3,7-二氨基-10H-吩噻嗪化合物盐及其用途的制作方法
本申请与2006年3月29日提交的美国专利申请60/786,690相关;在此整体引入美国专利申请60/786,690的内容,作为参考。
技术领域
本发明总体上涉及吩噻嗪化合物领域;更具体地涉及一些经稳定还原的吩噻嗪化合物,特别是涉及某些3,7-二氨基-10H-吩噻嗪(DAPTZ)化合物,例如N,N,N’,N’-四甲基-10H-吩噻嗪-3,7-二胺双(盐酸)化合物和N,N,N’,N’-四甲基-10H-吩噻嗪-3,7-二胺双(氢碘酸)化合物。这些化合物用作药物,例如用于治疗τ蛋白病(tauopathy),如阿尔茨海默氏病(Alzheimer’s disease),以及这些化合物也作为相应的氧化锍药物(例如,甲基锍氯化物,MTC)的前药。
背景技术:
为了更详细地描述和充分公开本发明及本发明相关领域的现状,本申请引用了多篇专利和出版物。各参考文献通过参考全文引入本说明书中,每篇参考文献应视为被单独专门地引入作为参考。
在整个说明书(包括随附权利要求书)中,除非另有说明,术语“包含(或包括)”应理解为包括所指出的整数或步骤,或整数组或步骤组,同时也不排除任何其它整数或步骤,或整数组或步骤组。
必须指出的是,除非另有说明,否则说明书和随附权利要求书中出现的名词包括其单数和复数形式。因此,“药物载体”包括两种或多种这种载体的混合物,依此类推。
本文中的范围通常表达为从“约”一特定数值和/或至“约”另一数值。当用这样的范围表达时,另一个实施方案包括从一特定值和/或至另一特定值。类似地,当数值前用“约”表示其为近似值时,前缀“约”的使用应该理解为该具体数值形成了另一个实施方案。
痴呆病的症状以蛋白结构(例如β-淀粉样蛋白斑和神经原纤维缠结(NFTs))在受感染的患者的脑中细胞内和/或细胞外沉淀物的进行性累积为特征。这些损伤的出现大部分与病理性神经元纤维变性和脑萎缩有关,也和认知缺损有关(参见如Mukaetova-Ladinska,E.B等人,2000,Am.J.Pathol.,卷157,No.2,623-636页)。据披露,甲基锍氯化物(MTC)和其它二氨基吩噻嗪类化合物在这类疾病中作为蛋白聚集抑制剂,所述疾病即其中蛋白发生病理性聚集的疾病(参见,例如WO 96/30766和WO 02/055720)。
甲基锍氯化物(MTC)目前用于治疗高铁血红蛋白血症(methemoglobinemia)(这是一种当血液不能向身体需要氧的地方输送氧时发生的疾病)。MTC也用作医学染料(例如,在手术前或手术过程中对身体特定部位进行染色);用于诊断(例如用作指示染料检测尿液中存在的特定化合物);温和的泌尿器官抗菌剂;粘液表面刺激物;用于治疗和预防肾结石;用于诊断和治疗黑素瘤。
MTC已经用来治疗疟疾,用于单独治疗(参见如Guttmann,P.和Ehrlich,P.,1891,“Uber die wirkung des methylenblau bei malaria,”Berl.Klin.Woschenr.,第28卷,第953-956页)或与氯喹联合用于治疗疟疾(参见如Schirmer,H.等人,2003,“Methylene blue as an antimalarial agent,”Redox Report,第8卷,第272-275页;Rengelshausen,J.等人,2004,“Pharmacokinetic interaction ofchloroquine and methylene bluecombination against malaria,”European Journal of Clinical Pharmacology,第60卷,第709-715页)。人类患有的疟疾是由如下疟原虫属的四种原生动物中的一种导致的:恶性疟原虫(P.falciparum)、间日疟原虫(P.vivax)、卵形疟原虫(P.ovale)或三日疟原虫(P.malariae)。所有种类疾病都是通过受感染的雌性阿诺菲力滋蚊(Anopheles mosquito)的叮咬传播的。偶尔会通过血液传播、器官移植传播、针头共用传播或先天地由母亲传染给胎儿。世界范围内有300-500百万疟疾感染者,每年约有1百万人死于该疾病。然而,耐药性是主要的因素,并且对于恶性疟原虫来说耐药性尤其显著,恶性疟原虫是导致几乎所有疟疾-相关死亡的主要疟原虫种类。目前推荐使用的、用于预防疟疾的药物或药物联合包括氯喹/盐酸氯胍、甲氟喹、多西环素和伯氨喹。
MTC(商品名Bioenvision Inc.,New York)具有强效的体外杀病毒活性。具体地,在实验室级测试中能有效抗病毒如HIV和西尼罗河病毒(West Nile Virus)。西尼罗河病毒感染(WNV)是感染中枢神经系统的潜在严重疾病。大部分感染者没有呈现出明显的症状或轻度流感样症状如发热或头痛。150人中约有1人会发展成严重症状包括震颤、惊厥、肌无力、视觉丧失、麻木、瘫痪或昏迷。通常,WNV是由感染蚊子叮咬传播的,但也可通过输血传播、器官移植传播、母乳喂养传播或妊辰期间由母亲传染给孩子。
目前也出于临床试验阶段,用于治疗慢性丙型肝炎(Hepatitis C)。慢性丙型肝炎是肝脏的病毒感染。HCV病毒是急性肝炎和慢性肝病的主要病因,所述肝病包括肝硬化和肝癌。HCV主要是通过与人血液直接接触传播的。全世界范围内的HCV感染的主要病因是未经筛选的输血、没有经充分灭菌的针头和注射器的重复使用。世界卫生组织已经声明丙型肝炎是全球健康问题,世界人口的约3%感染了HCV,并且因地域的不同而发生显著变化。据估计,该疾病在美国的发病率为1.3%或约350万人患有该疾病。埃及约有6200万人口,是世界上丙型肝炎发病率最高的国家,据估计该国家的约6200万人中超过20%感染了该疾病。
当MTC与光联合使用时,MTC能阻止核酸(DNA或RNA)的复制。血浆、血小板和红细胞不含有核DNA或RNA。当将MTC引入到血液成分中时,其穿过细菌细胞壁或病毒膜到达核酸结构的内部。经光活化后,该化合物随之与病毒或细菌病原体的核酸结合,从而阻止DNA或RNA的复制。由于MTC可使病原体灭活,因此其具有减少试验仍然检测不出来的病原体传播带来的危险的潜力。
MTC的口服和非肠道制剂可在美国通过商业渠道获得,商品名通常为Urolene
技术实现要素:
本发明一方面涉及特定化合物,特别是涉及特定3,7-二氨基-10H-吩噻嗪(DAPTZ)化合物。
本发明另一方面涉及含有本申请所述DAPTZ化合物和可药用载体或稀释剂的组合物。
本发明另一方面涉及含有本申请所述DAPTZ化合物和可药用载体或稀释剂的药物组合物。
本发明另一方面涉及制备药物组合物的方法,包括将本申请所述的DAPTZ和可药用载体或稀释剂混合。
本发明另一方面涉及逆转和/或抑制蛋白聚集(aggregation of protein)的方法(如τ蛋白、突触核蛋白等),例如,逆转和/或抑制与神经变性疾病和/或临床痴呆有关的蛋白聚集,所述方法包括将所述蛋白与有效量的本申请所述DAPTZ化合物接触。所述方法可在体内进行在或体外进行。
本发明另一方面涉及治疗或预防受试者的疾病的方法,包括向受试者给药预防有效量的或治疗有效量的本申请所述DAPTZ化合物,优选采用药物组合物形式。
本发明另一方面涉及本申请所述的DAPTZ化合物在通过治疗而用于治疗或预防人体或动物体(例如疾病症状)的方法中的用途。
本发明另一方面涉及本申请所述的DAPTZ化合物在制备用于治疗或预防疾病的药物中的用途。
在一个实施方案中,所述疾病为蛋白聚集疾病(disease of protein aggregation)。
在一个实施方案中,所述疾病为τ蛋白病,如神经病变性τ蛋白病,如阿尔茨海默氏病。
在一个实施方案中,所述疾病为皮肤癌如黑素瘤。
在一个实施方案中,所述疾病为病毒、细菌或原生动物疾病症状,如丙型肝炎、HIV、西尼罗河病毒(WNV)或疟疾。
本发明另一方面涉及使样品(如血液或血浆样品)中的病原体灭活的方法,包括如下步骤:将本发明所述的DAPTZ化合物加到所述样品中,然后将所述样品暴露到光下。
本发明另一方面涉及试剂盒,该试剂盒含有(a)置于合适的容器和/或合适的包装中的本申请所述的DAPTZ化合物,优选为药物组合物;和(b)使用说明书,例如指示如何给药所述化合物的书面说明书。
本领域技术人员能够理解的是,本发明某一方面涉及的特征和优选实施方案也与本发明另一方面相关。
附图说明
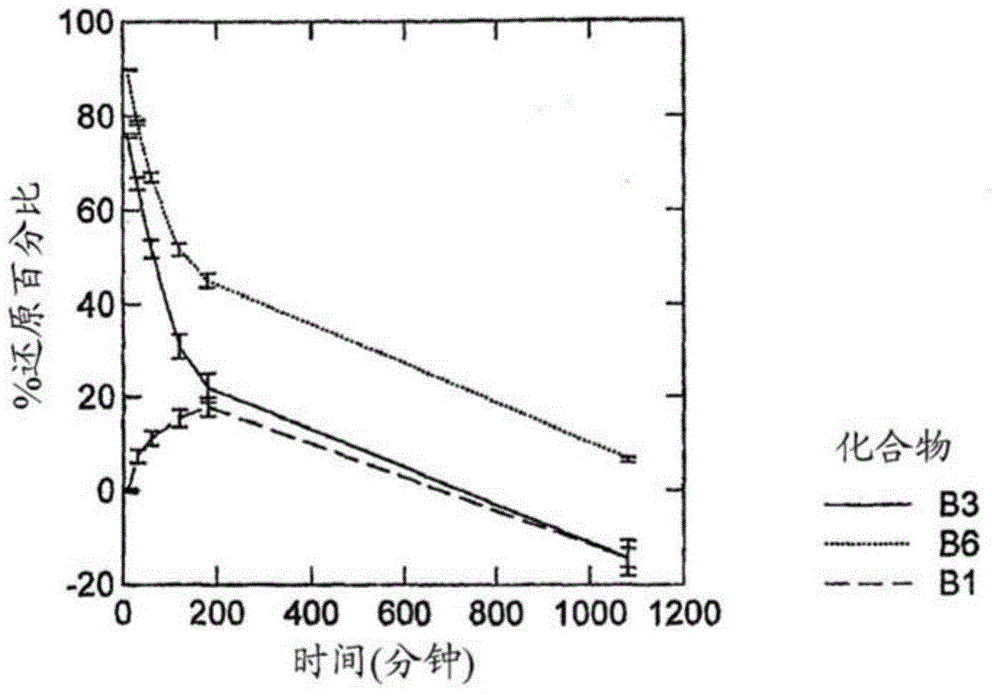
图1是如下三种化合物各自的还原形式的百分比(%)对时间(分钟)的曲线图,所述三种化合物为:B1(MTC)、B3(N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(盐酸))和B6(N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢碘酸)),利用665nm处的吸光度来测定。
图2为三种化合物B1、B3和B6各自的还原形式的百分比(%)对时间(分钟)的曲线图,利用610nm处的吸光度来测定。
图3A显示了20分钟后三种化合物各自水溶液样品的紫外/可见光吸收波谱,所述化合物为:B1(空心圆点(opencircles),最大在605nm)、B3(空心方块(open squares),最大在660nm处)和B6(空心三角形(open triangles),最大在660nm处)。
图3B显示了3个小时后三种化合物各自水溶液样品的紫外/可见光吸收波谱,所述化合物为:B1(空心圆点,最大在605nm处)、B3(空心方块,最大在605nm处)和B6(空心三角形,最大在605nm处)。
图3C显示了28个小时后三种化合物各自水溶液样品的紫外/可见光吸收波谱,所述化合物为:B1(空心圆点,最大在605nm处)、B3(空心方块,最大在605nm处)和B6(空心三角形,最大在605nm处)。
图4显示了N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢溴酸)的晶体结构。
图5显示了N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢溴酸)的侧面视图。
图6显示了晶体中N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢溴酸)分子的一个螺旋柱(helical column)部分。
具体实施方式
甲基锍氯化物(MTC)(也已知称作次甲蓝(Methylene blue,MB);美蓝(methylthionine chloride);四甲基硫堇氯化物(tetramethylthionine chloride);3,7-双(二甲基氨基)吩噻嗪-5-氯化物;C.I.碱性蓝9;四甲基硫堇氯化物;3,7-双(二甲基氨基)吩噻嗪氯化物;亚甲蓝(Swiss blue);C.I.52015;C.I.亚甲蓝8(C.I.Solvent Blue 8);苯胺紫(aniline violet);和Urolene ),其为具有如下结构式的低分子量(319.86)、水溶性、三环有机化合物:
甲基锍氯化物(MTC)(也称次甲蓝)(可能是最公知的吩噻嗪染料和氧化还原指示剂),其也被用作生物物理学系统的光学探针(optical probe)、用作纳米多孔材料的插入剂(intercalator)、用作氧化还原调节剂和用在光电成像(photoelectrochomic imaging)中。
当提到10H-吩噻嗪化合物即N,N,N’,N’-四甲基-10H-吩噻嗪-3,7-二胺时,相应的吩噻嗪-5-盐(即,MTC)可方便地认为是“氧化形式(oxidized form)”,而10H-吩噻嗪化合物本身可方便地认为是“还原形式(reduced form)”。
所述“还原形式”(“leuko form”)已知是不稳定的,能被很容易地、快速地氧化形成其相应的“氧化”形式。
May等人(Am J Physiol Cell Physiol,2004,第286卷,第C1390-C1398页)已经显示了人红细胞能顺序地还原和吸收MTC;MTC本身不能被细胞吸收;MTC的还原形式能通过细胞膜;吸收速度为酶依赖性的;并且显示了MTC和经还原的MTC在细胞内富集(经还原的MTC一旦进入细胞就形成MTC,从而再平衡(re-equilibrates))。
MTC和相似的药物在肠道内被吸收并进入血流。未被吸收的药物沿着消化道渗滤到达肠道末端(distal gut)。一个重要的但不希望的副反应是未经吸收的药物在肠道末端引起的影响,例如肠道末端的敏化和/或未被吸收药物对肠道末端菌群的抗微生物作用,这些都导致腹泻。因此,希望能使渗滤到达肠道末端的药物量最小化。通过增加药物在肠道内的更新(即增加药物的生物利用度),剂量得到减少,以及使不希望的副反应如腹泻得到改善。
既然能被细胞吸收的是MTC的还原形式,因此希望给药所述还原形式。这也减少对酶还原这一限速步骤速率的依赖。
本发明人已经识别出一系列能被认为是MTC“还原形式”的化合物,这些化合物具有出人意料且令人惊讶的稳定性。因此,所述化合物可称作MTC之类化合物的“经稳定的还原形式”。
这些化合物本身作为药物,也可用作前药,所述前药一经氧化即得到相应的氧化化合物(例如MTC),所述氧化化合物也可作为药物。
这类化合物的一个代表性化合物如下所示:
这类化合物的另一代表性化合物如下所示:
化合物
总的来说,本发明涉及具有如下结构式的特定3,7-二氨基-10H-吩噻嗪及其可药用盐、溶剂化物和水合物(本申请中统称为“二氨基-吩噻嗪化合物”和/或“DAPTZ化合物”):
其中:
R1和R9各自独立地选自:-H、C1-4烷基、C2-4烯基和卤代C1-4烷基;
R3NA和R3NB各自独立地选自:-H、C1-4烷基、C2-4烯基和卤代C1-4烷基;
R7NA和R7NB各自独立地选自:-H、C1-4烷基、C2-4烯基和卤代C1-4烷基;
HX1和HX2各自独立地为质子酸(protic acid)。
尽管不受任何具体理论的束缚,但本发明人认为所述化合物可能以下面的形式存在:
尽管所述DAPTZ化合物本身即是盐,但它们也可以以混盐的形式提供(即与另一种盐混合的DAPTZ)。这些混盐通过术语“及(或)其可药用盐”来涵盖。除非另有说明,否则当提到某特定化合物时,也包括其盐。
所述DAPTZ化合物也可以溶剂化物或水合物的形式提供。本申请使用的术语“溶剂化物”具有常规含义,是指溶质(如化合物、化合物的盐)与溶剂的复合物(complex)。如果溶剂是水,则溶剂化物很可便利地为水合物,例如一水合物、二水合物、三水合物等。除非另有说明,否则当提到特定化合物时,也包括其溶剂化物。
在一个实施方案中,所述C1-4烷基选自:直链C1-4烷基如-Me(甲基)、-Et(乙基)、-nPr(正丙基)、-iPr(异丙基)和-nBu(正丁基);支链C3-4烷基,如-iPr(异丙基)、-iBu(异丁基)、-sBu(仲丁基)和-tBu(叔丁基);和环状C3-4烷基如-cP(环丙基)和-cBu(环丁基)。
在一个实施方案中,所述C2-4烯基选自直链C1-4烯基如-CH=CH2(乙烯基)和-CH2-CH=CH2(烯丙基(allyl))。
在一个实施方案中,所述卤代C1-4烷基选自:-CF3、-CH2CF3和-CF2CF3。
R1和R9基团
在一个实施方案中,R1和R9各自独立地为-H、-Me、-Et或-CF3。
在一个实施方案中,R1和R9各自独立地为-H、-Me或-Et。
在一个实施方案中,R1和R9相同。
在一个实施方案中,R1和R9不同。
在一个实施方案中,R1和R9各自独立地为-H。
在一个实施方案中,R1和R9各自独立地为-Me。
在一个实施方案中,R1和R9各自独立地为-Et。
R3NA和R3NB基团
R3NA和R3NB各自独立地选自:-H、C1-4烷基、C2-4烯基和卤代C1-4烷基。
在一个实施方案中,R3NA和R3NB各自独立地选自:C1-4烷基、C2-4烯基和卤代C1-4烷基。
在一个实施方案中,R3NA和R3NB各自独立地为:-Me、-Et、-nPr、-nBu、-CH2-CH=CH2或-CF3。
在一个实施方案中,R3NA和R3NB各自独立地为:-Me、-nPr、-nBu、-CH2-CH=CH2或-CF3。
在一个实施方案中,R3NA和R3NB各自独立地为:-Me或-Et。
在一个实施方案中,R3NA和R3NB相同。
在一个实施方案中,R3NA和R3NB不同。
在一个实施方案中,R3NA和R3NB各自独立地为-Me。
在一个实施方案中,R3NA和R3NB各自独立地为-Et。
R7NA和R7NB基团
R7NA和R7NB各自独立地选自:-H、C1-4烷基、C2-4烯基和卤代C1-4烷基。
在一个实施方案中,R7NA和R7NB各自独立地选自:C1-4烷基、C2-4烯基和卤代C1-4烷基。
在一个实施方案中,R7NA和R7NB各自独立地为:-Me、-Et、-nPr、-nBu、-CH2-CH=CH2或-CF3。
在一个实施方案中,R7NA和R7NB各自独立地为:-Me、-nPr、-nBu、-CH2-CH=CH2或-CF3.
在一个实施方案中,R7NA和R7NB各自独立地为:-Me或-Et。
在一个实施方案中,R7NA和R7NB相同。
在一个实施方案中,R7NA和R7NB不同。
在一个实施方案中,R7NA和R7NB各自独立地为-Me。
在一个实施方案中,R7NA和R7NB各自独立地为-Et。
在一个实施方案中,R3NA、R3NB、R7NA和R7NB相同。
在一个实施方案中,R3NA、R3NB、R7NA和R7NB如本申请所述,条件是,R3NA、R3NB、R7NA和R7NB中的至少一个不是-Et。
任选的条件
在一个实施方案中,所述化合物如本申请所述,但是条件是R3NA、R3NB、R7NA和R7NB各自不是-Et。
在一个实施方案中,所述化合物如本申请所述,但是条件是如果R1和R9各自为-H;则
R3NA、R3NB、R7NA和R7NB各自不是-Et。
-N(R3NA)(R3NB)和-N(R7NA)(R7NB)
在一个实施方案中:
R3NA和R3NB各自独立地为C1-4烷基、C2-4烯基或卤代C1-4烷基;
R7NA和R7NB各自独立地为C1-4烷基、C2-4烯基或卤代C1-4烷基;
任选地,条件是R3NA、R3NB、R7NA和R7NB中的至少一个不是-Et。
在一个实施方案中:
R3NA和R3NB各自独立地为-Me、-Et、-nPr、-nBu、-CH2-CH=CH2或-CF3;
R7NA和R7NB各自独立地为-Me、-Et、-nPr、-nBu、-CH2-CH=CH2或-CF3;
任选地,R3NA、R3NB、R7NA和R7NB中的至少一个不是-Et。
在一个实施方案中,
R3NA和R3NB各自独立地为-Me或-Et;
R7NA和R7NB各自独立地为-Me或-Et;
任选地,条件是R3NA、R3NB、R7NA和R7NB中的至少一个不是-Et。
在一个实施方案中,-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团相同。
在一个实施方案中,-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团不同。
在一个实施方案中,-N(R3NA)(R3NB)和-N(R7NA)(R7NB)各自独立地选自:-NMe2、-NEt2、-N(nPr)2、-N(Bu)2、-NMeEt、-NMe(nPr)和-N(CH2CH=CH2)2。
在一个实施方案中,-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团相同,并各自独立地选自:-NMe2、-NEt2、-N(nPr)2、-N(Bu)2、-NMeEt、-NMe(nPr)和-N(CH2CH=CH2)2。
在一个实施方案中,-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团相同,并各自独立地选自:-NMe2和-NEt2。
在一个实施方案中,-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自为:-NMe2+。
在一个实施方案中,-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团中的至少一个不是-NEt2。
在一个实施方案中,每一个基团-N(R3NA)(R3NB)和-N(R7NA)(R7NB)的不是-NEt2。
例如,在一个实施方案中,-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)相同,并选自:-NMe2、-N(nPr)2、-N(Bu)2、-NMeEt、-NMe(nPr)和-N(CH2CH=CH2)2。
HX1和HX2
HX1和HX2各自独立地为质子酸。
质子酸的例子包括如无机酸如氢卤酸(例如HCl、HBr、HI)、硝酸(HNO3)、硫酸(H2SO4)和有机酸如碳酸(H2CO3)和醋酸(CH3COOH)。
在一个实施方案中,HX1和HX2各自独立地为单质子酸。
在一个实施方案中,HX1和HX2各自独立地为氢卤酸(即hydrohalic acid)。
在一个实施方案中,HX1和HX2各自独立地选自HCl、HBr和HI。
在一个实施方案中,HX1和HX2相同。
在一个实施方案中,HX1和HX2不同。
在一个实施方案中,HX1和HX2相同,并独立地选自HCl、HBr和HI。在这种情况下,化合物(二氨基-吩噻嗪化合物)可便利地称作“二氨基-吩噻嗪双(氢卤酸)盐”。
在一个实施方案中,HX1和HX2各自为HCl。在这种情况下,所述化合物可便利地称作“二氨基-吩噻嗪双(盐酸)盐”。
在一个实施方案中,HX1和HX2各自为HBr。在这种情况下,所述化合物可便利地称作“二氨基-吩噻嗪双(氢溴酸)盐”。
在一个实施方案中,HX1和HX2各自为HI。在这种情况下,所述化合物可便利地称作“二氨基-吩噻嗪“二氨基-吩噻嗪双(氢碘酸)盐”。
一些优选的组合
在一个实施方案中:
R1和R9各自独立地为-H、-Me或-Et;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2或-NEt2。
在一个实施方案中:
R1和R9各自独立地为-H、-Me或-Et;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2。
在一个实施方案中:
R1和R9各自独立地为-H;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2或-NEt2。
在一个实施方案中:
R1和R9各自独立地为-H;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2。
在一个实施方案中:
R1和R9各自独立地为-H、-Me或-Et;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2或-NEt2;和
HX1和HX2各自独立地选自HCl、HBr和HI。
在一个实施方案中:
R1和R9各自独立地为-H、-Me或-Et;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2;和
HX1和HX2各自独立地选自HCl、HBr和HI。
在一个实施方案中:
R1和R9各自独立地为-H;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2或-NEt2;和
HX1和HX2各自独立地选自HCl、HBr和HI。
在一个实施方案中:
R1和R9各自独立地为-H;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2;和HX1和HX2各自独立地选自HCl、HBr和HI。
在一个实施方案中:
R1和R9各自独立地为-H;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2;和HX1和HX2各自为HCl。
在一个实施方案中:
R1和R9各自独立地为-H;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2;和HX1和HX2各自为HBr。
在一个实施方案中:
R1和R9各自独立地为-H;和
-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自独立地为-NMe2;和HX1和HX2各自为HI。
同位素变化
在一个实施方案中,所述化合物中的一个或多个碳原子为11C或13C或14C。
在一个实施方案中,所述化合物中的一个或多个碳原子为11C。
在一个实施方案中,所述化合物中的一个或多个碳原子为13C。
在一个实施方案中,所述化合物中的一个或多个碳原子为14C。
在一个实施方案中,所述化合物中的一个或多个氮原子为15N。
在一个实施方案中,R3NA、R3NB、R7NA、R7NB、R1、R9和R10中的一个或多个或所有基团的一个或多个或所有碳原子为11C(或13C)(或14C)。
在一个实施方案中,R3NA、R3NB、R7NA和R7NB中的一个或多个或所有基团的一个或多个或所有碳原子为11C(或13C)(或14C)。
在一个实施方案中,-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团相同,并且为:-N(11CH3)2(或-N(13CH3)2.)(或-N(14CH3)2.)。
相容组合
上述实施方案的所有相容组合在本申请中明确公开,就如同每种组合视为被专门、单独记载了那样。
一些优选的实施方案
在一个实施方案中,所述化合物选自下述化合物及其可药用盐、溶剂化物和水合物。
在一个实施方案中,所述化合物选自下述化合物及其可药用盐、溶剂化物和水合物。
在一个实施方案中,所述化合物选自下述化合物及其可药用盐、溶剂化物和水合物:
纯度
本发明的DAPTZ化合物可被便利地描述为处于“经稳定的还原形式”。该化合物氧化(例如自氧化)形成相应的氧化形式。因此,很有可能的是,尽管不是必然的,含有本发明DAPTZ化合物的组合物含有至少一些相应氧化化合物(作为杂质)。
因此,本发明另一方面涉及本申请所述的DAPTZ化合物,其为基本纯化形式(substantially purified form)和/或为基本不含杂质(如相应的氧化化合物、其它杂质)形式的。
在一个实施方案中,所述基本纯化形式为至少50wt%纯,例如至少60wt%纯,例如至少70wt%纯,例如至少80wt%纯,例如至少90wt%纯,例如至少95wt%纯,例如至少97wt%纯,例如至少98wt%纯,例如至少99wt%纯。
在一个实施方案中,杂质的含量不高于50wt%,例如不高于40wt%,例如不高于30wt%,例如不高于20wt%,例如不高于10wt%,例如不高于5wt%,例如不高于3wt%,例如不高于2wt%,例如不高于1wt%。
由方法限定的产物
在一个实施方案中,所述DAPTZ化合物是由本申请描述的方法所或的化合物或者可由本申请描述的方法获得的化合物。
化学合成
本申请中描述了本发明DAPTZ化合物的化学合成方法。这些和/或其它已知的方法可通过已知方式改进和/或调整,以有利于合成在本发明范围内的其它DAPTZ化合物。
例如,可利用例如亚硝酸钠和醋酸及氯仿,将合适的吩噻嗪转化为相应的3,7-二硝基-吩噻嗪。然后采用例如醋酸酐和吡啶对所述环上的氨基进行保护,例如通过形成醋酸酯的形式进行保护。然后利用例如二氯化锡(II)和乙醇,将所述硝基还原成氨基。所述氨基接着被取代,例如被二取代(例如被甲基二取代),例如用例如碘甲烷、氢氧化钠、DMSO和四正丁基溴化铵进行。然后对所述氨基进行脱保护,用例如浓盐酸水溶液将N-乙酰基除去。接着制备成相应的盐,用例如浓盐酸水溶液,在例如脱保护的同时进行。该方法的实例如下方案所示:
方案1
因此,本发明另一方面涉及制备如下结构式的3,7-二氨基-10H-吩噻嗪(DAPTZ)化合物的方法:
其中,R1、R9、R3NA、R3NB、R7NA、R7NB、HX1和HX2如本申请所定义(例如,其中HX1和HX2各自为HCl),所述方法包括如下步骤:
(vi)形成盐(SF)。
在一个实施方案中,所述方法包括如下步骤:
(v)环上的氨基脱保护(DP);和
(vi)形成盐(SF)。
在一个实施方案中,所述方法包括如下步骤:
(iv)胺取代(AS),
任选的(v)环上的氨基脱保护(DP),和
(vi)形成盐(SF)。
在一个实施方案中,所述方法包括如下步骤:
(iii)硝基还原(NR),
(iv)胺取代(AS),
(v)环上的氨基脱保护(DP),和
(vi)形成盐(SF)。
在一个实施方案中,所述方法包括如下步骤:
任选的(ii)环上的氨基保护(AP),
(iii)硝基还原(NR),
(iv)胺取代(AS),
(v)环上的氨基脱保护(DP),和
(vi)形成盐(SF)。
在一个实施方案中,所述方法包括如下步骤:
(i)硝化(NO),
(ii)环上的氨基保护(AP),
(iii)硝基还原(NR),
(iv)胺取代(AS),
(v)环上的氨基脱保护(DP),和
(vi)形成盐(SF)。
在一个实施方案中,所述步骤是以所列顺序进行的(即列表中任一步骤与列表中的前一步骤同时进行或相继进行)。
在一个实施方案中,步骤(v)环上的氨基脱保护(DP)和步骤(vi)形成盐(SF)同时进行(即作为一个步骤)。
在一个实施方案中,所述硝化(NO)步骤为:
(i)硝化(NO),其中将10H-吩噻嗪转化成3,7-二硝基-10H-吩噻嗪,例如:
在一个实施方案中,硝化是利用亚硝酸盐进行的,例如用亚硝酸钠,例如亚硝酸钠及醋酸和氯仿进行。在一个实施方案中,R10为-H。
在一个实施方案中,环上的氨基保护(AP)步骤为:
(ii)环上的氨基保护(AP),其中将3,7-二硝基-10H-吩噻嗪的环上的氨基(-NH-)转化为经保护的环上的氨基(-NR保护),例如:
在一个实施方案中,环上的氨基保护采用醋酸酯形式,例如利用醋酸酐形成醋酸酯形式,例如采用醋酸酐及吡啶进行保护。
在一个实施方案中,所述硝基还原(NR)步骤为:
(iii)硝基还原(NR),其中将经保护的3,7-二硝基-10H-吩噻嗪的每一个硝基(-NO2)转化为氨基(-NH2),例如:
在一个实施方案中,硝基还原可利用如二氯化锡(II),例如利用二氯化锡(II)及乙醇进行还原。
在一个实施方案中,胺取代(AS)步骤为:
(iv)胺取代(AS),其中,将经保护的3,7-二氨基-10H-吩噻嗪中的氨基(-NH2)转化为二取代的氨基,例如:
在一个实施方案中,胺取代是利用烷基卤化物进行的,例如利用烷基碘化物如碘甲烷进行的,如利用碘甲烷及氢氧化钠、DMSO和四正丁基溴化铵进行胺取代。
在一个实施方案中,所述环上的氨基脱保护(DP)步骤是:
(v)环上的氨基脱保护(DP),其中将保护基团R保护除去,例如:
在一个实施方案中,环上的氨基脱保护可利用酸如盐酸进行,用利用浓盐酸水溶液进行。
在一个实施方案中,形成盐步骤为:
(vi)形成盐(SF),其中形成相应的盐,例如:
在一个实施方案中,形成盐可利用酸如盐酸来进行,如利用浓盐酸水溶液进行。
在一个实施方案中,环胺脱保护步骤和形成盐步骤同时进行(即作为一步),例如在一步反应,由化合物(2)得到化合物(1)。
在另一方法中,将合适的锍氯化物(例如甲基锍氯化物,MTC,也已知称作次甲蓝)转化为相应的卤化物,例如通过与碘化钾(如碘化钾水溶液)反应进行转化。然后将所得的锍碘化物进行还原,例如用碘乙烷和乙醇还原,形成相应的盐。类似的方法公开在Drew,H.D.K,和Head,F.S.H.,“Derivatives of Methylene-blue,”Journal of the Chemical Society,1933,248-253页中。这种方法的实例如下方案所示:
方案2
因此,本发明另一方面涉及制备如下结构式的3,7-二氨基-10H-吩噻嗪(DAPTZ)化合物的方法:
其中,R1、R9、R3NA、R3NB、R7NA、R7NB、HX1和HX2如本申请所定义(例如,其中HX1和HX2各自为HI),所述方法包括如下步骤:
(ii)还原并形成碘化物盐(RISF)。
在一个实施方案中,所述方法包括如下步骤:
(i)碘交换(iodide exchange,IE);和
(ii)还原并形成碘化物盐(RISF)。
在一个实施方案中,所述步骤是以所列顺序进行的(即列表中任一步骤与列表中的前一步骤同时进行或相继进行)。
在一个实施方案中,所述碘交换(IE)步骤为:
(i)碘交换(IE),其中将3,7-二(二取代的氨基)-锍盐转化为相应的3,7-二(二取代的氨基)-锍碘化物,例如(其中Y-为阴离子反离子,例如为卤素离子,例如为氯离子或为溴离子):
在一个实施方案中,碘交换(IE)是通过与碘化钾反应进行的,例如利用碘化钾水溶液进行碘交换。
在一个实施方案中,还原并形成碘化物盐(RISF)步骤为:
(ii)还原并形成碘化物盐(RISF),其中3,7-二(二取代的氨基)-锍碘化物被还原并转化为相应的3,7-二氨基-10H-吩噻嗪碘化物,例如:
在一个实施方案中,还原和形成碘化物盐(RISF)是通过与碘乙烷反应进行的,例如采用碘乙烷及乙醇进行反应。
在另一方法中,合适的锍盐,例如乙基锍-半(二氯化锌)同时被还原且环上的氨基被保护,例如通过与苯肼、乙醇、醋酸酐和吡啶进行反应。然后制备成相应的盐,例如利用浓盐酸水溶液制备成相应的盐,例如在脱保护的同时进行。这种方法的实例如下方案所示:
方案3
因此,本发明另一方面涉及制备如下结构式的3,7-二氨基-10H-吩噻嗪(DAPTZ)化合物的方法:
其中,R1、R9、R3NA、R3NB、R7NA、R7NB、HX1和HX2如本申请所定义(例如,其中HX1和HX2各自为HI),所述方法包括如下步骤:
(iv)形成盐(SF)。
在一个实施方案中,所述方法包括如下步骤:
(iii)环上的氨基脱保护(DP),和
(iv)形成盐(SF)。
在一个实施方案中,所述方法包括如下步骤:
(ii)环上的氨基保护(AP),
(iii)环上的氨基脱保护(DP),和
(iv)形成盐(SF)。
在一个实施方案中,所述方法包括如下步骤:
(i)还原(RED)
(ii)环上的氨基保护(AP),
(iii)环上的氨基脱保护(DP),和
(iv)形成盐(SF)。
在一个实施方案中,所述步骤是以所列顺序进行的(即列表中任一步骤与列表中的前一步骤同时进行或相继进行)
在一个实施方案中,(i)还原(RED)步骤和(ii)环上的氨基保护(AP)步骤同时进行(即作为一步)。
例如,在一个实施方案中,还原(RED)步骤和环上的氨基保护(AP)步骤的结合如下:
(i)还原(RED)和环上的氨基保护(AP),其中将3,7-二(二取代的氨基)-锍盐还原得到相应的3,7-二(二取代的氨基)-10H-吩噻嗪,将3,7-二(二取代的氨基)-10H-吩噻嗪的环上的氨基(-NH-)转化为经保护的氨基(-R保护),得到相应经保护的3,7-二(二取代的氨基)-10H-吩噻嗪,例如
在一个实施方案中,Y代表Cl-。
在一个实施方案中,还原(RED)步骤和环上的氨基保护(AP)步骤的结合是利用苯肼和醋酸酐进行的,例如利用苯肼、乙醇、醋酸酐和吡啶进行还原步骤和环上的氨基保护步骤。
在一个实施方案中,(iii)环上的氨基脱保护(DP)步骤和(iv)形成盐(SF)步骤是同时进行的(即作为一个步骤)。
例如,在一个实施方案中,环上的氨基脱保护(DP)步骤和形成盐(SF)步骤的结合如下:
(ii)环上的氨基脱保护(DP)和形成盐(SF),其中将经保护的3,7-二(二取代的氨基)-10H-吩噻嗪的保护基团脱除,得到3,7-二(二取代的氨基)-10H-吩噻嗪,形成相应的盐,例如:
在一个实施方案中,环上的氨基脱保护(DP)步骤和形成盐(SF)步骤的结合可利用酸如盐酸进行,例如利用浓盐酸水溶液进行环上的氨基脱保护步骤和形成盐步骤。
在相似的方法中,合适的锍氯化物(例如甲基锍氯化物,乙基锍氯化物)首先被还原和乙酰化,得到相应的1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮,例如通过与肼(NH2NH2)、甲肼(MeNHNH2)或硼氢化钠(NaBH4)反应进行还原;以及与醋酸酐((H3CCO)2O)反应进行乙酰化;例如在合适碱如吡啶(C5H5N)或许尼希碱(二异丙基乙胺,C8H19N)的存在下,在合适的溶剂如乙醇和乙腈中进行反应。然后对该被还原和乙酰化的化合物进行脱保护(通过脱除乙酰基),例如在任选地添加有合适的醚,例如乙醚的情况下,通过与合适的氢卤酸如盐酸或氢溴酸,在合适的溶剂如乙醇中反应来进行脱保护。
组合物
本发明另一方面涉及含有本申请所述DAPTZ化合物和可药用载体或稀释剂的组合物。
用途
逆转和/或抑制蛋白聚集
本发明一个方法是本发明所述的DAPTZ化合物的用途,用于调节(例如逆转和/或抑制)蛋白聚集,例如调节(如逆转和/或抑制)与神经变性疾病和或临床痴呆有关的蛋白聚集。该聚集可在体外或体内发生,并与下述疾病状态(disease state)有关。
因此,本发明一个方面涉及调节(例如逆转和/或抑制)蛋白聚集如与神经变性疾病和或临床痴呆有关的蛋白聚集的方法,包括将所述蛋白与有效量的本发明所述DAPTZ化合物接触。该方法可在体外或体内进行。
相似地,本发明一方面涉及调节(例如逆转和/或抑制)哺乳动物的脑中蛋白聚集的方法,所述聚集与本发明描述的疾病状态有关,所述治疗包括向所述需要该治疗的哺乳动物给药预防有效量或治疗有效量的本申请所述DAPTZ化合物,所述DAPTZ化合物是所述聚集的抑制剂。
治疗方法
本发明另一方面涉及治疗方法,包括向需要这种治疗的患者给药治疗预防有效量或治疗有效量的本申请所述DAPTZ化合物,所述化合物优选采用药物组合物形式。
治疗方法中的用途
本发明另一方面涉及本申请所述的DAPTZ化合物,所述化合物通过治疗而用于在治疗人体或动物体的方法(例如治疗疾病)中。
制备药物中的用途
本发明另一方面涉及本申请所述的DAPTZ化合物在制备用在治疗(例如治疗疾病)的药物中的用途。
在一个实施方案中,所述药物含有DAPTZ化合物。
被治疗的疾病状态-蛋白聚集疾病
本发明所述的DAPTZ化合物用于治疗或预防蛋白聚集疾病(disease of protein aggregation)。
因此,在一个实施方案中,所述疾病为蛋白聚集疾病,并且所述治疗例如利用本申请所述的DAPTZ化合物,其量足以抑制与所述疾病状态有关的蛋白聚集。
总的来说,所述蛋白聚集是由经诱导的构象聚合相互作用(induced conformational polymerisation interaction)引起的,即其中蛋白或其片段的构象变化导致了模板结合(templated binding),以及导致了更多(前体)蛋白分子以自传播的方式(self-propagating manner)聚集。一旦引发成核,就会随之发生聚集级联(aggregation cascade),这种聚集级联涉及更多蛋白质分子发生诱导的构象聚合,从而导致形成聚集体中的有毒产物片段,这些有毒产物片段基本上耐进一步的蛋白质水解。这样形成的蛋白质聚集体(protein aggregates)被认为是以下疾病状态的最接近病因,所述疾病状态表现为神经变性、临床痴呆和其它病理症状。
下表列出了给这疾病相关的聚集蛋白质和相应的蛋白聚集疾病。
针对上表的参考文献:
Abrahamson,M.,Jonsdottir,S.,Olafsson,I.&Grubb,A.(1992)Hereditarycystatin C amyloid angiopathy identification of the disease-causing mutation and specific diagnosis by polymerasechain reaction based analysis.Human Genetics 89,377-380.
Booth,D.R.,Sunde,M.,Bellotti,V.,Robinson,C.V.,Hutchinson,W.L.,Fraser,P.E.,Hawkins,P.N.,Dobson,C.M.,Radford,S.E.,Blake,C.C.F.&Pepys,M.B.(1997)Instability,unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis.Nature 385,787-793.
Carrell,R.W.&Gooptu,B.(1998)Conformationalchanges and disease-serpins,prions and Alzheimer's.Current Opinion in Structural Biology 8,799-809.
Chiti,F.,Webster,P.,Taddei,N.,Clark,A.,Stafani,M.,Ramponi,G.&Dobson,C.(1999)Designingconditions for in vitro formation of amyloid protofilaments and fibrils.Proceedings of the National Academy of Sciences,USA 96,3590-3594.
Czech,C.,Tremp,G.&Pradier,L.(2000)Presenilins and Alzheimer's disease:biological functions and pathogenic mechanisms.Progress in Neurobiology 60,363-384.
Davis,R.L.,Shrimpton,A.E.,Holohan,P.D.,Bradshaw,C.,Feiglin,D.,Collins,G.H.,Sonderegger,P.,Kinter,J.,Becker,L.M.,Lacbawan,F.,Krasnewich,D.,Muenke,M.,Lawrence,D.A.,Yerby,M.S.,Shaw,C.-M.,Gooptu,B.,Elliott,P.R.,Finch,J.T.,Carrell,R.W.&Lomas,D.A.(1999)Familial dementiacaused by polymerization of mutant neuroserpin.Nature 401,376-379.
DiFiglia,M.,Sapp,E.,Chase,K.O.,Davies,S.W.,Bates,G.P.,Vonsattel,J.P.&Aronin,N.(1997)Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain.Science 277,1990-1993.
Dische,F.E.,Wernstedt,C.,Westermark,G.T.,Westermark,P.,Pepys,M.B.,Rennie,J.A.,Gilbey,S.G.&Watkins,P.J.(1988)Insulin as an amyloid-fibril protein at sites of repeated insulin injections in a diabetic patient.Diabetologia 31,158-161.
Gasset,M.,Bladwin,M.A.,Lloyd,D.H.,abriel,J.-M.,Holtzman,D.M.,Cohen,F.E.,Fletterick,R.&Prusiner,S.B.(1992)Predicted a-helical region of the prion protein when synthesized as peptides form amyloid.Proceedings of the National Academy of Sciences,USA 89,10940-10944.
Glenner,G.G.&Wong,C.W.(1984)Alzheimer's disease:initial report of the purification andcharacterisation of a novelcerebrovascular amyloid protein.Biochemical and Biophysical Research Communications 120,885-890.
Goate,A.,Chartier-Harlin,M.-C.,Mullan,M.,Brown,J.,Crawford,F.,Fidani,L.,Giuffra,L.,Haynes,A.,Irving,N.,James,L.,Mant,R.,Newton,P.,Rooke,K.,Roques,P.,Talbot,C.,Pericak-Vance,M.,Roses,A.,Williamson,R.,Rossor,M.,Owen,M.&Hardy,J.(1991)Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease.Nature 349,704-706.
Gorevic,P.D.,Casey,T.T.,Stone,W.J.,DiRaimondo,C.R.,Prelli,F.C.&Frangione,B.(1985)b-2 Microglobulin is an amyloidogenic protein in man.Journal of Clinical Investigation 76,2425-2429.
Gustavsson,A.,U.&Westermark,P.(1991)Normal transthyretin and synthetic transthyretin fragments form amyloid-like fibrils in vitro.Biochemical and Biophysical Research Communications 175,1159-1164.
Hutton,M.,Lendon,C.,Rizzu,P.,Baker,M.,Froelich,S.,Houlden,H.,Pickering-Brown,S.,Chakraverty,S.,Isaacs,A.,Grover,A.,Hackett,J.,Adamson,J.,Lincoln,S.,Dickson,D.,Davies,P.,Petersen,R.C.,Stevens,M.,deGraaf,E.,Wauters,E.,van Baren,J.,Hillebrand,M.,Joosse,M.,Kwon,J.M.,Nowotny,P.,Che,L.K.,Norton,J.,Morris,J.C.,Reed,L.A.,Trojanowski,J.Q.,Basun,H.,Lannfelt,L.,Neystat,M.,Fahn,S.,Dark,F.,Tannenberg,T.,Dodd,P.R.,Hayward,N.,Kwok,J.B.J.,Schofield,P.R.,Andreadis,A.,Snowden,J.,Craufurd,D.,Neary,D.,Owen,F.,Oostra,B.A.,Hardy,J.,Goate,A.,van Swieten,J.,Mann,D.,Lynch,T.&Heutink,P.(1998)Association of missense and5'-splice-site mutations in tau with the inherited dementia FTDP-17.Nature 393,702-705.
Johansson,B.,Wernstedt,C.&Westermark,P.(1987)Atrial natriuretic peptide deposited as atrial amyloid fibrils.Biochemical and Biophysical Research Communications 148,1087-1092.
Lomas,D.A.,Evans,D.L.,Finch,J.T.&Carrell,R.W.(1992)The mechanism of Z a1-antitrypsin accumulation in the liver.Nature 357,605-607.
Maury,C.P.&Baumann,M.(1990)Isolation andcharacterization ofcardiac amyloid in familial amyloid polyneuropathy type IV(Finnish):relation of the amyloid protein to variant gelsolin.Biochimica et Biophysica Acta 1096,84-86.
Paulson,H.L.(1999)Human genetics'99:trinucleotide repeats.American Journal of Human Genetics 64,339-345.
Pepys,M.B.,Hawkins,P.N.,Booth,D.R.,Vigushin,D.M.,Tennent,G.A.,Soutar,A.K.,Totty,N.,Nguyen,O.,Blake,C.C.F.,Terry,C.J.,Feest,T.G.,Zalin,A.M.&Hsuan,J.J.(1993)Human lysozyme gene mutationscause hereditary systemic amyloidosis.Nature 362,553-557.
Polymeropoulos,M.H.,Lavedan,C.,Leroy,E.,Ide,S.E.,Dehejia,A.,Dutra,A.,Pike,B.,Root,H.,Rubenstein,J.,Boyer,R.,Stenroos,E.S.,Chandrasekharappa,S.,Athanassiadou,A.,Papaetropoulos,T.,Johnson,W.G.,Lazzarini,A.M.,Duvoisin,R.C.,Di Iorio,G.,Golbe,L.I.&Nussbaum,R.L.(1997)Mutation in the a-synuclein gene identified in families with Parkinson’s disease.Science 276,2045-2047.
Prusiner,S.B.,Scott,M.R.,DeArmond,S.J.&Cohen,F.E.(1998)Prion protein biology.Cell 93,337-348.
Shibata,N.,Hirano,A.,Kobayashi,M.,Siddique,T.,Deng,H.X.,Hung,W.Y.,Kato,T.&Asayama,K.(1996)Intense superoxide dismutase-1immunoreactivity in intracytoplasmic hyaline inclusions of familial Amyotrophic lateral sclerosis with posteriorcolumn involvement.Journal of Neuropathology and Experimental Neurology 55,481-490.
Sletten,K.,Westermark,P.&Natvig,J.B.(1976)Characterization of amyloid fibril proteins from medullarycarcinoma of the thyroid.Journal of Experimental Medicine 143,993-998.
Spillantini,M.G.,Crowther,R.A.,Jakes,R.,Hasegawa,M.&Goedert,M.(1998)a-synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies.Proceedings of the National Academy of Sciences,USA 95,6469-6473.
Uemichi,T.,Liuepnicks,J.j.&Benson,M.D.(1994)Hereditary renal amyloidosis with a novel variant fibrinogen.Journal of Clinical Investigation 93,731-736.
Westermark,P.,Engstrom,U.,Johnson,K.H.,Westermark,G.T.&Betsholtz,C.(1990)Islet amyloid polypeptide:pinpointing amino acid residues linked to amyloid fibril formation.Proceedings of the National Academy of Sciences,USA87,5036-5040.
Westermark,P.,Johnson,K.H.,O'Brien,T.D.&Betsholtz,C.(1992)Islet amyloid polypeptide-a novelcontroversy in diabetes research.Diabetologia 35,297-303.
Westermark,P.,Johnson,K.H.&Pitkanen,P.(1985)Systemic amyloidosis:A review with emphasis on pathogenesis.Applied Physiology 3,55-68.
Wischik,C.M.,Novak,M.,H.C.,Edwards,P.C.,Runswick,M.J.,Jakes,R.,Walker,J.E.,Milstein,C.,M.,R.&Klug,A.(1988)Isolation of a fragment of tau derived from thecore of the paired helical filament of Alzheimer's disease.Proceedings of the National Academy of Sciences,USA 85,4506-4510.
如在WO 02/055720和2006年3月29日提交的美国专利申请号60/786,700(题目:Inhibitors of Protein Aggregation)所公开的那样,二氨基吩噻嗪类化合物能用于抑制这些蛋白聚集疾病。
因此,应当理解的是,除非文中给出相反说明,对与τ蛋白或类τ蛋白(例如MAP2)有关的实施方案的描述应当被视为能同样适于本申请所述的其它蛋白质(例如β-淀粉样蛋白、突触核蛋白、朊病毒等)或其它蛋白质,这些蛋白质由于在对聚集传播至关重要的结构域种的构象变化,可引发或历经相似的病理性聚集,或者所述蛋白使由此形成的聚集体具有蛋白水解稳定性(参见例如Wischik等人在“Neurobiology of Alzheimer’disease”的文章中所述的那样,第2版,2000,Eds.Dawbarn,D.and Allen,S.J.,The Molecular and Cellular Neurobiology Series,Bios Scientific Publishers,Oxford)。所有这样的蛋白质都可在本申请中称作“聚集疾病蛋白质”。
同样,当本申请中提到“τ-τ聚集”或相似表述时,其也可视为适用于其它“聚集蛋白质的聚集”,如β-淀粉样蛋白聚集、朊病毒蛋白聚集、突触核蛋白聚集等。同样适用于“τ蛋白水解性降解”等。
优选的聚集疾病蛋白质
本发明优选的实施方案是基于τ蛋白。本申请所述的术语“τ蛋白”总体上是指τ蛋白家族的任何一种蛋白质。τ蛋白的特征为作为蛋白质家族众多成员中的一个,其在组装和拆散(disassembly)这样的重复循环中与微管共纯化(Shelanski等人(1973)Proc.Natl.Acad.Sci.USA,70.,765-768),并已知为微管-相关蛋白(microtubule-associated-proteins,MAPs)。τ家族的成员具有共同的特征,即具有特征性N-端片段、特征性串联重复区域和C-端尾部,其中约50个氨基酸的序列插在N-端片段中,该片段在脑部随发育经逐步调节,并且其中所述串联重复区域由3至4个31-32氨基酸的串联重复(序列)组成。
MAP2是在胞体树突室(somatodendritic compartment)中主导的微管相关蛋白(参见如Matus,A.,in“Microtubules”[Hyams and Lloyd,Eds.],第155-166页,John Wiley and Sons,New York,USA)。MAP2同工型在串联重复域中与τ蛋白几乎相同,但在N-端域的序列和长度方面存在较大差别(参加如Kindler和Garner,1994,Mol.Brain Res.,第26卷,第218-224页)。然而,在串联重复域(tandem-repeat region)的聚集对于τ重复域来说不是选择性的。因此,可以理解的是,本申请中任何有关τ蛋白或τ-τ聚集的讨论应当被视为也与τ-MAP2聚集、MAP2-MAP2聚集等等相关。
在一个实施方案中,所述蛋白为τ蛋白。
在一个实施方案中,所述蛋白为突触核蛋白,如α-突触核蛋白或β-突触核蛋白。
当所述蛋白为τ蛋白时,本发明的一个实施方案提供了一种抑制哺乳动物的脑中蛋白质聚集体(例如双股螺旋形细丝(PHFs)形式,任选在神经原纤维缠结(NFTs)中)产生的方法,治疗如上所述。
优选的蛋白聚集疾病
显然,τ蛋白(及其异常功能或加工)不光在阿尔茨海默氏病(AD)中扮演重要角色。神经变性疾病如匹克病(Pick’s disease)和进行性核上性麻痹(Progressive Supranuclear Palsy,PSP)的发病机理看起来与病理性截短的τ聚集体分别在新皮质的齿状回(dentate gyrus)和星状锥体细胞中的聚集相互关联。其它痴呆病包括额颞痴呆(fronto-temporal dementia,FTD);与染色体相关的帕金森综合征(parkinsonism linked tochromosome 17,FTDP-17);去抑制-痴呆-帕金森-萎缩综合征(disinhibition-dementia-parkinsonism-amyotrophycomplex,DDPAC);核-脑桥-黑质变性(pallido-ponto-nigral degeneration,PPND);ALS-关岛痴呆综合征(Guam-ALS syndrome);核-黑质-苍白球变性(pallido-nigro-luysian degeneration,PNLD);皮质-基底变性(cortico-basal degeneration,CBD)及其它(参见(参见例如Wischik等人在“Neurobiology of Alzheimer’disease”的文章中所述的那样,第2版,2000,Eds.Dawbarn,D.and Allen,S.J.,The Molecular and Cellular Neurobiology Series,Bios Scientific Publishers,Oxford,特别是表5.1)。所有这些疾病都称为“τ蛋白病”或称作“τ蛋白聚集疾病”,该疾病的特征主要或部分为异常τ聚集。
因此,在一个实施方案中,所述疾病为τ蛋白病。
在一个实施方案中,所述疾病为神经病变性τ蛋白病。
在一个实施方案中,所述疾病为阿尔茨海默氏病。
在一个实施方案中,治疗(例如神经病变性τ蛋白病如阿尔茨海默氏病的治疗)可任选地与一种或多种其它制剂联合使用,所述其它制剂如一种或多种胆碱酯酶抑制剂(例如多奈哌齐(也已知为AriceptTM)、利凡斯的明(也已知为ExelonTM)、加兰他敏(也已知为ReminylTM)、NMDA受体拮抗剂(例如美金刚(也已知为EbixaTM、NamendaTM)、毒蕈碱受体激动剂和/或淀粉样前体蛋白加工的抑制剂,该淀粉样前体蛋白加工导致β-淀粉样蛋白生成的增强。
治疗的疾病-其它疾病
在一个实施方案中,所述疾病为皮肤癌。
在一个实施方案中,所述疾病为黑素瘤。
在一个实施方案中,所述疾病为病毒、细菌或原生动物疾病状态。
在一个实施方案中,所述(原生动物类)疾病为疟疾。
在这个实施方案中,治疗可与一种或多种抗微生物制剂联合,例如氯喹和/或阿托伐醌(atovaquone)。
在一个实施方案中,所述(病毒)疾病是由丙型肝炎、HIV或西尼罗病毒(WNV)导致的。
其它用途
本发明另一方面涉及本申请所述的DAPTZ化合物在灭活样品(例如血液或血浆样品)中病原体的方法中的用途,包括将所述DAPTZ化合物引入到样品中,以及将样品暴露于光的步骤。
例如,在一个实施方案中,所述方法包括将DAPTZ化合物引入到样品中,然后将该样品暴露于光的步骤。
作为配体的用途
所述能抑制τ蛋白聚集的DAPTZ化合物也能作为配体或τ蛋白(或聚集的τ蛋白)的标记。因此,在一个实施方案中,所述DAPTZ化合物为τ蛋白(或聚集的τ蛋白)配体。
这些DAPTZ化合物(配体)可以与其它化学基团结合(incorporate)、连接(conjugated)、螯合(chelated)或联合(associated),例如与稳定的或不稳定的可检测同位素、放射性同位素、正电子发射原子(positron-emitting atoms)、磁共振标记(magnetic resonance labels)、染料、荧光标记物、抗原基团(antigenic groups)、治疗部分(therapeutic moieties)或其它任何有助于预防、诊断或治疗用途的基团或原子结合、连接、螯合或联合。
例如,在一个实施方案中,所述DAPTZ化合物如本申请所述,但是附加的限定是将所述化合物与一个或多个(例如1、2、3、4个等)可检测标记如同位素、放射性同位素、正电子发射原子、磁共振标记、染料、荧光标记物、抗原基团、治疗部分结合、连接、螯合或联合。
在一个实施方案中,所述DAPTZ化合物为配体和标记,如针对τ蛋白(或聚集的τ蛋白)的标记,并且与一个或多个(例如1、2、3、4个等)可检测标记结合、连接、螯合或联合。
例如,在一个实施方案中,所述DAPTZ如上所述,但是附加的限定是将所述化合物与一个或多个(例如1、2、3、4个等)可检测标记结合、连接、螯合或联合。
可以通过任何合适的手段显现或检测经标记的DAPTZ化合物(例如,当配合到τ蛋白或聚集的τ蛋白时),本领域技术人员能够理解的是可使用任何本领域已知的合适检测手段。
例如,所述DAPTZ化合物(配体-标记)可以合适地如下检测:结合正电子发射原子(例如11C)(例如作为一个或多个烷基取代基(如甲基取代基)的碳原子),并用本领域已知的正电子发射断层扫描术(PET)对所述化合物进行检测。
可采用本申请公开的已知的方法制备这些11C标记的DAPTZ化合物,例如与在WO 02/075318(参见图11a、11b、12)和WO 2005/030676中公开的方法类似的方法。
因此,本发明另一方面涉及一种标记τ蛋白(或聚集的τ蛋白)的方法,包括如下步骤:将所述τ蛋白(或聚集的τ蛋白)与DAPTZ化合物接触,该DAPTZ化合物与一个或多个(例如1、2、3、4个等)可检测标记结合、连接、螯合或联合。
本发明另一方面提供一种检测τ蛋白(或聚集的τ蛋白)的方法,包括如下步骤:(i)将τ蛋白(或聚集的τ蛋白)与DAPTZ化合物接触,该DAPTZ化合物与一个或多个(例如1、2、3、4个等)可检测标记结合、连接、螯合或联合,和(ii)检测与τ蛋白(或聚集的τ蛋白)结合的所述化合物的存在和/或其量。
本发明另一个方面提供诊断或预测(prognosis)被认为患有τ蛋白病的受试者(subject)该疾病的方法,包括如下步骤:
(i)向受试者中引入能够标记τ蛋白或聚集的τ蛋白,特别是τ蛋白的DAPTZ化合物(例如与一个或多个(例如1、2、3、4个等)可检测标记结合、连接、螯合或联合的DAPTZ化合物),
(ii)测定与受试者脑中τ蛋白或聚集的τ蛋白结合的所述化合物的存在和/或其量,
(iii)将(ii)中的测定结果与受试者的疾病状态相关联。
本发明另一个方面涉及能够标记τ蛋白或聚集的τ蛋白的DAPTZ化合物(例如与一个或多个(例如1、2、3、4个等)可检测标记结合、连接、螯合或联合的DAPTZ化合物),用在诊断或预测τ蛋白病的方法中。
本发明另一个方面涉及能够标记τ蛋白或聚集的τ蛋白,特别是τ蛋白的DAPTZ化合物(例如与一个或多个(例如1、2、3、4个等)可检测标记结合、连接、螯合或联合的DAPTZ化合物)在制备用于诊断或预测τ蛋白病的诊断剂或预测剂中的用途。
本领域技术人员能理解的是,DAPTZ化合物配体物/标记物可以以前体形式给药,通过所述受试者中存在的或服用的活化剂而转化为活性形式(例如配合形式、标记形式),而不是直接给药这些二氨基吩噻嗪化合物配体物/标记物。
本发明披露的配体物可用作诊断方法或预测方法的一部分。可以用其选择进行治疗的病人或用来评估用在所述受试者的治疗或疗法(例如τ蛋白聚集抑制剂)的有效性。
治疗
用在本发明治疗疾病语境中的术语“治疗”总体上与治疗和疗法有关,不管是针对人类还是动物(例如兽用),其中获得某些所需的治疗效果,例如抑制病症发展,并包括减慢发展速度、使发展停止、病症消退、病症改善和病症治愈。也包括作为预防措施(即预防、防治)的治疗。
本文使用的术语“治疗有效量”涉及DAPTZ化合物的量或含有DAPTZ化合物的物质、组合物或剂型(dosage from)的量,当根据所述的治疗方案给药后所述量足以产生某些所需的治疗效果,与合理的益处/风险比例相称。
相似地,本文中使用的术语“预防有效量的”涉及DAPTZ化合物的量或含有DAPTZ化合物的物质、组合物或剂型(dosage from)的量,当根据所需的治疗方案给药后所述量足以产生某些所需的预防效果,与合理的益处/风险比例相称。
术语“处置或治疗”包括联合治疗和联合疗法,其中将两种或多种治疗剂或疗法例如连续或同时联合。治疗和疗法的实例包括但不限于化学疗法(给药活性制剂包括如药物、抗体(例如在免疫疗法中那样)、前药(例如光敏疗法中那样、GDEPT、ADEPT等);外科手术;放射疗法和基因疗法。
例如,将利用本申请所述的DAPTZ化合物的处置或治疗与一种或多种(例如1、2、3、4种)其它制剂或治疗剂联合治疗可能是有益的。
具体的联合在主治医师的判断能力之内,医师会根据其掌握的常识和技术人员已知的剂量方案选择剂量。
所述药剂(即本申请所述的DAPTZ化合物,加上一种或多种其它药剂)可同时或顺序给药,并可经单独变化的剂量方案通过不同的途径给药。例如当顺序给药时,所述制剂可以较短间隔(例如5-10分钟内)或以较长的时间间隔(例如1、2、3、4个小时或更长间隔,或者甚至根据需要间隔时间更长)给药,精确的剂量方案与治疗剂的性质对应。
所述药剂(即本申请所述的DAPTZ化合物,加上一种或多种其它药剂)可配制在单一剂型中,或者可分别配制各个药剂,以试剂盒的形式装在一起,任选地带有针对这些制剂的使用说明书。
给药途径
可采用任何方便的给药途径将所述DAPTZ化合物或含有该化合物的药物组合物给药到受试者/病人,无论全身/外周或局部给药(即在所希望起作用的部位)都是可以的。
给药途径包括但不限于口服(例如摄入(ingestion));含服;舌下;经皮(包括如通过贴剂(patch)、膏剂(plaster)等);透粘膜(包括如通过贴剂、膏剂等);鼻内(例如通过鼻腔喷雾);眼部(例如通过滴眼剂);肺部(例如通过例用如气溶胶的吸入或吹入疗法,如通过如嘴或鼻);直肠(例如通过栓剂或灌肠剂);阴道(例如通过阴道栓剂);非肠道方式如通过注射包括皮下注射、皮内注射、肌内注射、静脉注射、动脉内注射、心内注射、膜内注射、椎内注射、囊内注射、囊下注射、眼眶内注射、腹膜内注射、气管内注射、表皮下注射、关节内注射、蛛网膜下注射和胸骨内注射(包括如内腔式注射到脑内);通过埋入库(depot)或贮器(reservoir),例如连续或同时埋入。
受试者/患者
受试者/患者可以是动物、哺乳动物、胎生哺乳动物、啮齿目动物(例如豚鼠、仑鼠、大鼠、小鼠)、鼠科动物(例如小鼠)、兔形目动物(例如家兔)、禽类(例如鸟)、犬类(例如狗)、猫科动物(例如猫)、马科动物(例如马)、猪科动物(例如猪)、羊族动物(例如绵羊)、牛族动物(例如母牛)、灵长类(动物)、猿类(例如猴子或猿)、猴子(例如狨猴、狒狒)、猿(例如大猩猩、黑猩猩、猩猩(orangutang)、长臂猿(gibbon)),或人类。
进一步,受试者/患者可以处于其成长的任何阶段,例如胎儿。
在一个优选的实施方案中,受试者/患者为人类。
在一个优选的实施方案中,受试者/患者不是人类。
制剂
尽管可单独使用(例如给药)DAPTZ化合物,但通常优选的是将其用在组合物或制剂中。
在一个实施方案中,所述组合物为含有本发明所述DAPTZ化合物和可药用载体、稀释剂或赋型剂的药物组合物(例如制剂(formulation)、制品(preparation)、药物(medicament))。
在一个实施方案中,所述组合物为含有至少一个本发明所述DAPTZ化合物和一种或多种其它本领域技术人员已知的可药用成分的药物组合物,所述可药用成分包括但不限于可药用载体、稀释剂、赋型剂、助剂、填料、缓冲剂、防腐剂、抗氧化剂、润滑剂、稳定剂、助溶剂、表面活性剂(例如润湿剂)、掩蔽剂、着色剂、调味剂和甜味剂。
在一个实施方案中,所述组合物还含有其它活性制剂如其它治疗剂或预防剂。
合适的载体、稀释剂、赋型剂等可在标准药物教材中找到。参见,例如Handbook of Pharmaceutical Additives,第二版(eds.M.Ash和I.Ash),2001(Synapse Information Resources,Inc.,Endicott,New York,USA),Remington's Pharmaceutical Sciences,第20版,pub.Lippincott,Williams&Wilkins,2000;and Handbook of Pharmaceutical Excipients,第二版,1994。
本发明其它方法涉及制备药物组合物的方法,包括将至少一种本发明所述的[11C]-放射标记的DAPTZ化合物嗪化合物与一种或多种其它本领域技术人员已知的可药用成分混合,所述可药用成分如载体、稀释剂、赋型剂等。如果以单个单位(例如片剂等)进行配制,每个单位含有预定量(剂量)的DAPTZ化合物。
本发明使用的术语“可药用的”涉及化合物、成分、物质、组合物、剂型等,其在合理的医学判断范围内适用于与出问题的受试者(例如人)的器官接触,而不会产生过量的毒性、刺激、过敏性应答或其它问题或病症,与合理的益处/风险比例相称。每一种载体、稀释剂、赋型剂等在与制剂的其它成分相容方面必须也是“可用的”。
所述制剂可以通过制药领域已知的任何方法进行制备。这些包括如下步骤:将活性化合物与作为一种或多种附加成分的载体混合。通常,所述制剂是通过将DAPTZ化合物与载体(例如液体载体、细颗粒固体载体,等)均匀而又紧密(intimately)混合在一起,然后根据需要将产物成型。
所述制剂可经制备实现快速或缓慢释放;即释、延迟释放、定时释放或持续释放;或它们的组合。
适用于非肠道给药(例如通过注射)的制剂包括含水或不含水、等渗、无热原、无菌液体、(例如溶液、悬浮液),其中将所述DAPTZ化合物溶解、悬浮或以其它方式提供(例如采用微脂粒或其它微粒形式)。这些液体还含有其它可药用成分,如抗氧化剂、缓冲剂、防腐剂、稳定剂、抑菌剂、悬浮剂、增稠剂和使制剂与被给药者的血液(或其它相关体液)等渗的溶质。赋型剂的例子包括如水、醇、多元醇、甘油、植物油等。用于这些制剂中的合适等渗载体包括氯化钠注射液、林格氏溶液或乳酸林格氏(注射液)。
典型地,DAPTZ化合物在液体中的浓度为约1ng/ml至约10μg/ml,例如从约10ng/ml至约1μg/ml。所述制剂可置于单位剂量或多剂量密封容器中,例如置于安瓿瓶(ampoule)和小瓶(vial)中,并且可储存在冷冻干燥(冻干的)的条件下,此时仅需要在使用之前即时加入无菌液体载体,例如加入注射用水。临时注射溶液和悬浮液可由无菌粉末、颗粒和片剂制备。
一些优选制剂的实施例
本发明一个方面涉及含有20至300mg本发明披露的DAPTZ化合物和可药用载体、稀释剂或赋型剂的剂量单位(例如药物片剂或胶囊)(例如,通过本发明方法获得,或可通过本发明得到;具有本发明披露的纯度;等等)。
在一个实施方案中,所述剂量单位为片剂。
在一个实施方案中,所述剂量单位为胶囊。
在一个实施方案中,所述量为30至200mg。
在一个实施方案中,所述量为约30mg。
在一个实施方案中,所述量为约60mg。
在一个实施方案中,所述量为约100mg。
在一个实施方案中,所述量为约150mg。
在一个实施方案中,所述量为约200mg。
在一个实施方案中,所述可药用载体、稀释剂或赋型剂为如下物质或包括如下物质的一种或两种都包括:甘油酯(例如Gelucire月桂酰聚乙二醇-32甘油酯PhEur,USP)和硅胶(例如2%AerosilColliodal Silicon Dioxide PhEur,USP)。
剂量
本领域技术人员能够理解的是,DAPTZ化合物和含有该DAPTZ化合物的组合物的合适剂量会根据病人的不同而不同。最佳剂量的确定通常包括平衡治疗益处对任何风险或有害副反应的水平。尽管总的来说,所选的剂量水平是为了能在作用部位实现局部浓度,该浓度能起到所需的效果而不产生大量有害或毒副反应,但所选择的剂量水平会依赖于各种因素,包括但不限于特定化合物的活性、给药途径、给药时间、化合物的代谢速度、治疗的持续时间、联合使用的其它药物,化合物和/或物质、疾病的严重程度和物种、患者的性别、年龄、体重、病状、一般健康状况和先前治疗史。化合物的量和给药途径最终由医生、兽医或临床医师决定。
在整个治疗阶段可采用一次剂量连续或间歇给药(例如以合适的间隔分份剂量)。确定最有效途径和给药剂量的方法是本领域技术人员已知的,并随用于治疗的制剂、治疗目的、进行治疗的目标细胞和进行治疗的受试者而变化。单一或多次给药可以根据由医生、兽医或临床医师选择的剂量水平和模式进行。
总的来说,DAPTZ化合物的合适剂量为每千克受试者体重每天约100ng至约25mg(特别是约1μg至约10mg)。
在一个实施方案中,将所述DAPTZ化合物按照下述剂量方案给药到人类患者:约100mg,每天3次。
在一个实施方案中,将所述DAPTZ化合物按照下述剂量方案给药到人类患者:约150mg,每天2次。
在一个实施方案中,将所述DAPTZ化合物按照下述剂量方案给药到人类患者:约200mg,每天2次。
本发明具体涉及以下方面:
1.化合物或其可药用盐、溶剂化物或水合物,其中所述化合物选自下式表示的化合物:
其中:
R1和R9各自独立地选自:-H、C1-4烷基、C2-4烯基和卤代C1-4烷基;
R3NA和R3NB各自独立地选自:-H、C1-4烷基、C2-4烯基和卤代C1-4烷基;
R7NA和R7NB各自独立地选自:-H、C1-4烷基、C2-4烯基和卤代C1-4烷基;以及
HX1和HX2各自独立地为质子酸。
2.项1所述的化合物,其中R1和R9各自独立地为-H、-Me、-Et或-CF3。
3.项1所述的化合物,其中R1和R9各自独立地为-H、-Me或-Et。
4.项1-3任一所述的化合物,其中R1和R9相同。
5.项1所述的化合物,其中R1和R9各自独立地为-H。
6.项1-5任一所述的化合物,其中R3NA和R3NB各自独立地为-Me、-Et、-nPr、-nBu、-CH2-CH=CH2或-CF3。
7.项1-6任一所述的化合物,其中R3NA和R3NB相同。
8.项1-7任一所述的化合物,其中R7NA和R7NB各自独立地为-Me、-Et、-nPr、-nBu、-CH2-CH=CH2或-CF3。
9.项1-8任一所述的化合物,其中R7NA和R7NB相同。
10.项1-9任一所述的化合物,条件是R3NA、R3NB、R7NA和R7NB中的至少一个不是-Et。
11.项1-9任一所述的化合物,条件是:
如果:R1和R9各自为-H;
则:R3NA、R3NB、R7NA和R7NB各自不是-Et。
12.项1-5任一所述的化合物,其中-N(R3NA)(R3NB)基团
和-N(R7NA)(R7NB)基团各自独立地选自-NMe2、-NEt2、-N(nPr)2、-N(Bu)2、-NMeEt、-NMe(nPr)和-N(CH2CH=CH2)2。
13.项1-5任一所述的化合物,其中-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团相同,并选自:-NMe2和-NEt2。
14.项1-5、11和12任一所述的化合物,其中-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团相同。
15.项1-5和11-13任一所述的化合物,条件是:N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自不是-NEt2。
16.项1-5和11-13任一所述的化合物,其中-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团相同,并选自:-NMe2、-N(nPr)2、-N(Bu)2、-NMeEt、-NMe(nPr)和-N(CH2CH=CH2)2。
17.项1-5任一所述的化合物,其中-N(R3NA)(R3NB)基团和-N(R7NA)(R7NB)基团各自为:-NMe2。
18.项1-17任一所述的化合物,其中HX1和HX2各自独立地为单质子酸。
19.项1-17任一所述的化合物,其中HX1和HX2各自独立地为氢卤酸。
20.项1-17任一所述的化合物,其中HX1和HX2各自独立地选自HCl、HBr和HI。
21.项1-17任一所述的化合物,其中HX1和HX2各自为HCl。
22.项1-17任一所述的化合物,其中HX1和HX2各自为HBr。
23.项1-17任一所述的化合物,其中HX1和HX2各自为HI。
24.项1所述的化合物或其可药用盐、溶剂化物或水合物,其中所述化合物选自下式表示的化合物:
25.项1所述的化合物或其可药用盐、溶剂化物或水合物,其中所述化合物选自下式表示的化合物:
26.项1所述的化合物或其可药用盐、溶剂化物或水合物,其中所述化合物选自下式表示的化合物:
27.项1-26任一所述的化合物,该化合物为基本纯化形式。
28.项1-27任一所述的化合物,其中所述化合物上的一个或多个碳原子为11C。
29.一种组合物,该组合物含有项1-28任一所述的化合物和可药用载体或稀释剂。
30.一种药物组合物,该药物组合物含有项1-28任一所述的化合物和可药用载体或稀释剂。
31.一种制备药物组合物的方法,包括将根据项1-28任一所述的化合物与可药用载体或稀释剂混合。
32.根据项1-27任一所述的化合物,所述化合物通过治疗而用于治疗或预防人体或动物体的方法中。
33.根据项1-27任一所述的化合物,所述化合物用在治疗或预防蛋白聚集疾病的方法中。
34.根据项1-27任一所述的化合物,所述化合物用在治疗或预防τ蛋白病的方法中。
35.根据项1-27任一所述的化合物,所述化合物用在治疗或预防神经病变性τ蛋白病的方法中。
36.根据项1-27任一所述的化合物,所述化合物用在治疗或预防阿尔茨海默氏病的方法中。
37.根据项1-27任一所述的化合物,所述化合物用在治疗或预防皮肤癌或黑素瘤的方法中。
38.根据项1-27任一所述的化合物,所述化合物用在治疗或预防病毒、细菌或原生动物疾病状态的方法中。
39.根据项1-27任一所述的化合物,所述化合物用在治疗或预防丙型肝炎、HIV或西尼罗病毒(WNV)的方法中。
40.根据项1-27任一所述的化合物,所述化合物用在治疗或预防疟疾的方法中。
41.项1-27任一所述的化合物在制备用于治疗或预防蛋白聚集疾病的药物中的用途。
42.项1-27任一所述的化合物在制备用于治疗或预防的药物中的用途。
43.项1-27任一所述的化合物在制备用于治疗或预防τ蛋白病的药物中的用途。
44.项1-27任一所述的化合物在制备用于治疗或预防神经病变性τ蛋白病的药物中的用途。
45.项1-27任一所述的化合物在制备用于治疗或预防阿尔茨海默氏病的药物中的用途。
46.项1-27任一所述的化合物在制备用于治疗或预防皮肤癌或黑素瘤的药物中的用途。
47.项1-27任一所述的化合物在制备用于治疗或预防病毒、细菌或原生动物疾病状态的药物中的用途。
48.项1-27任一所述的化合物在制备用于治疗或预防丙型肝炎、HIV或西尼罗病毒(WNV)的药物中的用途。
49.项1-27任一所述的化合物在制备用于治疗或预防疟疾的药物中的用途。
50.一种治疗或预防受试者的蛋白聚集疾病的方法,包括向所述受试者给药预防有效量或治疗有效量的项1-27任一所述的化合物。
51.一种治疗或预防受试者的τ蛋白病的方法,包括向所述受试者给药预防有效量或治疗有效量的项1-27任一所述的化合物。
52.一种治疗或预防受试者的神经病变性τ蛋白病的方法,包括向所述受试者给药预防有效量或治疗有效量的项1-27任一所述的化合物。
53.一种治疗或预防受试者的阿尔茨海默氏病的方法,包括向所述受试者给药预防有效量或治疗有效量的项1-27任一所述的化合物。
54.一种治疗或预防受试者的皮肤癌或黑素瘤的方法,包括向所述受试者给药预防有效量或治疗有效量的项1-27任一所述的化合物。
55.一种治疗或预防受试者的病毒、细菌或原生动物疾病状态的方法,包括向所述受试者给药预防有效量或治疗有效量的项1-27任一所述的化合物。
56.一种治疗或预防受试者的丙型肝炎、HIV或西尼罗病毒(WNV)的方法,包括向所述受试者给药预防有效量或治疗有效量的项1-27任一所述的化合物。
57.一种治疗或预防受试者的疟疾的方法,包括向所述受试者给药预防有效量或治疗有效量的项1-27任一所述的化合物。
58.一种逆转和/或抑制蛋白聚集的方法,包括将所述蛋白质与有效量的项1-27任一所述的化合物接触。
59.一种逆转和/或抑制哺乳动物的脑中蛋白聚集的方法,所述聚集与疾病状态有关,所述治疗包括向需要该治疗的哺乳动物给药预防有效量的或治疗有效量的项1-27任一所述的化合物的步骤。
60.一种灭活样品中的病原体的方法,包括将项1-27任一所述的化合物引入到所述样品中,然后将所述样品暴露于光下。
实施例
如下所述,下述合成仅出于解释目的而不是对本发明保护范围的限定。
化学合成
合成1
3-硝基-10H-吩噻嗪
将亚硝酸钠(20.00g,210mmol)加到10H-吩噻嗪(20.00g,50mmol)、氯仿(100cm3)和醋酸(20cm3)的混合物中,将所得混合物在室温搅拌1小时。然后向混合物中加入醋酸(20cm3),再搅拌18小时。过滤所得悬浮液,用醋酸、乙醇、水和最后用乙醇分别洗涤得到紫/棕色固体。将残余物溶解在热DMF中,冷却后过滤,得到二硝基化合物,为紫色固体状。浓缩所述DMF溶液,用水和甲醇洗涤沉淀物,得到标题所示的单-硝基化合物(15g,~50%),为棕色固体;νmax(KBr)/cm-1:3328(NH),3278(NH),3229(NH),3119(CH),3049(CH),1557(NO2),1531(NO2);δH(250MHz;DMSO):6.64(5H,m,ArH),7.68(1H,d,J2.5,ArH),7.79-7.84(1H,dd,J 2.75,6.5,ArH);δC(62.9MHz;DMSO):113.3(ArC),115.3(ArC),116.9(ArC),121.8(ArC),123.6(ArC),123.7(ArC),124.6(ArC),126.4(ArC),128.1(ArC),138.8(ArC),141.0(ArC),147.8(ArC)。
合成2
3,7-二硝基-10H-吩噻嗪
在合成3-硝基-10H-吩噻嗪的步骤之后进行该步骤,利用3-硝基-10H-吩噻嗪(10.00g,41mmol)、氯仿(40cm3)、醋酸(2x 10cm3)和亚硝酸钠(11.86g,173mmol)进行反应。所得残余物用DMF重结晶,得到标题所示的二-硝基化合物(6.60g 56%),为紫色针状物;νmax(KBr)/cm-1:3331(NH),3294(NH),3229(NH),3101(CH),3067(CH),1602(NO2),1558(NO2);δH(250MHz;DMSO):6.73-6.76(2H,d,J 9,ArH),7.78(2H,s,ArH),7.89-7.85(2H,d,J 9,ArH)。
合成3
1-(3,7-二硝基-吩噻嗪-10-基)-乙酮
将3,7-二硝基-10H-吩噻嗪(3.00g,10.37mmol)、醋酸酐(15.88g,155.50mmol)和吡啶(30cm3)形成的溶液搅拌回流18小时。然后将所得热溶液小心倒入冰水中。过滤,得到形成的沉淀,并将其溶解在二氯甲烷中,用硫酸镁干燥,过滤、浓缩,得到棕/橙色固体,该固体经柱色谱纯化(SiO2,乙酸乙酯:石油醚=2:3,以二氯甲烷溶液形式加载)得到浅黄色固体状的标题化合物(2.46g,71%),用丙酮重结晶,得到浅黄色针状物;νmax(KBr)/cm-1:3091(CH),3063(CH),1680(C=O),1575(NO2),1510(NO2);δH(250MHz;CDCl3):2.28(3H,s,CH3),7.65-7.69(2H,d,J 9,ArH),8.22-8.26(2H,dd,J 2.75,8.75,ArH),8.33-8.32(2H,d,J 2.5,ArH);δC(62.9MHz;CDCl3):168.2(C=O),146.3(ArC),143.3(ArC),133.6(ArC),127.8(ArC),123.4(ArC),122.9(ArC),23.1(CH3);m/z(ES)331.0(80%,[M]+)。
合成4
1-(3,7-二氨基-吩噻嗪-10-基)-乙酮
将1-(3,7-二硝基-吩噻嗪-10-基)-乙酮(2g,6.04mmol)、二氯化锡(II)二水合物(14.17g,62.8mmol)和乙醇(50cm3)形成的混合物加热回流并在该温度搅拌5小时。然后将所得混合物冷却到室温并倒入冰水中。用5%碳酸氢钠将pH调节到pH7,然后用乙酸乙酯(3x 50cm3)萃取产物。萃取物用盐水洗涤并经硫酸镁干燥,过滤浓缩,得到标题化合物(1.64g,100%),为蓝紫色固体;νmax(KBr)/cm-1:3445(NH),3424(NH),3368(NH),3322(NH),3203(NH),3054(CH),2995(CH),1706(C=O),1650(NO2),1590(NO2);δH(250MHz;CDCl3):2.01(3H,s,CH3),5.09-5.43(4H,brd s,NH),6.47-6.51(2H,dd,J 1.5,8.25,ArH),6.61(2H,s,ArH),7.11-7.15(2H,d,J 8,ArH);δC(62.9MHz;CDCl3):169.1(C=O),147.2(ArC),128.1(ArC),127.6(ArC),127.3(ArC),112.3(ArC),111.5(ArC),22.6(CH3);m/z(ES)293.9(95%,[M+H,Na]+),272.0(20%,[M+H]+),227.9(100%,[M+H,-Ac]+)。
合成5
3,7-二氨基-吩噻嗪双(盐酸)(B4)
将1-(3,7-二氨基-吩噻嗪-10-基)-乙酮(0.25g,0.921mmol)溶解在盐酸(5N,10cm3)中,将所得溶液加热回流并搅拌30分钟。浓缩该反应混合物,得到标题化合物,为淡蓝色固体。δH(250MHz;D2O):6.60(2H,brd d,ArH),7.07(4H,brd s,ArH)。
合成6
1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮
将1-(3,7-二氨基-吩噻嗪-10-基)-乙酮(0.25g,0.92mmol)溶解在DMSO(3cm3)中。加入甲苯(10cm3)、碘甲烷(1.96g,13.8mmol)、四丁基溴化铵(50mg),最后加入氢氧化钠水溶液(50%,1.25cm3)。将所得混合物在室温搅拌2小时。然后再加入氢氧化钠水溶液(50%,1.25cm3)和碘甲烷(1.96g,13.8mmol)。在室温将所得混合物再搅拌3小时,然后加入第三份氢氧化钠水溶液(50%,1.25cm3)和碘甲烷(1.96g,13.8mmol),将所得混合物再搅拌18小时。所得粘稠的悬浮液用水(3x 75cm3)洗,收集甲苯萃取物。将所述水洗物用二氯甲烷(3x 50cm3)萃取,所得萃取物与甲苯合并,经硫酸镁干燥,过滤浓缩,得到深紫色固体。残余物用柱色谱纯化(SiO2,乙酸乙酯:石油醚=2:3,以二氯甲烷溶液形式加载),得到标题化合物产物(0.12g,40%),为淡紫色固体;νmax(KBr)/cm-1:2910(CH),2876(CH),2856(CH),2799(CH),1659(C=O),1596(NO2),1502(NO2);δH(250MHz;CDCl3):2.16(3H,s,CH3),2.93(12H,s,NCH3),6.59-6.62(2H,d,J 8.5,ArH),6.69-6.71(2H,d,J2.75,ArH),7.08-7.47(2H,brd s,ArH);δC(62.9MHz;CDCl3):170.3(C=O),148.9(ArC),127.2(ArC),127.1(ArC),127.0(ArC),110.9(ArC),110.7(ArC),40.7(NCH3),22.9(CH3)。
合成7
N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(盐酸)(B3)
将1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮(0.5g,1.84mmol)溶解在盐酸水溶液(5N,15cm3)中,将所得溶液加热至回流温度并搅拌30分钟。浓缩反应混合物,得到标题化合物,为绿/蓝色固体;δH(250MHz;D2O):3.18(12H,s,NCH3),6.67(2H,d,J 8.5,ArH),7.16(4H,brd s,ArH);δC(62.9MHz;D2O):144.3(ArC),138.9(ArC),122.4(ArC),120.8(ArC),120.7(ArC),117.6(ArC),48.9(NCH3)。
合成8
甲基锍碘化物
向圆底烧瓶中加入甲基锍氯化物(MTC,次甲蓝)(2g,6.25mmol)和水(50cm3),将所得混合物搅拌10分钟,或搅拌直到固体溶解。然后向所述混合物中加入碘化钾(1.56g,9.4mmol),形成黑绿色悬浮液。将反应加热到沸腾,然后自然冷却得到题化合物(2.03g,79%),为亮绿色针状物。分析:C16H18N3SI的计算值:C,46.72;H,4.41;N,10.22;S,7.80;I,30,85。实测值:C,46.30;H,4.21;N,10.14;S,7.86;I,29.34。
合成9
N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢碘酸)(B6)
向圆底烧瓶中加入甲基锍碘化物(2g,4.86mmol)、乙醇(100cm3)和碘乙烷(75.8g,486mmol),将所得混合物加热回流18小时,其间颜色发生变化,从绿/蓝色变为棕色,并伴有黄色沉淀物出现。一经冷却到室温就将混合物过滤,用乙醚(20cm3)洗涤,得到标题化合物(1.99g,76%),为淡绿色固体。δH(250MHz;D2O):3.20(12H,s,NCH3),6.76(2H,d,J 8.5,ArH),7.22(2H,brd s,ArH);δC(62.9MHz;D2O):145.0(ArC),139.3(ArC),122.6(ArC),121.1(ArC),120.9(ArC),117.9(ArC),48.9(NCH3)。
合成10
1-(3,7-双-二乙基氨基-吩噻嗪-10-基)-乙酮
向干燥的25cm3圆底烧瓶中加入乙基锍氯化锌(0.5g,1.13mmol)和乙醇(10cm3)。然后氮气气氛下滴入苯肼(0.134g,1.24mmol)。将所得混合物在25℃搅拌1小时,高真空浓缩。然后加入吡啶(50cm3)和醋酸酐,将所得混合物在60℃搅拌18小时。将所得溶液倒入冰/水(250cm3)中,将有机物萃取到乙酸乙酯(3x 50cm3)中。所述萃取物用饱和硫酸铜溶液洗涤,经硫酸镁干燥,过滤浓缩,得到棕色油状的粗品,将所述粗品用快速柱色谱纯化(用40%乙酸乙酯:60%石油醚(40-60℃)洗脱,硅胶为40-63μ),得到绿色玻璃状固体。δH(250MHz;CDCl3):7.0-7.5(2H,brds,ArH),6.64(2H,s,ArH),6.52(2H,d,ArH),3.35(8H,q,7,NCH2),2.18(3H,s,CH3),1.16(12H,t,7,CH3);δC(62.9MHz;CDCl3):12.5(CH3),22.9(CH3),44.6(NCH2),110.1(ArC),127.4(ArC),146.5(ArC),170.2(C=O)。
合成11
N,N,N',N'-四乙基-10H-吩噻嗪-3,7-二胺双(盐酸)
向25cm3圆底烧瓶中加入3,7-二乙基氨基-10-乙酰基-吩噻嗪(0.125g,0.33mmol)和盐酸水溶液(5M,5cm3)。将所述混合物在100℃加热2小时,然后冷却到室温,浓缩,得到标题化合物(0.11g,81%),为黄绿色玻璃状固体。δH(250MHz;CD3OD):7.07(4H,brd,ArH),6.65(2H,brd,ArH),3.35(8H,brd,NCH2),0.97(12H,brd,CH3);δC(62.9MHz;CD3OD):10.8(CH3),55.1(NCH2),116.6(ArC),120.4(ArC),121.5(ArC),123.6(ArC),132.6(ArC),144.5(ArC)。
合成12
1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮
利用甲肼/吡啶分两锅进行制备。向置于氩气气氛中的250cm3圆底烧瓶中加入甲基锍氯化物三水合物(26.74mmol,10g)、乙醇(100cm3)和甲肼(58.83mmol,2.71g)。将所得混合物加热到40℃,搅拌2小时。将所得黄/绿色悬浮液冷却到5℃,氩气气氛下过滤,用乙醇洗涤,干燥得到淡绿色固体状的无色次甲蓝(leuco-methylene blue)。向该无色产物中加入醋酸酐(40cm3)和吡啶(10cm3),将所得溶液在100℃加热18小时。然后将其冷却并小心倒入冰水中,同时搅拌,得到沉淀物,过滤该沉淀物,用水洗,60℃干燥2小时得到标题化合物(5.82g,66%),为亮棕色固体。Mp 137℃;νmax(KBr)/cm-12910(CH),2876(CH),2856(CH),2799(CH),1659(C=O),1596(NO2),1502(NO2);δH(250MHz;CDCl3)2.16(3H,s,CH3),2.93(12H,s,NCH3),6.59-6.62(2H,d,J 8.5,ArH),6.69-6.71(2H,d,J 2.75,ArH),7.08-7.47(2H,brd s,ArH);δC(62.9MHz;CDCl3)170.3(C=O),148.9(ArC),127.2(ArC),127.1(ArC),127.0(ArC),110.9(ArC),110.7(ArC),40.7(NCH3),22.9(CH3);m/z(ES)284.2(100%,[M–OAc]+),328.1(15%,[M+H]+),350.1(41%,[M+Na]+)。
合成13
1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮
利用甲肼/许尼希碱通过一锅法合成。氮气气氛下向5000cm3反应容器中加入甲基锍氯化物三水合物(0.54mol,200g)和乙腈(1000cm3)。向其中逐滴加入甲肼(1.07mol,49.36g),滴加速度为每分钟1.5mL。混合物的温度升高到32℃,搅拌20分钟。向所得的黄/绿色悬浮液中加入醋酸酐(5.35mol,541g),然后加入许尼希碱(二异丙基乙胺)(1.55mol,200g)。将混合物在90℃加热2小时。然后将冷却的混合物小心倒入分成10份、每份200cm3的冰水(2000cm3)中,同时搅拌,得到沉淀物。将所得沉淀物搅拌45分钟,过滤,用水洗(3x250cm3)。空气干燥30分钟。将所得粗品物质用热乙醇(2750cm3)重结晶,得到标题化合物(112.1g,64%),为浅灰色固体。Mp 137℃;νmax(KBr)/cm-12910(CH),2876(CH),2856(CH),2799(CH),1659(C=O),1596(NO2),1502(NO2);δH(250MHz;CDCl3)2.16(3H,s,CH3),2.93(12H,s,NCH3),6.59-6.62(2H,d,J 8.5,ArH),6.69-6.71(2H,d,J 2.75,ArH),7.08-7.47(2H,brd s,ArH);δC(62.9MHz;CDCl3),170.3(C=O),148.9(ArC),127.2(ArC),127.1(ArC),127.0(ArC),110.9(ArC),110.7(ArC),40.7(NCH3),22.9(CH3);m/z(ES)284.2(100%,[M-OAc]+),328.1(15%,[M+H]+),350.1(41%,[M+Na]+)。
合成14
1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮
利用甲肼/吡啶通过一锅法合成。氮气气氛下向250cm3圆底烧瓶中加入甲基锍氯化物三水合物(26.74mmol,10g)和乙腈(50cm3)。历时30分钟将甲肼(53.5mmol,2.46g)分成四等份加入。混合物的温度用冷水浴保持在35℃,搅拌30分钟。向所得黄/绿色悬浮液中加入醋酸酐(267mmol,27.3g)和吡啶(80.2mmol,6.35g)。将所得混合物在90℃加热2小时。然后将冷却的混合物分成10等份小心倒入冰水(200cm3)中,同时搅拌,得到沉淀物。将所述沉淀物搅拌30分钟,然后过滤,用水洗(3x 50cm3)并空气干燥30分钟。所得粗品物质用热乙醇(120cm3)重结晶,得到标题化合物(5.97g,68%),为浅灰色固体。Mp 137℃;νmax(KBr)/cm-12910(CH),2876(CH),2856(CH),2799(CH),1659(C=O),1596(NO2),1502(NO2);δH(250MHz;CDCl3)2.16(3H,s,CH3),2.93(12H,s,NCH3),6.59-6.62(2H,d,J 8.5,ArH),6.69-6.71(2H,d,J 2.75,ArH),7.08-7.47(2H,brd s,ArH);δC(62.9MHz;CDCl3)170.3(C=O),148.9(ArC),127.2(ArC),127.1(ArC),127.0(ArC),110.9(ArC),110.7(ArC),40.7(NCH3),22.9(CH3);m/z(ES)284.2(100%,[M-OAc]+),328.1(15%,[M+H]+),350.1(41%,[M+Na]+)。
合成15
1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮
利用硼氰化钠/吡啶通过一锅法合成。氮气气氛下向500cm3圆底烧瓶中加入甲基锍氯化物三水合物(0.134mol,50g)和乙腈(250cm3)。将硼氰化钠(0.174mol,6.6g)分成4等份历时30分钟内加入。混合物的温度用冷水浴保持在35℃,搅拌30分钟。向所得黄/绿色悬浮液中加入醋酸酐(0.535mol,55g)和吡啶(0.174mol,13.76g)。将所得混合物在90℃搅拌2小时。然后搅拌下将冷却的混合物分成10份小心倒入冰水(250cm3)中,得到沉淀物。将所述沉淀物搅拌30分钟,过滤,用水洗(3x 50cm3),空气干燥30分钟。所得粗品物质用热乙醇(500cm3)结晶,得到标题化合物(26.7g,61%),为浅灰色固体。Mp137℃;νmax(KBr)/cm-12910(CH),2876(CH),2856(CH),2799(CH),1659(C=O),1596(NO2),1502(NO2);δH(250MHz;CDCl3)2.16(3H,s,CH3),2.93(12H,s,NCH3),6.59-6.62(2H,d,J 8.5,ArH),6.69-6.71(2H,d,J 2.75,ArH),7.08-7.47(2H,brd s,ArH);δC(62.9MHz;CDCl3)170.3(C=O),148.9(ArC),127.2(ArC),127.1(ArC),127.0(ArC),110.9(ArC),110.7(ArC),40.7(NCH3),22.9(CH3);m/z(ES)284.2(100%,[M-OAc]+),328.1(15%,[M+H]+),350.1(41%,[M+Na]+)。
合成16
1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮
利用硼氰化钠/许尼希碱通过一锅法合成。氮气气氛下向500cm3圆底烧瓶中加入甲基锍氯化物三水合物(80.2mmol,30g)和乙腈(150cm3)。将硼氰化钠(104mmol,3.94g)分成4等份历时30分钟内加入。所得混合物的温度用冷水浴保持在35℃,搅拌30分钟。向所得黄/绿色悬浮液中加入醋酸酐(321mmol,32.75g)和许尼希碱(二异丙基乙胺)(120mmol,15.55g)。将所得混合物在90℃加热2小时。然后搅拌下将冷却的混合物分成10等份小心倒入冰水(200cm3)中,得到沉淀物。将所述沉淀物搅拌30分钟,过滤并用水洗(3x 50cm3),空气干燥30分钟。所得粗品物质用热乙醇结晶(300cm3)得到标题化合物(13.55g,52%),为浅灰色固体。Mp 137℃;νmax(KBr)/cm-12910(CH),2876(CH),2856(CH),2799(CH),1659(C=O),1596(NO2),1502(NO2);δH(250MHz;CDCl3)2.16(3H,s,CH3),2.93(12H,s,NCH3),6.59-6.62(2H,d,J 8.5,ArH),6.69-6.71(2H,d,J 2.75,ArH),7.08-7.47(2H,brd s,ArH);δC(62.9MHz;CDCl3)170.3(C=O),148.9(ArC),127.2(ArC),127.1(ArC),127.0(ArC),110.9(ArC),110.7(ArC),40.7(NCH3),22.9(CH3);m/z(ES)284.2(100%,[M-OAc]+),328.1(15%,[M+H]+),350.1(41%,[M+Na]+)。
合成17
1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮
利用一水合肼/吡啶通过一锅法合成。氮气气氛下向250cm3圆底烧瓶中加入甲基锍氯化物三水合物(26.74mmol,10g)和乙腈(50cm3)。向其中加入一水合肼(58.8mmol,2.95g),将所得对混合物加热回流并搅拌10分钟,然后冷却到25℃。向所得黄/绿色悬浮液中加入醋酸酐(424mmol,43.3g)和吡啶(124mmol,9.78g)。将所得混合物在90℃加热2小时。然后搅拌下将冷却的混合物分成10等份小心倒入水(100cm3)中,得到沉淀物。将所述沉淀物搅拌30分钟,过滤,用水洗(3x 50cm3),空气干燥30分钟。将所得粗品物质用热乙醇(100cm3)结晶,得到标题化合物(4.87g,56%),为浅灰色固体。Mp 137℃;νmax(KBr)/cm-12910(CH),2876(CH),2856(CH),2799(CH),1659(C=O),1596(NO2),1502(NO2);δH(250MHz;CDCl3)2.16(3H,s,CH3),2.93(12H,s,NCH3),6.59-6.62(2H,d,J 8.5,ArH),6.69-6.71(2H,d,J 2.75,ArH),7.08-7.47(2H,brd s,ArH);δC(62.9MHz;CDCl3)170.3(C=O),148.9(ArC),127.2(ArC),127.1(ArC),127.0(ArC),110.9(ArC),110.7(ArC),40.7(NCH3),22.9(CH3);m/z(ES)284.2(100%,[M-OAc]+),328.1(15%,[M+H]+),350.1(41%,[M+Na]+)。
合成18
1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮
利用一水合肼/许尼希碱通过一锅法合成。氮气气氛下向250cm3圆底烧瓶中加入甲基锍氯化物三水合物(80.2mmol,30g)和乙腈(150cm3)。向其中加入一水合肼(176.5mmol,8.84g),将所得混合物加热回流并搅拌10分钟,然后冷却到25℃。向所得黄/绿色悬浮液中加入醋酸酐(794mmol,81.2g)和许尼希碱(二异丙基乙胺)(232mmol,29.97g)。将所得混合物在90℃加热2小时。然后搅拌下将冷却的混合分10等份小心倒入冰水(400cm3)中,得到沉淀物。将所述沉淀物搅拌30分钟,过滤,用水洗(3x 100cm3),空气干燥30分钟。将所得粗品物质用热乙醇(400cm3)结晶,得到标题化合物(17.15g,65%),为浅灰色固体。Mp 137℃;νmax(KBr)/cm-12910(CH),2876(CH),2856(CH),2799(CH),1659(C=O),1596(NO2),1502(NO2);δH(250MHz;CDCl3)2.16(3H,s,CH3),2.93(12H,s,NCH3),6.59-6.62(2H,d,J 8.5,ArH),6.69-6.71(2H,d,J 2.75,ArH),7.08-7.47(2H,brd s,ArH);δC(62.9MHz;CDCl3)170.3(C=O),148.9(ArC),127.2(ArC),127.1(ArC),127.0(ArC),110.9(ArC),110.7(ArC),40.7(NCH3),22.9(CH3);m/z(ES)284.2(100%,[M-OAc]+),328.1(15%,[M+H]+),350.1(41%,[M+Na]+)。
合成19
3,7-二硝基-10H-吩噻嗪
向10H-吩噻嗪(20.00g,100mmol)、二氯甲烷(100cm3)和醋酸(40cm3)的混合物中加入亚硝酸钠(20.07g,300mmol),将所得混合物在室温搅拌10分钟。接着加入另外的醋酸(40cm3)、二氯甲烷(100cm3)和亚硝酸钠(20.07g,300mmol)。还加入120cm3醋酸以尝试将粘稠的反应混合物破解。将所得混合物搅拌3小时。过滤所得悬浮液,用各为100cm3的乙醇、水和乙醇(最后一次)洗涤得到紫/棕色固体。将残余物在热DMF中搅拌,冷却后过滤所述二硝基产物,将所述产物用乙醇(150cm3)洗涤、干燥,得到标题化合物(24.88g,86%),为棕色固体;νmax(KBr)/cm-13331(NH),3294(NH),3229(NH),3101(CH),3067(CH),1602(NO2),1558(NO2);δH(250MHz;DMSO)6.73-6.76(2H,d,J9,ArH),7.78(2H,s,ArH),7.89-7.85(2H,d,J9,ArH)。
合成20
1-(3,7-双-二乙基氨基-吩噻嗪-10-基)-乙酮
氮气气氛下向250cm3圆底烧瓶中加入乙基锍硝酸盐一水合物(7.13mmol,3g)和乙腈(20cm3)。向其中加入一水合肼(16.4mmol,0.82g),将所得混合物加热回流并搅拌10分钟,然后冷却到25℃。向所得棕色溶液中加入醋酸酐(114mmol,11.65g)和许尼希碱(二异丙基乙胺)(21.4mmol,2.77g)。将所得混合物在90℃加热2小时。然后将冷却的混合物分成10份小心倒入冰水(40cm3)中,得到沉淀物。将所述沉淀物搅拌30分钟,然后过滤,用水洗(3x 25cm3)、空气干燥30分钟。将所得粗品物质用热乙醇(50cm3)结晶,得到标题化合物(1.73g,63%),为浅灰色固体。δH(250MHz;CDCl3)7.0-7.5(2H,brds,ArH),6.64(2H,s,ArH),6.52(2H,d,ArH),3.35(8H,q,7,NCH2),2.18(3H,s,CH3),1.16(12H,t,7,CH3);δC(62.9MHz;CDCl3)12.5(CH3),22.9(CH3),44.6(NCH2),110.1(ArC),127.4(ArC),146.5(ArC),170.2(C=O)。
合成21
N,N,N',N'-四乙基-10H-吩噻嗪-3,7-二胺双(盐酸)
向圆底烧瓶中加入1-(3,7-双-二乙基氨基-吩噻嗪-10-基)-乙酮(0.5g,1.30mmol)、乙醇(5cm3)和盐酸(37%,1.3cm3),将所得溶液在80℃搅拌1小时。一经冷却到室温就将混合物浓缩,得到标题化合物(0.54g,100%),为淡绿色玻璃状物。δH(250MHz;CD3OD)7.07(4H,brd,ArH),6.65(2H,brd,ArH),3.35(8H,brd,NCH2),0.97(12H,brd,CH3);δC(62.9MHz;CD3OD)10.8(CH3),55.1(NCH2),116.6(ArC),120.4(ArC),121.5(ArC),123.6(ArC),132.6(ArC),144.5(ArC)。
合成22
N,N,N',N'-四乙基-10H-吩噻嗪-3,7-二胺双(氢溴酸)
向圆底烧瓶中加入1-(3,7-双-二乙基氨基-吩噻嗪-10-基)-乙酮(0.5g,1.30mmol)、乙醇(5cm3)和氢溴酸(48%,0.75cm3),将所得溶液在80℃加热1小时。一经冷却到室温就将混合物浓缩,得到标题化合物(0.65g,100%),为亮黄色玻璃状物。δH(250MHz;D2O)7.05(4H,brd,ArH),6.79(2H,brd d,ArH),3.43(8H,brd,NCH2),1.05(12H,brd t,CH3);δC(62.9MHz;D2O)12.3(CH3),56.2(NCH2),117.9(ArC),121.4(ArC),122.4(ArC),124.5(ArC),133.5(ArC),145.1(ArC)。
合成23
N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(盐酸)
向圆底烧瓶中加入1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮(1g,3.05mmol)、乙醇(10cm3)和盐酸(37%,3cm3),将所得溶液在80℃加热1小时。一经冷却到室温就在搅拌下向其中加入乙醚直到得到恒定浊度的溶液。经一定时间后有沉淀物生成,过滤,得到所述沉淀物,用乙醚(10cm3)洗涤得到标题化合物(0.98g,90%),为淡绿色固体。Mp(分解)230℃;νmax(KBr)/cm-13500-3229(NH),3061(CH),3021(CH),2948(CH),2879(CH),2679(CH),2601(CH),1604(CH),1483(CH),1318(CH);δH(250MHz;D2O)3.18(12H,s,NCH3),6.67(2H,d,J 8.5,ArH),7.16(4H,brd s,ArH);δC(62.9MHz;D2O)144.3(ArC),138.9(ArC),122.4(ArC),120.8(ArC),120.7(ArC),117.6(ArC),48.9(NCH3);m/z(ES)286.1(100%,[M-H,2Cl]+),285.1(40%),284.1(41%,[M-3H,2Cl]+)。
合成24
N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢溴酸)
向圆底烧瓶中加入1-(3,7-双-二甲基氨基-吩噻嗪-10-基)-乙酮(1g,3.05mmol)、乙醇(10cm3)和氢溴酸(48%,4cm3),将所得溶液在80℃加热1小时。一经冷却到室温就有沉淀物产生,过滤,得到所述沉淀物,用乙醚(10cm3)洗涤得到亮芥色产物(1.22g,89%)。Mp(分解)230℃;νmax(KBr)/cm-13500-3229(NH),3061(CH),3021(CH),2948(CH),2879(CH),2679(CH),2601(CH),1604(CH),1483(CH),1318(CH);δH(250MHz;D2O)3.18(12H,s,NCH3),6.66(2H,d,J 8.75,ArH),7.15(4H,s,ArH);δC(62.9MHz;D2O)144.3(ArC),138.9(ArC),122.4(ArC),120.8(ArC),120.7(ArC),117.6(ArC),48.9(NCH3)。
合成25
N,N,N',N'-四乙基-10H-吩噻嗪-3,7-二胺双(氢溴酸)
向圆底烧瓶中加入1-(3,7-双-二乙基氨基-吩噻嗪-10-基)-乙酮(1.0g,2.60mmol)、甲醇(10cm3)和氢溴酸(48%,2.94cm3),将所得溶液在80℃加热1小时。一经冷却到5℃就向混合物中加入乙醚,得到混浊溶液。将所述浑浊溶液搅拌30分钟得到标题化合物(0.83g,63%),为亮黄色固体。δH(250MHz;D2O)7.05(4H,brd,ArH),6.79(2H,brd d,ArH),3.43(8H,brd,NCH2),1.05(12H,brd t,CH3);δC(62.9MHz;D2O)12.3(CH3),56.2(NCH2),117.9(ArC),121.4(ArC),122.4(ArC),124.5(ArC),133.5(ArC),145.1(ArC)。
稳定性研究
本发明所述的DAPTZ化合物可被稳定地还原(即处于经稳定的还原形式)。例如,这些化合物在固态形式时是稳定的,例如至少能稳定存在1周,如稳定存在至少2周,如稳定存在至少1个月,如稳定存在至少2个月,如稳定存在至少1年(例如在室温,如18-25℃,如在密封容器中)。
已经发现,固态形式的化合物B3(N,N,N’,N’-四甲基-10H-吩噻嗪-3,7-二胺双(盐酸))的样品甚至在储存2年后还可被基本还原。
一旦将DAPTZ化合物溶解在水中(即采用水溶液形式),这些化合物被缓慢氧化(溶液颜色变蓝),通常在1-3小时内被氧化。
研究了本发明的两种DAPTZ化合物的稳定性,特别是B3(N,N,N’,N’-四甲基-10H-吩噻嗪-3,7-二胺双(盐酸))和B6(N,N,N’,N’-四甲基-10H-吩噻嗪-3,7-二胺双(氢碘酸))的稳定性。MTC用作标准品。
称重所述化合物,并装在通用容器中。加入足量水制成1mM溶液,搅拌所得混合物使固体溶解。在不同时间点,确定50μL每种溶液样品(一式三份)在610nm和665nm处的吸光度(absorbance)。起始时间点设在10分钟,因为化合物需要时间完全溶解。此外,在时间点20分钟、3小时和18小时记录紫外/可见光谱。
假定在10分钟时MTC的读数为0%还原,空白代表100%还原(无色),以这种方式来计算还原形式的百分比(%)。
图1是三种化合物,即B1、B3和B6各自的还原形式的百分比(%)对时间(分钟)的曲线图,根据665nm处的吸光度测定得到的。
图2是三种化合物,即B1、B3和B6各自的还原形式的百分比(%)对时间(分钟)的曲线图,根据610nm处的吸光度测定得到的。
图3A,3B和3C显示了三种化合物,即B1(空心圆点)、B3(空心方块)和B6(空心三角形)各自的水溶液样品的紫外/可见光吸收波谱,分别是在20分钟(图3A)、3小时(图3B)和18小时(图3C)后测得的。
这些数据证明了DAPTZ化合物(经稳定的还原形式)持续至少1小时保持基本稳定(>50%),化合物B6持续至少3小时保持基本稳定(>50%)。然而,在约18小时后,这些化合物与MTC没有明显区别。参见例如图3C,其中的波谱几乎难以区别。
此外,发现“碘化物”化合物(化合物B6)的自氧化速度比“氯化物”化合物(化合物B3)慢,这说明了自氧化的速度依赖于反离子。尽管速度差别很小,但其在药物配制中可能是具有重要意义的。其它盐可能更稳定地对抗氧化。
2006年4月制备了一批N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢溴酸)并经NMR分析。当在室温避光储存10个月后,再次对所述固体物质进行分析,NMR数据相同。固体的颜色在这段时间内保持一致。这表明,采用这种形式的分子在所述条件下及在所述时间内为稳定的。
生物研究
方法:体外实验以建立B50
这些方法详细地描述于WO96/30766中。简言之,对应于核心重复域的τ片段(其已经被吸附到固相基质上)能够捕获可溶性全长τ并能高亲和力地与τ结合。这种结合使聚集的τ分子具有抗蛋白水解性消化(proteolytic digestion)的稳定性。该过程是自传播的(self-propagating),可被原型药物制剂(prototype pharmaceutical agents)选择性阻断。
更具体地,将稀释在碳酸盐缓冲液中(pH9.6)的经截短的τ(残基297-390;dGA)结合在分析板上,水相中加入全长τ(T40)。该水相结合缓冲液含有0.05%Tween-20和1%明胶的磷酸盐缓冲盐水溶液(pH7.4)。用mAb 499检测结合的τ,该检测识别出水相全长τ中的N-端表位,但没有识别出固相结合的经截短的τ片段。
抑制50%τ-τ结合所需的化合物的浓度定义为B50值。
方法:基于细胞的实验以建立EC50
该方法详细描述于WO02/055720中。简言之,在诱导型启动子的控制下成纤维细胞(3T6)表达全长τ(“T40”)和低构成水平(constitutive levels)的PHF-核心τ片段(12kD片段)。当T40的表达经诱导后,其在细胞内历经聚集-依赖性的截短,N-端是在~αα295和C-端是在~αα390,从而产生较高水平的12kDPHF-核心域片段。12kD片段的产生可经τ-聚集抑制剂通过剂量依赖方式阻断。事实上,化合物对细胞内蛋白水解生成12kD片段的抑制活性的量可根据与描述体外τ-τ结合抑制使用的参数相同的参数进行完全描述。即,在细胞内蛋白水解生成12kD片段的程度可完全由通过重复域的τ-τ结合的程度来确定。相关蛋白酶在细胞内的利用率是非限定性的。
结果表达为12kD片段生成的50%受到抑制时的浓度。将这定义为EC50值。
方法:细胞内毒性–LD50和治疗指数(RxI)
在用来测定EC50的基于细胞的实验中,测试本发明披露的化合物的毒性。毒性测定如下:在将剩余细胞溶解后根据制造商的说明,利用乳酸脱氢酶试剂盒TOX-7(Sigma Biosciences),使细胞与化合物接触24小时,然后确定细胞数,由此测定毒性。或者,使用Promega UK(CytoTox 96)的试剂盒,同样根据制造商的说明进行操作。
治疗指数(RxI)计算如下:RxI=LD50/EC50。
数据总结在下表中。
B3:N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(盐酸)。
B4:10H-吩噻嗪-3,7-二胺双(盐酸)。
B6:N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢碘酸)。
MTC:甲基锍氯化物。
分配系数研究
已知药物在有机相/水系统(典型地为正辛醇/水系统)中的分配系数通常记录为对数(即log10P)形式,该分配系数能很好的说明药物的生物活性。参见如Hansch,C.等人,1964,J.Am.Chem.Soc.,卷86,1616-1626页;Kubinyi,H.,1977,J.Med.Chem.,第20卷,第625-629页。一般认为,出现这种现象的原因是因为化合物的吸收依赖于其在生物或和水相之间的分配。分配系数也用于分离技术和用于预测药物溶解度中。
在药物类物质的情况下,疏水性与吸收、生物利用度、疏水药物-受体相互作用、代谢作用和毒性有关。低亲水性就会具有高log10P值,这可能导致较差的吸收或渗透。对很可能被很好吸收的化合物而言,已经显示出,这些化合物的log10P值一定不大于5.0。3000多种市售药物经计算的log10P值的分布证实了这个事实。
许多测定分配系数的方法是公知的。在该研究中,所选化合物的水溶液在每相中与正辛醇一起振荡,每相中各等份的浓度用可见光分光光度法测定。然后,利用下述公式针对每个化合物计算log10P:
log10P=log10[药物]辛醇-log10[药物]水=log10([药物]辛醇/[药物]水)
数据总结在下表中。结果表明,本发明的DAPTZ化合物具有预期针对类药物分子的log10P值。
B3:N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(盐酸)。
B6:N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢碘酸)。
MTC:甲基锍氯化物。
晶体结构
图4显示了N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢溴酸)的晶体结构。该晶体结构显示了三种晶体学上不同的溴离子。Br1和Br2以二重对称的方式占据了特殊位置,而有机主要分子和Br3占据了通常位置。因此,整个化学计量为C16H21N3SBr2。
图5显示了N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢溴酸)的侧面图,并揭示了非平面性;外部苯环之间的双面角为11.0(3)度。
图6显示了晶体中N,N,N',N'-四甲基-10H-吩噻嗪-3,7-二胺双(氢溴酸)分子的一个螺旋柱的部分。
- 还没有人留言评论。精彩留言会获得点赞!