一种丙酸铵晶体及其粉剂的制备方法与流程
本发明属于化工原料制备领域,具体涉及一种丙酸铵晶体及其粉剂的制备方法。
背景技术:
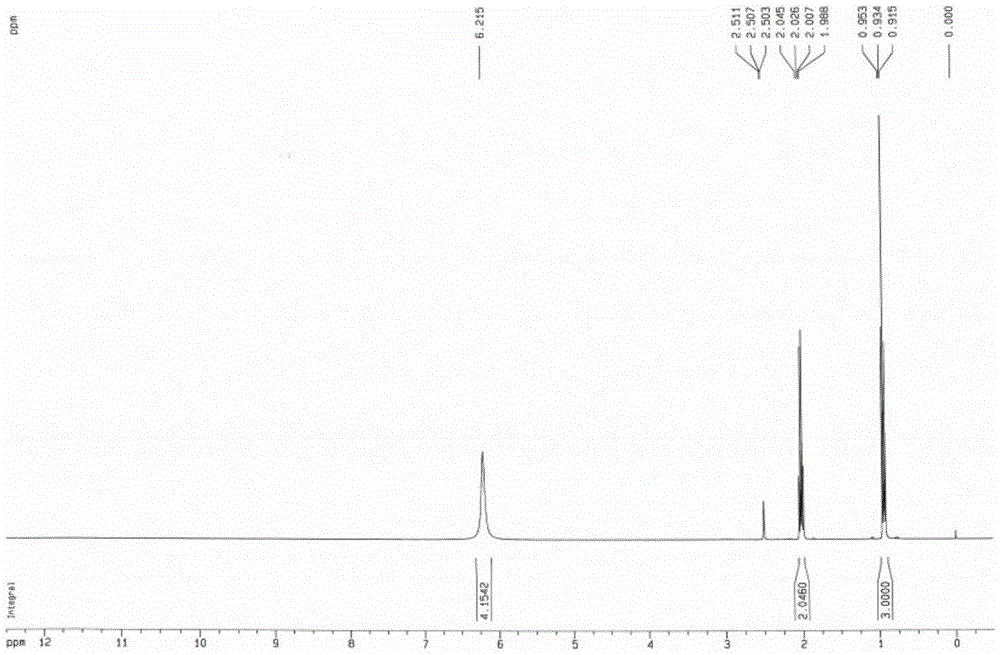
:随着饲料工业的迅猛发展、规模壮大,要求饲料具有更高的品质和新鲜度,但饲料在生产、运输、贮存和饲喂等过程中面临着如饲料水分不均、暴晒、暴雨;梅雨季、台风季等气候条件和频繁多变的流感因素的影响,导致饲料发霉变质,进而产生毒素、品质下降。为防止饲料发霉,最经济、简便和行之有效的办法是添加饲料防霉剂,但绝大多数防霉剂在应用上都有或多或少之弊端:毒性、腐蚀性、气味、影响适口性、性价比不高等。不断设计开发一种无毒性、无腐蚀性、无气味、不影响适口性、性价比高和安全环保的应用于饲料防霉保鲜的添加剂有着极为重要的现实意义。丙酸铵是一种吸湿性极强的物质,其主要用途是用作防腐、防霉剂,在酸性前提下产生游离丙酸,具有抗菌性,对防止黄曲霉菌素的产生有特效,而对酵母几乎无效,对各类霉菌、革兰氏阴性杆菌效果好,并且pH值越低防腐效果越好,对于饲料或谷物的防霉烂和防结块有良好的效果。对以上所述的市场和现实需要,丙酸铵具有重要的开发价值。但由于丙酸铵的易水解、吸潮和略高温即溶化特性,加之本身参与的是弱酸弱碱反应,到终点更加趋于困难,工艺繁琐、增加成本,并很难提纯、收率降低,因此寻找一种纯度高、收率高、且工艺简单、成本低廉的丙酸铵的制备工艺,是本领域目前刻不容缓的事情。技术实现要素:为此,本发明所要解决的技术问题在于克服现有技术中丙酸铵的制备工艺不成熟、工艺复杂、成本高昂且制得的产品很难提纯、收率很低的技术瓶颈,从而提出一种纯度高、收率高、且工艺简单、成本低廉的丙酸铵晶体及其粉剂的制备工艺。为解决上述技术问题,本发明的技术方案为:本发明提供一种丙酸铵晶体的制备方法,所述方法包括如下步骤:1)将丙酸依次加入顺次连接的第一反应器、第二反应器、第三反应器,并对第一反应器和第二反应器中的丙酸进行搅拌;2)将液氨经蒸发分离除杂质后进行预热和稳压;3)将步骤2)中经分离、预热和稳压的氨气通入第一反应器,与第一反应器内的丙酸在搅拌下发生反应;4)冷却所述第一反应器和第二反应器,控制反应温度,并再次进行稳压;5)稳压后,持续反应1h以上,第一反应器中氨气溢出至第二反应器,与第二反应器内的丙酸反应使第二反应器内温度升高,开启第三反应器,并进行冷却降温;6)继续反应2-2.5h后第二反应器内氨气溢出至第三反应器,与第三反应器内的丙酸反应使第三反应器内温度升高,测定第三反应器内丙酸液中丙酸铵的含量;7)停止通入氨气,继续搅拌一段时间后,取出产物、密封、冷却保存,得到丙酸铵晶体。作为优选,所述步骤1)中,所述搅拌处理的搅拌速度为45r/min-50r/min。作为优选,所述步骤2)中,氨蒸发分离压力为0.6MPa-1.2MPa;预热处理后温度达到40℃-60℃;稳压后,氨气压力为0.6MPa-1.2MPa。作为优选,所述步骤4)中,所述反应温度为110℃-130℃,稳压后氨气压力为0.6MPa-1.2MPa。作为优选,所述步骤6)中,丙酸液中丙酸铵的含量占所述丙酸液与丙酸铵混合物质量的10%-15%。作为优选,所述步骤6)之后还包括将所述第一反应器与所述第二反应器中溢出未参与反应而剩余的氨气引至新鲜丙酸溶液中进行下批生产的步骤。作为优选,所述步骤7)之后还包括将所述第二反应器中的丙酸液置换于所述第一反应器,将第三反应器中的丙酸液储存用于制备下批丙酸铵的步骤。作为优选,所述步骤2)中的预热过程的热源温度为75-85℃,其由自来水和参与冷却的高温水混合得到。作为优选,所述步骤1)中所述第一反应器中丙酸加入量为每1m3中加入600kg-700kg;所述第二反应器和第三反应器中丙酸加入量为每1m3中加入700kg-800kg。本发明还提供一种丙酸铵粉剂的制备方法,包括如下步骤:将60-75重量份丙酸铵晶体和25-40重量份活化二氧化硅载体搅拌混合均匀、干燥后再粉碎至80目-100目,过筛后即得。本发明的上述技术方案相比现有技术具有以下优点:(1)本发明提供的丙酸铵晶体的制备方法,该方法工艺简单、高效、环保,第一反应器、第二反应器和第三反应器中未反应的丙酸液可回收用于下批丙酸铵的生产,最终多余出的另一部分尾氨吸收丙酸液,经再合成调节可制作为低含量丙酸铵、高含量丙酸结构防霉剂,无损耗和浪费;本反应为放热反应,放出的热量又可作为供液氨蒸发、预热的热源;制得的丙酸铵纯度高、收率高、成本低廉。(2)本发明提供的丙酸铵粉剂的制备方法,制得的丙酸铵粉剂在作用时独有持续、缓释、彻底的优势特点,无气味、无腐蚀、适口好、效率高。附图说明为了使本发明的内容更容易被清楚的理解,下面根据本发明的具体实施例并结合附图,对本发明作进一步详细的说明,其中图1本发明制备的丙酸铵晶体的红外光谱图;图2本发明制备的丙酸铵晶体的核磁共振氢(1H)谱图;图3本发明制备的丙酸铵晶体的核磁共振氢碳(13C)谱图;图4本发明制备的丙酸铵化晶体的核磁共振氢DEPT135谱图;图5本发明制备的丙酸铵晶体的核磁共振氢DEPT90谱图;图6本发明制备的丙酸铵晶体的ESI质谱图。具体实施方式实施例1本实施例用于说明本发明提供的丙酸铵晶体的制备方法。步骤如下:1)将600kg丙酸加入容积为1m3的第一反应器、700kg丙酸加入容积为1m3的第二反应器、700kg丙酸加入容积为1m3的第三反应器中,所述第一反应器为主反应装置,所述第二反应器为一级氨尾气吸收装置,所述第三反应器为二级氨吸收装置,所述主反应装置、一级氨尾气吸收装置和二级氨尾气吸收装置顺次连接;并对主反应装置和一级氨尾气吸收装置中的丙酸进行搅拌;本实施例中,所述主反应装置为主反应釜,一级氨尾气吸收装置为一级氨尾气吸收釜、二级氨尾气吸收装置为二级氨尾气吸收釜;启动主反应釜和一级氨尾气吸收釜内搅拌电机电源,再分别通过变频器调节两釜内搅拌轴速度,使搅拌桨(轴流、分散搅拌桨)的速度在45r/min;2)将液氨罐里液氨经氨蒸发分离器在0.6MPa下分离出其中微量夹带的油类和机械杂质,再进入氨预热器预热至40℃,所述氨预热器的热源温度为75℃,其由自来水和参与冷却的高温水混合得到,并由调节阀调节氨气压力稳定在0.6MPa;3)经预分离、预热和稳压的氨气,再缓慢开通进入主反应釜的气阀,使氨气匀速进入主反应釜,并将氨气分散于丙酸液体相里,在45r/min的速度下 搅拌发生气液放热反应;4)同时开启主反应釜和一级氨尾气吸收釜配套的冷却塔,经冷却循环水冷却降温,使反应温度最终控制在110℃,再次调解氨气压力至0.6MPa;5)稳压后,持续反应1h以上,主反应釜中氨气溢出至一级氨尾气吸收釜,与一级氨尾气吸收釜内的丙酸反应使一级氨尾气吸收釜内温度升高,开启二级氨尾气吸收釜,并进行冷却降温;6)继续反应2h后一级氨尾气吸收釜内氨气溢出至二级氨尾气吸收釜,与二级氨尾气吸收釜内的丙酸反应使二级氨尾气吸收釜内温度升高,采用快速滴定法测定二级氨尾气吸收釜内丙酸液中丙酸铵的含量,本实施例中丙酸铵的含量占所述丙酸液与丙酸铵混合物质量的10%;停止通入氨气,防止氨气进主反应釜阀,再搅拌反应约5min,然后将所述主反应釜与所述一级氨尾气吸收釜中未参与反应而剩余的氨气引至新鲜丙酸溶液中进行下批生产;7)在冷却的条件下,主反应釜继续搅拌一段时间,取出产物,冷却保存、检测、包装,即得丙酸铵晶体;然后将所述一级氨尾气吸收釜中的丙酸液置换于所述主反应釜,将二级氨尾气吸收釜中的丙酸液储存用于制备下批丙酸铵。实施例2本实施例用于说明本发明提供的丙酸铵晶体的制备方法。步骤如下:1)将650kg丙酸加入容积为1m3的第一反应器、750kg丙酸加入容积为1m3的第二反应器、750kg丙酸加入容积为1m3的第三反应器中,所述第一反应器为主反应装置,所述第二反应器为一级氨尾气吸收装置,所述第三反应器为二级氨吸收装置,所述主反应装置、一级氨尾气吸收装置和二级氨尾气吸收装置顺次连接;并对主反应装置和一级氨尾气吸收装置中的丙酸进行搅拌;本实施例中,所述主反应装置为主反应釜,一级氨尾气吸收装置为一级氨尾气吸收釜、二级氨尾气吸收装置为二级氨尾气吸收釜;启动主反应釜和一级氨尾气吸收釜内搅拌电机电源,再分别通过变频器调节两釜内搅拌轴速度, 使搅拌桨(轴流、分散搅拌桨)的速度在48r/min;2)将液氨罐里液氨经氨蒸发分离器在1MPa下分离出其中微量夹带的油类和机械杂质,再进入氨预热器预热至50℃,所述氨预热器的热源温度为80℃,其由自来水和参与冷却的高温水混合得到,并由调节阀调节氨气压力稳定在1MPa;3)经预分离、预热和稳压的氨气,再缓慢开通进入主反应釜的气阀,使氨气匀速进入主反应釜,并将氨气分散于丙酸液体相里,在47r/min的速度下搅拌发生气液放热反应;4)同时开启主反应釜和一级氨尾气吸收釜配套的冷却塔,经冷却循环水冷却降温,使反应温度最终控制在120℃,再次调解氨气压力至1MPa;5)稳压后,持续反应1h以上,主反应釜中氨气溢出至一级氨尾气吸收釜,与一级氨尾气吸收釜内的丙酸反应使一级氨尾气吸收釜内温度升高,开启二级氨尾气吸收釜,并进行冷却降温;6)继续反应2.2h后一级氨尾气吸收釜内氨气溢出至二级氨尾气吸收釜,与二级氨尾气吸收釜内的丙酸反应使二级氨尾气吸收釜内温度升高,采用快速滴定法测定二级氨尾气吸收釜内丙酸液中丙酸铵的含量,本实施例中丙酸铵的含量占所述丙酸液与丙酸铵混合物质量的12%;停止通入氨气,防止氨气进主反应釜阀,再搅拌反应约5min,然后将所述主反应釜与所述一级氨尾气吸收釜中未参与反应而剩余的氨气引至新鲜丙酸溶液中进行下批生产;7)在冷却的条件下,主反应釜继续搅拌一段时间,取出产物,冷却保存、检测、包装,即得丙酸铵晶体;然后将所述一级氨尾气吸收釜中的丙酸液置换于所述主反应釜,将二级氨尾气吸收釜中的丙酸液储存用于制备下批丙酸铵。实施例3本实施例用于说明本发明提供的丙酸铵晶体的制备方法。步骤如下:1)将700kg丙酸加入容积为1m3的第一反应器、800kg丙酸加入容积为 1m3的第二反应器、800kg丙酸加入容积为1m3的第三反应器中,所述第一反应器为主反应装置,所述第二反应器为一级氨尾气吸收装置,所述第三反应器为二级氨吸收装置,所述主反应装置、一级氨尾气吸收装置和二级氨尾气吸收装置顺次连接;并对主反应装置和一级氨尾气吸收装置中的丙酸进行搅拌;本实施例中,所述主反应装置为主反应釜,一级氨尾气吸收装置为一级氨尾气吸收釜、二级氨尾气吸收装置为二级氨尾气吸收釜;启动主反应釜和一级氨尾气吸收釜内搅拌电机电源,再分别通过变频器调节两釜内搅拌轴速度,使搅拌桨(轴流、分散搅拌桨)的速度在50r/min;2)将液氨罐里液氨经氨蒸发分离器在1.2MPa下分离出其中微量夹带的油类和机械杂质,再进入氨预热器预热至60℃,所述氨预热器的热源温度为85℃,其由自来水和参与冷却的高温水混合得到,并由调节阀调节氨气压力稳定在1.2MPa;3)经预分离、预热和稳压的氨气,再缓慢开通进入主反应釜的气阀,使氨气匀速进入主反应釜,并将氨气分散于丙酸液体相里,在50r/min的速度下搅拌发生气液放热反应;4)同时开启主反应釜和一级氨尾气吸收釜配套的冷却塔,经冷却循环水冷却降温,使反应温度最终控制在130℃,再次调解氨气压力至1.2MPa;5)稳压后,持续反应1h以上,主反应釜中氨气溢出至一级氨尾气吸收釜,与一级氨尾气吸收釜内的丙酸反应使一级氨尾气吸收釜内温度升高,开启二级氨尾气吸收釜,并进行冷却降温;6)继续反应2h后一级氨尾气吸收釜内氨气溢出至二级氨尾气吸收釜,与二级氨尾气吸收釜内的丙酸反应使二级氨尾气吸收釜内温度升高,采用快速滴定法测定二级氨尾气吸收釜内丙酸液中丙酸铵的含量,本实施例中丙酸铵的含量占所述丙酸液与丙酸铵混合物质量的15%;停止通入氨气,防止氨气进主反应釜阀,再搅拌反应约5min,然后将所述主反应釜与所述一级氨尾气吸收釜中未参与反应而剩余的氨气引至新鲜丙酸溶液中进行下批生产;7)在冷却的条件下,主反应釜继续搅拌一段时间,取出产物,冷却保存、检测、包装,即得丙酸铵晶体;然后将所述一级氨尾气吸收釜中的丙酸液置换于所述主反应釜,将二级氨尾气吸收釜中的丙酸液储存用于制备下批丙酸铵。实施例4本实施例用于说明本发明提供的丙酸铵粉剂制备方法。步骤如下:将实施例1-3任一所制得的丙酸铵晶体60重量份与40重量份活化二氧化硅载体搅拌混合均匀、干燥后再粉碎至80目,过筛、检验、包装,即得直接可加入饲料的丙酸铵粉剂。实施例5将实施例1-3任一所制得的丙酸铵晶体70重量份与30重量份活化二氧化硅载体搅拌混合均匀、干燥后再粉碎至90目,过筛、检验、包装,即得直接可加入饲料的丙酸铵粉剂。实施例6将实施例1-3任一所制得的丙酸铵晶体75重量份与25重量份活化二氧化硅载体搅拌混合均匀、干燥后再粉碎至100目,过筛、检验、包装,即得直接可加入饲料的丙酸铵粉剂。实验例实验例1本实验例用于说明本发明提供的丙酸铵粉剂(由实施例4所述方法制得)与其他类型防霉剂效果对比试验。1.试验材料与方法1.1试验材料1.1.1试验用饲料:仔猪料水分含量13.25%(东莞康达尔饲料有限公司提供)表1试验用饲料成分玉米大麦麦夫皮菜籽饼绵籽饼贝壳粉盐合计37.034.59.08.010.01.00.51001.1.2试验用防霉剂表2试验用防霉剂配方组分及重量百分比1.2.1试验方法1.2.1试验分组与设计本实验分六组和一个对照组共七个组,每组用上述仔猪饲料10千克,添加防霉剂剂量为饲料重量的0.12%(即每组添加上述试验防霉剂1.2克),充分混合均匀,防止不同试验组的交叉感染,以不添加防霉剂为对照组,分别装入透明PE袋中密封,置于30-35℃,相对湿度75%左右的环境条件下静置(以防搬动翻腾饲料流动而不易观察或观察结果有误差),每周观察一次,记录结块及霉变时间。2.1试验结果记录见表3表3试验结果记录表天数714212835424956637077849198防霉剂A组-----------●+++++B组--------●±+++++☉C组----+●+++++☉D组---+●+++++☉E组--+●+++++☉F组---±●+++++☉G组-+●+++++☉2.2霉变程度测定依据见表4表4霉变程度测定结果和依据说明3.结果与讨论由上述试验结果得出,所试仔猪饲料在水分含量13.25%,温度为30-35℃,相对湿度为75%左右环境条件下贮存,加有0.12%剂量的防霉剂组明显比对照组延迟发霉,在上述试验的几类防霉剂中,从防霉效果、霉变速度和霉变程度看:最差的是E组天然植物类,21天开始发霉并结块,霉变速度快;其次D组有机酸盐类,28天开始结块并发霉,霉变速度快;再次是F组配有有机酸、有机酸盐及其他组分,28天后开始发霉结块;单一有丙酸的B组和丙酸与冰醋酸复配C组相对比,防霉时间略长,说明丙酸要好于冰醋酸,一旦饲料霉变,速度也加快。本发明A组实施例4试验结果与其他组对比,在防霉时间和霉变速度上, 都优于其他五组,在同等条件下饲料贮存A组实施例4保鲜77天开始结块,84天开始发霉,且霉变速度较慢。实验例2本发明实施例4-6所合成的丙酸铵粉剂,其卫生指标要求:总砷(As)≤0.0008%、重金属(以Pb计)≤0.0011%和氟(F)≤0.06%。对本发明所涉及制备物卫生指标检测结果如下:(1)总砷(As)的测定:(测定方法:GB/T13079-2006氢化物原子荧光光度法)仪器:原子荧光光谱仪(型号:AF-640A):北京北分瑞利分析仪器公司;未检出(<0.0001)。(2)重金属(以Pb计)的测定:(测定方法:GB/T13080-2004试样溶解—干灰化法)仪器:原子吸收分光光度计(型号:WFX-310):北京北分瑞利分析仪器公司;未检出(<0.0001)。(3)氟(F)的测定:(测定方法:GB/T13083-2002离子选择性电极法)仪器:酸度计(型号PHSJ-3F):上海仪电科学仪器公司;电极:232型甘汞电极、PF-1型氟离子选择电极(测量范围10-1mol/L~5×10-7mol/L)。测定结果0.00014%。经检测,结果符合国家卫生指标。实验例3本发明实施例2所合成的丙酸铵晶体,使用红外(IR)、核磁共振(NMR)和质谱(ESI)技术对其进行测试和表征,结果如下:(1)红外(IR)光谱测试(检测依据为:JY/T001-1996)仪器:傅立叶变换红外光谱仪(型号:WQF-520A)北京瑞利光谱仪器公司测试条件:KBr压片法测试结果如下:1、该样品的红外光谱图见附图1。2、谱图表征如下:a、1403cm-1,为NH4+弯曲振动吸收峰;b、1556cm-1,为羧酸根阴离子伸缩振动吸收峰;c、2983cm-1,为甲基亚甲基伸缩振动吸收峰;d、3149cm-1,为N-H伸缩振动吸收峰。(2)核磁共振(NMR)测试(检测依据为:JY/T007-1996)仪器:超导脉冲付里叶变换核磁共振谱仪,(型号:BrukerDRX-400,400MHz)德国。测试条件:溶剂:DMSO-d6;参考物:TMS;温度:20℃;频率:1H谱:400.130MHz;13C谱:100.613MHz。测试结果:1、该样品测试了核磁共振氢(1H)谱、碳(13C)谱、DEPT135和DEPT90谱,分别见附图2、附图3、附图4和附图5。2、谱图表征如下:a、氢(1H)谱;谱中δ0.934(3H,t,7.6)为3位的CH3质子峰,δ2.017(2H,q,7.6)为2位CH2质子峰,δ6.215(4H,s,9.2,)为活泼氢的质子峰(参见下面结构式)。b、碳(13C)谱、DEPT谱碳谱显示有3个碳峰,DEPT135谱出现了1个正峰和1个负峰,DEPT90谱无出峰,3个谱结合起来,可确定碳谱中的δ10.6为3位CH3 碳峰,δ30.0为2位CH2碳峰,δ177.7为1位羰碳峰(参见上面结构式)。(3)质谱(ESI)测试(检测依据为:JY/T003-1996)仪器:三重四极杆液相色谱-质谱联用仪(型号:TSQQuantumUltra)测试条件:喷雾电压:-3.0kV;源加热温度:200℃;毛细管温度:300℃;鞘气流速:30arb;辅助气流速:5arb。测试结果:1、该样品的ESI质谱图见附图6。2、谱图表征如下:a、M/Z91为M+·;b、M/Z73为CH3CH2COO-。显然,上述实施例仅仅是为清楚地说明所作的举例,而并非对实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。而由此所引伸出的显而易见的变化或变动仍处于本发明创造的保护范围之中。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1