图5所示,在图5中,泳道1~7为pET32b(+)-toxA-toxB在BL21(DE3)菌株的表达情况:1为30℃诱导1小时,3为37℃诱导前,2为诱导1小时,4为30℃诱导3小时,5为37℃诱导3小时,6为30℃诱导过夜,7为37℃诱导过夜;泳道8~14为pET32b(+)-toxA-toxB在Rosseta菌株的表达:8、9为诱导前,10为30℃诱导1小时,11为37℃诱导1小时,12为30℃诱导3小时,13为37℃诱导3小时,14为30℃诱导过夜;15为NEB公司的250KD蛋白质Marker。因目的蛋白约为110kD,电泳显示其在Marker第一条带之上,目的蛋白表达量在30%左右。保存菌株,命名为pET32b(+)-toxA-toxB/BL21(DE3)。由于融合蛋白较大,故在诱导表达时选择低温慢速长时间诱导,尽可能减少包涵体形成的可能。超声破碎条件选择在功率200W,超声4s,间歇8s的条件下,超声20min可以多数融合蛋白分布于上清中,方便于对融合蛋白进行可溶性蛋白的纯化操作。实施例4艰难梭菌毒素A/B的融合蛋白的纯化及酶切释放1、艰难梭菌毒素A/B的融合蛋白的纯化,包括如下具体步骤:(1)200ml诱导表达培养物离心获得的细胞,用10mlNTA-0重悬,置于冰浴中超声波破壁20min(200W,工作4s,间歇8s)。细胞破壁液于12000rpm,4℃离心20min,取上清置于烧杯(25ml)中。(2)加入1mlNi-NTA树脂混悬液(含50%V/V树脂,保存于20%V/V乙醇溶液中),置于冰上(或在4℃冰箱中)用脱色摇床120rpm振荡1h。(3)装入层析柱,移去层析柱底端的帽子,上清液通过NTA柱,收集流出部分,用于电泳分析。(4)加10mlNTA-20洗涤层析柱,洗去未结合的杂蛋白。(5)分别加入5mlNTA-60、4mlNTA-80以及2mlNTA-100梯度洗涤层析柱,尽可能洗去未结合的杂蛋白。(6)用2mlNTA-200洗脱目的蛋白,分4次洗脱,每次0.5ml。(7)柱子的再生,先用5mlNTA-500冲洗柱子,再用5ml20%V/V乙醇冲洗柱子,最后盖上柱子底端的帽子,用0.5ml20%V/V乙醇溶液重悬树脂,4℃保存。树脂可重复使用3~5次。纯化结果如图5所示,其中,融合蛋白在NTA20溶液开始有洗脱,NTA100溶液洗脱可以达到纯度约90%,NTA200溶液洗脱可以达到95%的纯度。2、艰难梭菌毒素A/B的融合蛋白的凝血酶酶切在载体构建时,将(His)6标签置于毒素A羧基端序列之前,方便后续的纯化操作,并在二者之间设有编码凝血酶酶切位点的序列,更方便了后续融合蛋白经凝血酶酶切后释放目的蛋白,即融合蛋白。凝血酶酶切条件为:1mg融合蛋白中加入凝血酶125单位,4℃酶切过夜,次日检测酶切效果,如图7所示,在图7中,泳道1、2、3为凝血酶处理样品;泳道4、5、6为未经处理样品;样品1、2为超声破碎上清,2、3、4、5为纯化样品。由图7可知,经凝血酶酶切后,艰难梭菌毒素A/B的融合蛋白可以释放无标签的融合蛋白,该蛋白大小约为110KD,凝血酶的酶切效率在95%左右。实施例5原核表达载体pET-N-toxA-toxB的构建与鉴定以实例2中构建的pET32b(+)-toxA-toxB重组质粒为模板,去除标签蛋白和可溶性蛋白片段399bp,启动密码子ATG位于艰难梭菌外毒素A羧基端和外毒素B羧基端重组编码序列之前,获得艰难梭菌外毒素A羧基端和外毒素B羧基端非融合蛋白。1、获得无标签的pET-N-toxA-toxB片段:设计引物,反向PCR方法扩增实例2中构建的pET32b(+)-toxA-toxB重组质粒,去除His和Trx标签。扩增用引物序列如下:正向引物3:GCCTCTACAGGATATACAAG,反向引物3:CATATGTATATCTCCTTC;2、连接反应:上述步骤获得的片段在T4DNALigase作用下,设计连接体系,其中体系中片段为50ng。温度设定为16℃,过夜连接,获得表达载体;3、转化及鉴定:氯化钙法将获得含有目的基因的表达载体转化大肠杆菌DH5α,均匀涂布于含Amp抗性的LB琼脂平板,37℃培养过夜。次日挑取24个单菌落进行试管培养,并提取质粒。对转化子质粒进行鉴定,菌落PCR鉴定,结果如图8所示,原核表达载体pET-N-toxA-toxB的PCR产物应该为三条带,分别为822bp、549bp、210bp,未去除标签和Trx的载体PCR扩增条带为1222bp、949bp、610bp,图中显示的含有822bp、549bp、210bp条带的样品为应该阳性;挑选阳性菌落送测序。选择鉴定显阳性的转化子送出测序,测序正确后正式命名为pET-N-toxA-toxB。实施例6艰难梭菌毒素A/B的融合蛋白大肠杆菌表达工程菌的制备1、诱导表达及鉴定:按照实施例3艰难梭菌毒素A/B的融合蛋白大肠杆菌表达工程菌的获得进行诱导表达及鉴定,结果如图9所示,其中,泳道1为浓缩后CAB,泳道2为NAB诱导前,泳道3~7为NAB20度0.1、0.3、0.5、0.8、1.0诱导2h;泳道8~12为NAB37度0.1、0.3、0.5、0.8、1.0诱导2h。从图9可以看出,因目的蛋白约为100kD,电泳显示其符合大小,目的蛋白表达量在30%左右。保存菌株,命名为pET-N-toxA-toxB/BL21(DE3)。由于非艰难梭菌毒素A/B的融合蛋白较大,故在诱导表达时选择低温慢速长时间诱导,尽可能减少包涵体形成的可能。超声破碎条件选择在功率200W,超声4s,间歇8s的条件下,超声20min可以多数非融合蛋白分布于上清中,方便于对非融合蛋白进行可溶性蛋白的纯化操作。实施例7非艰难梭菌毒素A/B的融合蛋白纯化1、非艰难梭菌毒素A/B的融合蛋白纯化(1)接菌种过夜培养,次日1:100比例转接LB,37度培养至OD600为0.8~1.0之间,加终浓度为0.3mM的IPTG,在20℃诱导4h,离心收菌,按照400ml菌液用100ml20mMTris(pH7.5)重悬,超声破碎至澄清,离心分离上清和包涵体。(2)用20mMTris(pH7.5)平衡Q树脂,上清用0.45m滤膜过滤后上样,用20mMTris(pH7.5)吸取非特异结合的蛋白,然后依次用含100mM、200mM、300mM、400mM、450mM、500mM、1000mM氯化钠的20mMTris(pH7.5)缓冲液洗脱,分别取样电泳检测,结果如图10所示,其中,泳道1为诱导前,泳道2为诱导4h,泳道3~11分别为上清、100、200、300、400-1、400-2、450、500、1000mM氯化钠洗脱样品。由图10可知,用450mM氯化钠的20mMTris(pH7.5)洗脱液纯化的条带符合目的条带大小,并且条带单一。实施例8艰难梭菌毒素A/B的融合蛋白的活性验证1、艰难梭菌毒素A/B的融合蛋白凝血活性测定实验取250μl2%的兔红细胞,每孔加25μl;配置外毒素A羧基端和外毒素B羧基端融合蛋白母液,浓度为0.1mg/ml,各孔加样质量依次为阴性对照、0.05μg、0.1μg、0.2μg、0.3μg、0.4μg、0.5μg融合蛋白、阳性对照;将96孔板放于4℃孵育4h,观察结果如图11所示,其中,1为阴性对照PBS;2~7分别为0.05μg、0.1μg、0.2μg、0.3μg、0.4μg、0.5μg融合蛋白;8为阳性对照。从图中可以看出,阴性对照和阳性对照结果成立,融合蛋白在0.2μg出现明显红细胞凝集,表明艰难梭菌毒素A/B的融合蛋白具有凝血活性。2、艰难梭菌外毒素A羧基端和外毒素B羧基端非融合蛋白凝血活性测定实验按照上述艰难梭菌毒素A/B的融合蛋白凝血活性测定实验,测定非融合蛋白凝血活性,1为阴性对照PBS;2~7分别为0.05μg、0.1μg、0.2μg、0.3μg、0.4μg、0.5μg融合蛋白;8为阳性对照。试验结果为,阴性对照和阳性对照结果成立,融合蛋白在0.2μg出现明显红细胞凝集,表明艰难梭菌毒素A/B的融合蛋白具有凝血活性。3、艰难梭菌毒素A/B的融合蛋白免疫活性验证取10只6~8周龄的BALB/C小鼠,分为两组,一组免疫融合蛋白组,二组为对照组;配制艰难梭菌毒素A/B的融合蛋白母液,加入佐剂,混匀后,肌肉注射免疫组小鼠,对照组注射相应量的PBS,第0、1、2、3周免疫小鼠,每次免疫前和第三周免疫后一周采血;Elisa方法测定血清中抗体滴度,具体步骤如下:不带his标签的重组蛋白浓度为0.02ug/ul,每孔包被2ug,37℃孵育1h,洗涤、封闭,加入血清样品,37℃孵育1h,洗涤,加入1:2000倍稀释的羊抗鼠IgG二抗,37℃孵育1h,洗涤,每孔加入TMB100ul,室温放置5-10分钟,终止液终止反应,450nm波长测定吸光值,结果如下1所示:表1从上表1可以看出,Elisa结果显示艰难梭菌毒素A/B的融合蛋白免疫BALB/C小鼠后血清中的抗体滴度很高,毒素A羧基端和外毒素B羧基端融合蛋白具有较强的抗原性,可以用于艰难梭菌疫苗。相比与现有技术的缺点和不足,本发明具有以下有益效果:本发明的艰难梭菌毒素A/B的融合蛋白不仅可以获得同时带有毒素A和毒素B免疫原性的融合蛋白,活性高,而且还减少CDAD疫苗研发和生产的难度以及成本。以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1