产黄曲霉毒素真菌多重PCR检测用引物组、试剂盒及检测方法与流程
本发明属于生物检测领域,涉及利用多重PCR技术对产毒真菌进行快速检测鉴定的引物组、试剂盒及检测方法。具体而言,本发明涉及针对产黄曲霉毒素真菌的多重PCR检测用引物组、试剂盒及检测方法。
背景技术:
黄曲霉毒素(Aflatoxin,AFT)是主要由黄曲霉(A.flavus)、寄生曲霉(A.parasiticus)及特曲霉(A.nomius)等真菌产生的一类化学结构类似的次级代谢产物,现已分离出AFB1、AFB2、AFG1、AFG2、AFM1、AFM2、AFB2a、AFG2a、AFBM2a及AFGM2a等多达18种不同的黄曲霉毒素,其中AFB1的毒性最强且化学性质最稳定,它具有很强的致癌性、致突变性和致畸性,是国际上公认的A类致癌物。大量的调查研究证实,AFB1主要作用的靶器官为肝脏,AFB1能够导致肝脏坏死或肝癌形成;此外,AFB1还会影响动物胚胎发育,造成免疫抑制和反复感染,导致动物的产奶量或产蛋量降低。黄曲霉毒素主要污染花生、玉米、大豆、小麦、坚果等农作物及其制品;此外,肉类、乳及乳制品、水产品、发酵食品等也存在被黄曲霉毒素污染的风险。黄曲霉毒素污染不仅导致食品自身营养价值和经济价值的降低,更对人畜生命安全造成巨大的潜在威胁。
目前,国内外研究的焦点主要集中在黄曲霉毒素的检测方法,制定了包含薄层层析法、液相色谱法、放射免疫分析方法、酶联免疫法、免疫层析法等在内的比较成熟的检测规范。然而,这些方法也存在例如周期长、程序复杂、成本高等诸多不足之处。针对产黄曲霉毒素真菌本身的检测技术鲜有报道,远落后于相关毒素检测技术的研究与应用。迄今为止,对产黄曲霉毒素真菌的检测主要沿用传统的培养鉴定方法,即,在分离得到病原菌单菌落的基础上,通过形态学观察和科赫法则来判断病原菌的种类,同时采用病原菌产毒能力检测来进行验证,但这一传统方法实施起来比较困难。例如,整个检测鉴定过程常常需要昂贵的设备,并且要求操作者具备专业的病原菌分离技术、形态学鉴定知识和丰富的 经验,费时费力,一般需要几天才能完成。此外,传统形态学鉴定方法由于耗时长而效率低,而且容易出现假阳性或假阴性结果,难以满足对产黄曲霉毒素真菌快速灵敏检测的实际需要。
近年来,借助于分子生物学技术,对产黄曲霉毒素真菌的快速检测有了很大发展。例如,检测标准SN/T 2582-2010建立了产黄曲霉毒素真菌的PCR检测方法,实现了分别针对真菌通用分子标识序列(内部转录间隔区序列ITS)及黄曲霉毒素生化合成过程中三个关键基因(产黄曲霉毒素调节基因aflR、柄曲霉素转甲氧基酶基因omt-1及杂色曲霉素A脱氢酶基因ver-1)的单重PCR检测。然而,操作相对繁琐、影响因素较多、检测通量相对不足等缺陷影响了这一单重PCR检测技术在产黄曲霉毒素真菌检测中的广泛应用与发展。
相比之下,多重PCR技术能在同一PCR反应体系内实现对多个目标基因的同步扩增,从而能够快速、灵敏、特异地进行检测。具体而言,多重PCR检测法采用将多个目标基因联用的方式,通过不同目标基因间检测结果的互相映证,可实现提高检测准确度、降低假阳性率的效果。
在影响多重PCR检测的诸多因素中,目标基因的数目和种类的选取至关重要。若目标基因的数目过多,PCR体系中加入的引物对数量过多,产生非预期反应的概率增大,导致电泳鉴定图中出现杂带或造成拖尾等,从而影响检测效率;而若将引物加入不同的PCR体系分别进行反应,势必又会降低通量并增加检测工作量。另外,目标基因种类的选取对于是否能够有效进行多重PCR检测而言也是关键因素之一:如果目标基因特异性过高(即种属分布过窄),会造成检测覆盖种类不足,从而造成漏检等问题;反之,则可能会引起假阳性率高等弊端。
进而,在选定目标基因后,针对多个目标基因进行的引物设计同样可能从根本上影响多重PCR检测的结果:由于需要在同一PCR体系中针对不同目标基因进行同步扩增,适宜各引物对的反应条件需能够彼此配合、且各引物对之间不会相互干扰,从而确保针对各目标基因的PCR反应均能够顺利、准确地进行。
目前,已发展出少量用于对产黄曲霉毒素真菌进行检测的多重PCR 技术。例如,秦文彦等(可污染食品及饲料的产黄曲霉毒素真菌的多重PCR检测,《菌物学报》26(3):448~454,2007)公开了根据黄曲霉毒素生化途径中的关键调控基因aflR、omt-1和ver-1的序列以及真菌共有的5.8S rDNA的ITS序列分别设计四对引物,并利用这些引物建立了以可能被真菌污染的饲料或食品为检测对象的多重PCR检测体系。当模板DNA浓度降至0.1ng/μL时,aflR基因扩增片段完全消失,表明该体系的总体灵敏度为2ng/μL(灵敏度计算方式参见下文的具体描述),考虑到黄曲霉毒素极强的毒性和致癌力,这一灵敏度仍亟待提高。此外,CN103103258A也公开了一种利用多重PCR技术分析花生粕中黄曲霉毒素的方法,然而,CN103103258A中并未提及该方法的灵敏度,因而难以知晓其是否能满足实际检测要求,事实上,CN103103258A中使用的四对引物与秦文彦等(可污染食品及饲料的产黄曲霉毒素真菌的多重PCR检测,《菌物学报》26(3):448~454,2007)中使用的四对引物完全相同,因而推测其灵敏度同样至多为2ng/μL。
可见,现有技术并不能完全满足产黄曲霉毒素真菌检测的需求。因此,建立一种快速、灵敏、特异、准确、高效的产黄曲霉毒素真菌多重PCR检测技术显得尤为迫切。这是解决产黄曲霉毒素真菌侵染的检测问题的重要途径,此类方法具有良好的应用与发展前景。
技术实现要素:
针对上述技术问题,本发明组合利用了真菌通用分子标识序列(内部转录间隔区序列ITS)及黄曲霉毒素生化合成过程中三个关键基因(产黄曲霉毒素调节基因aflR、柄曲霉素转甲氧基酶基因omt-1及杂色曲霉素A脱氢酶基因ver-1),采用多重PCR技术对产黄曲霉毒素真菌进行快速检测。本发明在比对分析产黄曲霉毒素真菌PCR检测方法标准《SN/T 2582-2010产黄曲霉毒素真菌PCR检测方法》的基础上优化设计,筛选出覆盖范围广、特异性强、灵敏度高的相应引物对及引物组,优化组合建立了多重PCR检测体系,并开发了产黄曲霉毒素真菌多重PCR检测用试剂盒;同时,建立了检测覆盖范围广、特异性强且灵敏度高的检测方法,由此完成了本发明。
在第一方面,本发明提供了一种用于快速检测产黄曲霉毒素真菌的 多重PCR检测用引物组,所述多重PCR检测用引物组由特异性针对真菌内部转录间隔区序列ITS的ITS引物对、特异性针对产黄曲霉毒素调节基因aflR的aflR引物对、特异性针对柄曲霉素转甲氧基酶基因omt-1的omt-1引物对及特异性针对杂色曲霉素A脱氢酶基因ver-1的ver-1引物对组成,其中,
所述ITS引物对的序列为:
上游引物ITS-600-F:5’-TCCGTAGGTGAACCTGCG-3’(SEQ ID NO:1);
下游引物ITS-600-R:5’-TCCTCCGCTTATTGATATGC-3’(SEQ ID NO:2),
所述aflR引物对的序列为:
上游引物aflR-421-F:5’-AGCAAAGCACCCTGTCTTC-3’(SEQ ID NO:3);
下游引物aflR-421-R:5’-TTGAGAACGATAAGGACGACC-3’(SEQ ID NO:4),
所述omt-1引物对的序列为:
上游引物omt-1-317-F:5’-TCATTTTCGAGGAGGTTGCC-3’(SEQ ID NO:5);
下游引物omt-1-317-R:5’-AGCTGTTTCTCGCATTTCAGC-3’(SEQ ID NO:6),
所述ver-1引物对的序列为:
上游引物ver-1-201-F:5’-TTCGGTCACCTGAAAGACG-3’(SEQ ID NO:7);
下游引物ver-1-201-R:5’-TGACGCAAGCGGTGTTAG-3’(SEQ ID NO:8)。
在第二方面,本发明提供了一种用于快速检测产黄曲霉毒素真菌的多重PCR检测用试剂盒,所述试剂盒中包含如本发明第一方面所述的多重PCR检测用引物组。
在第三方面,本发明还提供了一种使用本发明的多重PCR检测用引物组或多重PCR检测用试剂盒对产黄曲霉毒素真菌进行快速检测的方 法,所述方法包括以下步骤:
a)提取待检测样品的总DNA作为模板DNA的步骤;
b)使用如本发明第一方面所述的多重PCR检测用引物组或如本发明第二方面所述的多重PCR检测用试剂盒对所述模板DNA进行多重PCR扩增的步骤;
c)对扩增产物进行检测的步骤;
从而对所述待检测样品中产黄曲霉毒素真菌的存在与否进行分析。
本发明所提供的针对产黄曲霉毒素真菌的多重PCR检测用引物组、试剂盒及检测方法具有检测适用面广、对产黄曲霉毒素真菌种属覆盖全面、特异性强、灵敏度高等优势,从而有效解决了现行检测标准和文献中报道的针对产黄曲霉毒素真菌的PCR检测方法中由检测覆盖种类不足、引物特异性差及灵敏度不强等因素造成的漏检、假阳性等问题。此外,本发明提供的针对产黄曲霉毒素真菌的多重PCR用检测引物组、试剂盒及检测方法还具备检测耗时短、检测通量高、操作较简单、设备要求低等特点,可以节省大量的人力、物力和财力,适合高通量快速检测的要求,具有广泛的推广应用市场前景。
具体实施方式
根据本发明的一些实施方式,除本发明的多重PCR检测用引物组外,本发明的多重PCR检测用试剂盒中可进一步包含反应缓冲液、4种脱氧核苷三磷酸、DNA聚合酶等成分。
根据本发明一些优选的实施方式,除本发明的多重PCR检测用引物组外,本发明的多重PCR检测用试剂盒中可进一步包含具有如下成分的PCR反应基础溶液:Tris-HCl;KCl;MgCl2;dATP、dTTP、dGTP及dCTP;以及Taq DNA聚合酶。
根据本发明一些优选的实施方式,在本发明的多重PCR检测用试剂盒中,各成分以如下浓度包含于50μL的PCR最终反应溶液中:
其中,所述ITS引物对的序列为:
上游引物ITS-600-F:5’-TCCGTAGGTGAACCTGCG-3’;
下游引物ITS-600-R:5’-TCCTCCGCTTATTGATATGC-3’,
所述aflR引物对的序列为:
上游引物aflR-421-F:5’-AGCAAAGCACCCTGTCTTC-3’;
下游引物aflR-421-R:5’-TTGAGAACGATAAGGACGACC-3’,
所述omt-1引物对的序列为:
上游引物omt-1-317-F:5’-TCATTTTCGAGGAGGTTGCC-3’;
下游引物omt-1-317-R:5’-AGCTGTTTCTCGCATTTCAGC-3’,
所述ver-1引物对的序列为:
上游引物ver-1-201-F:5’-TTCGGTCACCTGAAAGACG-3’;
下游引物ver-1-201-R:5’-TGACGCAAGCGGTGTTAG-3’。
根据本发明一些更为优选的实施方式,在本发明的多重PCR检测用试剂盒中,各成分以如下浓度包含于50μL的PCR最终反应溶液中:
其中,所述ITS引物对的序列为:
上游引物ITS-600-F:5’-TCCGTAGGTGAACCTGCG-3’;
下游引物ITS-600-R:5’-TCCTCCGCTTATTGATATGC-3’,
所述aflR引物对的序列为:
上游引物aflR-421-F:5’-AGCAAAGCACCCTGTCTTC-3’;
下游引物aflR-421-R:5’-TTGAGAACGATAAGGACGACC-3’,
所述omt-1引物对的序列为:
上游引物omt-1-317-F:5’-TCATTTTCGAGGAGGTTGCC-3’;
下游引物omt-1-317-R:5’-AGCTGTTTCTCGCATTTCAGC-3’,
所述ver-1引物对的序列为:
上游引物ver-1-201-F:5’-TTCGGTCACCTGAAAGACG-3’;
下游引物ver-1-201-R:5’-TGACGCAAGCGGTGTTAG-3’。
根据本发明一些优选的实施方式,本发明的多重PCR检测用试剂盒中还含有阴性对照DNA和阳性对照DNA。
根据本发明一些进一步优选的实施方式,本发明的多重PCR检测用试剂盒中的所述阴性对照DNA来自黑曲霉(Aspergillus niger)菌株CGMCC 3.0316,所述阳性对照DNA来自黄曲霉(Aspergillus flavus)菌株CGMCC 3.4410。
根据本发明的一些实施方式,在提取待检测样品的总DNA之前,可根据待检测样品的种类对待检测样品进行粉碎等处理。为获得多重PCR检测的模板DNA,可利用可商购的DNA提取试剂盒获取待检测样品中的总DNA;或者,也可通过水煮裂解法获取待检测样品中的总DNA。
根据本发明的一些实施方式,在使用本发明的多重PCR检测用试剂盒对产黄曲霉毒素真菌进行检测的方法中,进行所述多重PCR扩增的反应体系为:以总体积为50μL计,加入浓度为10pg/μL~100ng/μL的模板DNA 1μL;反应条件为:94℃预变性2min;94℃变性20s、55℃退火30s、72℃延伸40s,共进行35个循环;72℃延伸7min。
在本发明的实施方式中,可依照如下方式对扩增产物进行分析:利用1.5%(w/v)的琼脂糖凝胶电泳检测多重PCR扩增产物,经含有0.01%(v/v)的EB染色液染色后,观测凝胶中DNA条带大小。阳性对照中应出现全部4个扩增片段(ITS、aflR、omt-1和ver-1基因扩增片段);阴性对照中应只出现ITS扩增片段(约600bp);空白模板对照中应无任何 扩增片段。当检测样品中ITS、aflR、omt-1和ver-1基因扩增片段没有全部出现时,判定检测结果为阴性,即无产黄曲霉毒素真菌侵染。当检测样品同时出现全部4个扩增片段时,判定检测结果为可疑阳性,即可能存在产黄曲霉毒素真菌侵染。对于可疑阳性样品,应在产毒纯化后按照《SN/T 1035-2011进出口食品中产毒青霉属、曲霉属及其毒素的检测方法》和《GB/T 4789.16-2003食品卫生微生物学检验常见产毒霉菌的鉴定》进行黄曲霉毒素测定,以进行进一步确认,如确认结果为阳性,判定结果为阳性,否则判定为阴性。
在本发明中,多重PCR反应的检测灵敏度依照如下方式进行计算:在50μL的多重PCR反应体系内分别加入不同浓度的模板DNA 1μL,针对四个目标基因,各自所能检出的模板DNA的最低浓度即为该多重PCR反应对于各目标基因的检测灵敏度,取针对四个目标基因的检测灵敏度的最大值(即,最差的灵敏度)作为该多重PCR反应的检测灵敏度。例如,当在50μL的多重PCR反应体系内分别加入1pg/μL、10pg/μL、100pg/μL、1ng/μL及10ng/μL的阳性对照(黄曲霉菌株CGMCC 3.4410)模板DNA 1μL时,若能够在加入1pg/μL、10pg/μL、100pg/μL、1ng/μL及10ng/μL的阳性对照模板DNA 1μL时检测到ITS的目的条带,则对ITS而言,所能检出的模板DNA的最低浓度为1pg/μL,该多重PCR反应对于ITS的检测灵敏度为1pg/μL;对aflR、omt-1和ver-1基因的检测灵敏度评判标准与此类似;相应地,例如,在该多重PCR反应对于aflR、omt-1和ver-1基因的检测灵敏度分别为1pg/μL、1pg/μL和10pg/μL时,认为该多重PCR反应的检测灵敏度为10pg/μL。
附图说明
图1示出了本发明同检测标准SN/T 2582-2010关于产黄曲霉毒素真菌单重PCR检测特异性的比较。
图2示出了本发明同检测标准SN/T 2582-2010关于产黄曲霉毒素真菌单重PCR检测灵敏度的比较。
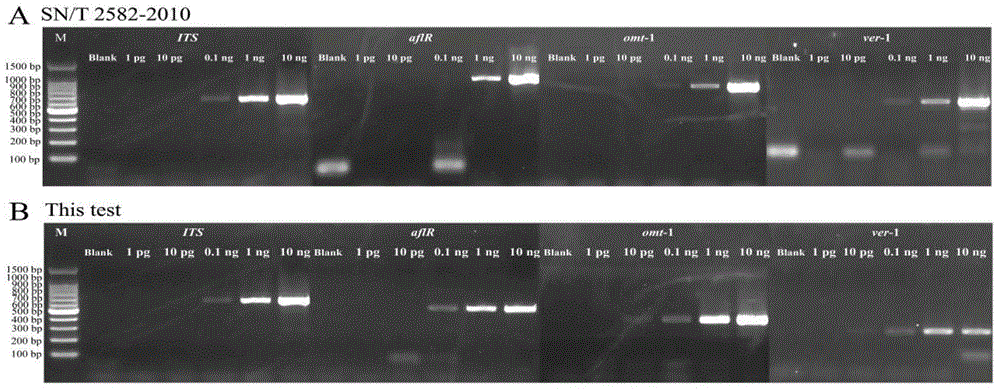
图3示出了本发明同检测标准SN/T 2582-2010关于产黄曲霉毒素真菌多重PCR检测灵敏度的比较。
实施例
以下实施例和比较例的目的在于对本发明的产黄曲霉毒素真菌多重PCR检测用引物组、试剂盒及检测方法的原理及应用作进一步的说明,但并不以任何形式限制本发明的范围。
实施例1:单重PCR反应体系及反应条件的建立
1.引物设计及筛选
根据真菌中黄曲霉毒素合成途径及相应酶编码基因信息,选择真菌黄曲霉毒素生化合成过程中三个关键基因-产黄曲霉毒素调节基因aflR、柄曲霉素转甲氧基酶基因omt-1及杂色曲霉素A脱氢酶基因ver-1作为研究对象。通过NCBI(www.ncbi.nlm.nih.gov)的GenBank数据库获取相应序列,将aflR、omt-1及ver-1序列同检测标准SN/T 2582-2010中的PCR扩增片段进行序列相似性比对分析,选取扩增片段中更为保守的序列区段,采用Primer Premier 5.0软件设计分析引物,优化筛选出特异性高的3个引物对并与ITS PCR检测用引物对进行组合,序列如表1所示。
表1 ITS、aflR、omt-1及ver-1基因的PCR扩增用引物对序列
2.单重PCR反应体系及反应条件的建立
本研究进行了分别针对ITS、aflR、omt-1及ver-1基因的单重PCR检测反应体系及反应条件优化,确定了最佳的反应体系及反应条件。
最佳PCR反应体系为(以50μL的PCR最终反应溶液计):
针对ITS基因的最佳PCR反应条件为:94℃预变性2min;94℃变性20s、55℃退火30s、72℃延伸40s,共进行35个循环;72℃延伸7min;
针对aflR、omt-1及ver-1基因的最佳PCR反应条件为:94℃预变性2min;94℃变性20s、55℃退火30s、72℃延伸30s,共进行35个循环;72℃延伸7min。
实施例2:单重PCR检测中各引物对检测特异性的验证
对于实施例2和比较例1而言,在进行检测前对实验所采用的各菌株进行培养,抽提基因组DNA,用超微量分光光度计NanoDrop 2000检测DNA浓度,分别以1μL浓度为10ng/μL的DNA作为模板进行PCR扩增,其它反应体系及反应条件如实施例1中所述。
采用所设计的PCR检测用引物组中的4对特异性PCR引物对(如表1所示),对11株标准菌株或已经过鉴定的菌株(如表2所示)进行PCR检测。对于是否产AFT的检测,按照《GB/T 4789.16-2003食品卫生微生物学检验常见产毒霉菌的鉴定》进行。正交试验结果如图1B所示。图1B中,Blank表示空白对照的单重PCR检测;M表示100bp ladder Marker,各条带大小依次为1500bp、1000bp、900bp、800bp、700bp、600bp、500bp、400bp、300bp、200bp及100bp;泳道1、2、3、4中所加入的样品分别为ITS、aflR、ver-1和omt-1基因的扩增产物。对于经产黄曲霉毒素能力鉴定表现为阳性的4株菌株(编号分别为Sample 5、Sample 6、Sample 8及Sample 10),利用本发明的针对ITS、aflR、omt-1和ver-1基 因的引物对分别进行单重PCR扩增均能得到扩增片段,可判定结果为可疑阳性;而经鉴定为无黄曲霉毒素产毒能力的其它7株菌株(编号分别为Sample 1、Sample 2、Sample 3、Sample 4、Sample 7、Sample 9及Sample 11)的PCR扩增产物中均只出现ITS的目的扩增片段(泳道1),可判定结果为阴性。该结果表明,本发明设计出的各引物对在单重PCR检测中对产黄曲霉毒素真菌具有较强的特异性。
比较例1:单重PCR检测中各引物对检测特异性的比较
为对使用本发明的引物对进行的检测方法的特异性与检测标准SN/T 2582-2010的特异性进行比较,利用检测标准SN/T 2582-2010所提供的引物对,按照与实施例2相同的方法进行单重PCR和琼脂糖凝胶电泳分析,结果如图1A所示。图1A中,M表示与实施例2相同的100bp ladder Marker;泳道1、2、3、4中所加入的样品分别为ITS、aflR、ver-1和omt-1基因的扩增产物。对于经产黄曲霉毒素能力鉴定表现为阳性的4株菌株(编号分别为Sample 5、Sample 6、Sample 8及Sample 10),利用检测标准SN/T 2582-2010所提供的针对ITS、aflR、omt-1和ver-1基因的引物对分别进行单重PCR扩增均能得到扩增片段,可判定结果为可疑阳性;而经鉴定为无黄曲霉毒素产毒能力的其它7株菌株(编号分别为Sample 1、Sample 2、Sample 3、Sample 4、Sample 7、Sample 9及Sample 11)的PCR扩增产物中均只出现ITS的目的扩增片段(泳道1),可判定结果为阴性。
可见,本发明的针对ITS、aflR、omt-1和ver-1基因的各产黄曲霉毒素真菌单重PCR检测用引物对及相应的检测方法与检测标准SN/T 2582-2010具有相同的检测特异性,具备特异性检测产黄曲霉毒素真菌的能力。
表2 所使用的菌株列表
实施例3:单重PCR检测中各引物对检测灵敏度的验证
对于实施例3和比较例2而言,在进行检测前培养黄曲霉菌株CGMCC 3.4410,抽提基因组DNA,用超微量分光光度计NanoDrop 2000检测DNA浓度,进行10倍梯度稀释,利用表1中所示的各引物对,分别以1μL不同浓度的DNA作为模板进行PCR扩增,其它反应体系及反应条件如实施例1中所述。对PCR产物进行电泳分析,结果如图2B所示。
图2A和图2B中,M表示100bp ladder Marker,各条带大小依次为1500bp、1000bp、900bp、800bp、700bp、600bp、500bp、400bp、300bp、200bp及100bp;ITS、aflR、omt-1及ver-1表示分别以ITS、aflR、omt-1及ver-1为目标检测基因的单重PCR检测,Blank表示空白对照的单重PCR检测。从左至右,各泳道所标注的1pg、10pg、0.1ng、1ng及10ng分别表示在50μL的PCR反应体系中,所加入的1μL模板DNA的浓度分别为1pg/μL、10pg/μL、0.1ng/μL、1ng/μL及10ng/μL。
从图2B可以看出,采用本发明提供的产黄曲霉毒素真菌单重PCR检测用引物对及检测方法,对ITS、aflR、omt-1及ver-1基因的PCR检 测反应灵敏度分别达到0.1ng/μL、0.1ng/μL、10pg/μL及10pg/μL。
比较例2:单重PCR检测中各引物对检测灵敏度的比较
为对使用本发明的引物对的检测方法的灵敏度与检测标准SN/T 2582-2010的灵敏度进行比较,利用检测标准SN/T 2582-2010所提供的引物对,按照与实施例3相同的方法进行单重PCR和琼脂糖凝胶电泳分析,结果如图2A所示。
从图2A可以看出,采用检测标准SN/T 2582-2010提供的产黄曲霉毒素真菌PCR检测用引物对及检测方法,对ITS、aflR、omt-1及ver-1基因的单重PCR检测反应灵敏度分别达到0.1ng/μL、1ng/μL、0.1ng/μL及0.1ng/μL;而采用本发明提供的产黄曲霉毒素真菌单重PCR检测用引物对及检测方法,对ITS、aflR、omt-1及ver-1基因的单重PCR检测反应灵敏度分别达到0.1ng/μL、0.1ng/μL、10pg/μL及10pg/μL,也就是说,就aflR、omt-1及ver-1基因的单重PCR检测反应灵敏度而言,本发明提供的检测引物对及检测方法均优于检测标准SN/T 2582-2010提供的检测引物对及检测方法。这表明本发明提供的PCR检测用引物对及检测方法能够获得较高的检测灵敏度,具备更好地满足产黄曲霉毒素真菌PCR检测需求的优势。
实施例4:多重PCR反应体系及反应条件的建立
基于单重PCR检测的反应体系及反应条件,本发明对多重PCR检测中的各参数进行了优化,确定了最佳的反应体系及反应条件。
最佳多重PCR反应体系为(以50μL的PCR最终反应溶液计):
上述多重PCR反应的反应条件为:
94℃预变性2min;94℃变性20s、55℃退火30s、72℃延伸40s,共进行35个循环;72℃延伸7min。
实施例5:多重PCR检测用引物组的检测特异性的验证
在进行检测前对实验所采用的各菌株进行培养,抽提基因组DNA,用超微量分光光度计NanoDrop 2000检测DNA浓度,分别以1μL浓度为10ng/μL的DNA作为模板进行PCR扩增。利用实施例4中建立的多重PCR反应体系及反应条件,通过正交反应多次试验,对11株实验菌株(如表2所示)进行多重PCR检测,结果表明:
对于经产黄曲霉毒素能力鉴定表现为阳性的4株菌株(编号分别为Sample 5、Sample 6、Sample 8及Sample 10),利用本发明的针对ITS、aflR、omt-1和ver-1基因的引物对进行多重PCR扩增可得到全部4个扩增片段(ITS、aflR、omt-1和ver-1基因扩增片段),而经鉴定为无黄曲霉毒素产毒能力的其它7株菌株(编号分别为Sample 1、Sample 2、Sample 3、Sample 4、Sample 7、Sample 9及Sample 11)的多重PCR扩增产物中均只出现ITS的目的扩增片段。该结果同检测标准SN/T 2582-2010提供的产黄曲霉毒素真菌PCR检测方法的检测结果完全一致,表明本发明设计出的由表1中列出的4个引物对构成的多重PCR检测用引物组对产黄曲霉毒素真菌具有较强的特异性。
实施例6:多重PCR检测用引物组的检测灵敏度的验证
对于实施例6和比较例3而言,在进行检测前培养黄曲霉菌株CGMCC 3.4410,抽提基因组DNA,用超微量分光光度计NanoDrop 2000检测DNA浓度,进行10倍梯度稀释,利用表1中所示的各引物对,分别以1μL不同浓度的DNA作为模板进行PCR扩增,其它反应体系及反应条件如实施例4中所述。对PCR产物进行电泳分析,结果如图3所示。
图3中,M表示100bp ladder Marker,各条带大小依次为1500bp、 1000bp、900bp、800bp、700bp、600bp、500bp、400bp、300bp、200bp及100bp;Blank表示空白对照的多重PCR检测。Test表示依据本发明提供的多重PCR反应体系及反应条件的检测结果,SN/T 2582-2010表示依据检测标准《SN/T 2582-2010产黄曲霉毒素真菌PCR检测方法》构建的多重PCR反应体系及反应条件的检测结果。从左至右,各泳道所标注的1pg、10pg、0.1ng、1ng及10ng分别表示在50μL的PCR反应体系中,所加入的1μL模板DNA的浓度分别为1pg/μL、10pg/μL、0.1ng/μL、1ng/μL及10ng/μL。
从图3可以看出,采用本发明提供的产黄曲霉毒素真菌多重PCR检测用引物组及检测方法,对产黄曲霉毒素真菌中的ITS、aflR、omt-1及ver-1基因进行的多重PCR检测反应灵敏度达到0.1ng/μL。
比较例3:多重PCR检测用引物组的检测灵敏度的比较
将使用本发明多重PCR检测用引物组的检测方法的特异性与依据检测标准《SN/T 2582-2010产黄曲霉毒素真菌PCR检测方法》构建的多重PCR检测方法的灵敏度进行比较,结果如图3所示。
采用依据检测标准《SN/T 2582-2010产黄曲霉毒素真菌PCR检测方法》构建的多重PCR检测方法对产黄曲霉毒素真菌ITS、aflR、omt-1及ver-1基因的多重PCR检测反应的灵敏度达到1ng/μL;而采用本发明提供的产黄曲霉毒素真菌多重PCR检测用引物组及检测方法,对产黄曲霉毒素真菌ITS、aflR、omt-1及ver-1基因的多重PCR检测反应的灵敏度可进一步提升至0.1ng/μL。这表明本发明提供的多重PCR检测用引物组及检测方法能够获得较高的检测灵敏度,具备更好地满足产黄曲霉毒素真菌多重PCR检测需求的优势。
实施例7:应用于实际样品的检测试验
利用实施例4中建立的多重PCR反应体系及反应条件,采用本发明提供的产黄曲霉毒素真菌多重PCR检测用引物组及检测方法,对应用表2所列菌株构建的人工侵染的玉米(11份)、花生(11份)及饲料(11份)样品中的产黄曲霉毒素真菌进行实际检测。
在33份检测样品中,本方法检测出12份样品被产黄曲霉毒素真菌 侵染,这与预期结果完全一致,说明本发明所提供的针对产黄曲霉毒素真菌的PCR检测用引物组及检测方法在应用于实际样品的检测试验中的准确度高达100%。
以上结果表明,本发明所提供的针对产黄曲霉毒素真菌的多重PCR检测用引物组及检测方法具有出色的准确性和实用性,并且检测耗时短,具有广泛的市场推广应用前景。
虽然,上文中已经用一般性说明及具体实施例对本发明作了详尽的描述,但在本发明描述的基础上,可以对之作一些修改或改进,这对本领域技术人员而言是容易实现的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!