阿魏酸转化为香草醛的方法与流程
发明领域
本发明属于生物技术领域并且涉及一种通过遗传工程改造的微生物将阿魏酸转化为香草醛的改善的发酵方法。
现有技术
香草醛以多于16000吨的全球年需求量是最重要的香料化合物之一。其被广泛用于各种应用,如生产香料、药物、食品和饮料。大量(99%)工业使用的香草醛是由木脂素和原油衍生物化合物合成的,因为由扁叶香果兰(vanillaplanifolia)的荚提取香草醛非常昂贵并且是劳动密集的。尽管化学合成的香草醛可以以非常诱人的价格生产,人们对基于生物技术方法的天然香草醛的生产非常感兴趣。由于消费者日益关注“天然”、“健康”和“有机”产品,与化学产生的香料相比天然香料具备更高的经济价值。这导致广泛研究基于可再生物质以环境友好的方法生产有价值的化合物。
香草醛和其他结构相关的酚类化合物除了用作香料制剂,表现出抗微生物和抗氧化性能,并因此也可用于各种制药应用和用作防腐剂。特别地该生物活性功能取决于正确的立体化学。生物催化是高度立体、区域和化学特异性的,而合成生产会导致外消旋混合物和非预期的副产物。此外,化学方法通常面对环境问题的反应条件以及产生有害废物的问题。
放线菌拟无枝酸菌属(amycolatopsissp.)atcc39116在发酵过程中对于香草醛具备优异的忍受性,因此可工业应用于由阿魏酸生产天然香草醛,以及其他结构相关的化合物。阿魏酸是植物世界中最丰富的羟基肉桂酸,因为它是高等植物细胞壁的主要成分。在这种情况下香草醛可以被标记为“天然香料”,因为它是由植物源提取或“通过适当的物理、酶或微生物方法由植物、动物或微生物源材料得到”。
不幸的是,由于有机体代谢作用中的解毒作用或分解,微生物生产的香草醛通常会快速的转化以提供碳源和能量源。因此,为了在不能降解香草醛的模型有机体如大肠杆菌(e.coli)或酿酒酵母(s.cerevisiae)中创建香草醛生产途径,进行了大量研究。然而,这些方法并不能工业应用,因为细菌对产品没有足够的忍受性。
为了使微生物生产具备经济吸引力,首先努力调节拟无枝酸菌属atcc39116的代谢实现香草醛的高效积累[fleigeetal.“investigationoftheamyolatopsissp.strainatcc39116vanillindehydrogenaseanditsimpactonthebiotechnicalproductionofvanillin”,appl.environ.microbiol.79,p81‐90(2013)]。
阿魏酸在拟无枝酸菌属atcc39116以及许多生产香草醛的菌株中的分解代谢,通过与两步酶法得到的香草醛一致的依赖辅酶a的非β‐氧化途径进行。这两种反应分别通过阿魏酰辅酶a合成酶(fcs)和烯酰辅酶a水合酶/醛缩酶(ech)催化(2、6、7、14‐17)。香草醛随后通过香草醛脱氢酶(vdh)进一步转化为香草酸。拟无枝酸菌属atcc39116缺失编码基因(vdh)导致显著提高的香草醛积累,以及生物转化实验中减少多于90%的分解代谢。
参考appl.environment.microbiol.79(1),p81‐90(2012),其公开了拟无枝酸菌属atcc39116的vdh缺失突变体用于将阿魏酸转化为香草醛。然而该突变体不表现出任何香草醛脱氢酶活性,但表现出对卡那霉素的抗性。
为了得到拟无枝酸菌属atcc39116的高效基因调控,研发了新的分子工具[fleigeetal.“constructionofexpressionvectorsformetabolicengineeringofthevanillin‐producingactinomyceteamycolatopsissp.strainhr167genes…”,appl.microbiol.biotechnol.10.1007/s00253‐014‐5724‐5(2014)]。使用自杀质粒和表达载体实现了这种有前途的生产菌株的基因组的遗传修饰。
因此,本发明解决的问题是研发一种将阿魏酸转化为香草醛的改善的发酵方法,其具备更高的产率。更特别地本发明涉及拟无枝酸菌属微生物新陈代谢的改进,从而设计一种用于天然香草醛的有效生产菌株。
技术实现要素:
本发明的第一个目标是将阿魏酸转化为香草醛的拟无枝酸菌属atcc39116菌株–突变体f33或突变体f84。该培养物根据以下方法得到,其包括以下步骤:
(a1)删除拟无枝酸菌复制起点的主要部分并得到含有用于在大肠杆菌中复制的oriv、安普霉素抗性基因和一些用于克隆的独特限制位点的片段(3575bp);
(a2)重新接合(religating)片段并用得到的质粒p6sui转化大肠杆菌;
(a3)由psetgus分离葡萄糖醛酸苷酶基因作为spei/ecorv片段并克隆至nhei/drai‐线性化自杀质粒p6sui,得到p6suigusa;
(a4)通过同源重组删除vdh基因,其中使用以下低聚核苷酸扩增拟无枝酸菌属atcc39116基因组中vdh的上游和下游侧翼区:
上游区域:
vdhlf_for3(5’‐tcgtacttcgcggtgatctcgtgg‐3’)
和
vdhlf_rev_rf(5’‐gcgcatcaggagtgacacgcgcggcgggggcg‐3’);
下游区域:
vdhrf_for_lf(5’‐cgcccccgccgcgcgtgtcactcctgatgcgc‐3’)和
vdhrf_rev3(5’‐ttgcagtcctttgtagagcgacacg‐3’);
(a5)纯化得到的扩增物并在随后的使用引物vdhlf_for3和vdhrf_rev3的融合pcr中结合;
(a6)将得到的包括vdh上游和下游区域的片段δvdh克隆至自杀质粒p6suigusa的ecorv‐位点;
(a7)由大肠杆菌移除得到质粒p6suigusa::δvdh并将载体转运至拟无枝酸菌属atcc39116;以及
(a8)根据正确基因型选择拟无枝酸菌属atcc39116的转化体并培养得到第二重组子。或:
(b1)用naei/sacii消化大肠杆菌‐拟无枝酸菌穿梭载体p6apra以消除拟无枝酸菌复制起点的主要部分,得到含有用于在大肠杆菌中复制的oriv、安普霉素抗性基因和一些用于克隆的独特限制位点的片段(3575bp);
(b2)重新接合片段并用得到的质粒p6sui转化大肠杆菌;
(b3)由psetgus分离葡萄糖醛酸苷酶基因gusa作为spei/ecorv片段并克隆至nhei/drai‐线性化自杀质粒p6sui,得到p6suigusa;
(b4)使用低聚核苷酸由拟无枝酸菌属atcc39116基因组dna一起扩增ech和fcs基因
ech_for_saci(5’‐aaaagagctctaaggaggtgacaactgctggccgcgctcg‐3’)
和
fcs_rev_xbai(5’‐aaaatctagaccacagagtagccgcagcggg‐3’);
(b5)用saci和xbai消化pcr产物并随后将其克隆至saci/xbai‐线性化pbluescriptsk‐,
(b6)将得到的质粒psk::echfcs转运至大肠杆菌mach‐1t;
(b7)用ecoicri和hindiii消化psk::echfcs,并用与perme*共线的fspai/hindiii‐线性化表达载体p6perme连接得到的含有两个基因的片段;
(b8)使用低聚核苷酸由得到的质粒p6perme::echfcs扩增含有启动子perme*以及ech和fcs基因的片段
p6apramcs_for(5’‐gcgagcaccggaggcagg‐3’)和
p6apramcs_rev(5’‐cattcatccggggtcagcac‐3’)
(b9)将平端pcr产物克隆至hincii‐线性化自杀质粒p6suigusa::δvdh,实现在δvdh‐位点的整合;
(b10)将得到的质粒p6suiδvdh::perme*::echfcs转运至拟无枝酸菌属atcc39116δvdh;
(b11)根据正确基因型选择拟无枝酸菌属atcc39116的转化体并培养得到第二重组子。
本发明的另一个目的涉及制备香草醛的方法,包括将阿魏酸供给至拟无枝酸菌属atcc39116‐突变体f33或突变体f84或其将阿魏酸转化为香草醛的分离酶,持续充足时间使所述阿魏酸转化为香草醛,并回收形成的香草醛。
优选所述的阿魏酸是天然阿魏酸。
同样优选的是一种方法,其中所述微生物用于将阿魏酸转化为香草醛,并且该微生物位于含有碳源的培养基中。特别优选的是选自组包括糖、糖醇、有机酸和复杂混合物的碳源。使用进一步含有氮源的培养基同样有益。
本发明的另一个目的涉及一种包括使用香草醛作为香料的方法,其中改进包括使用由上述方法制备的香草醛作为所述香草醛,优选包括由天然阿魏酸制得的香草醛。
根据现有技术已知的使用常规拟无枝酸菌属atcc39116菌株的方法,可以得到香草醛的最终浓度为13.9g/l,摩尔产率为75%。令人惊异的是,已经观察到使用改善的菌株实现了17.5g/l(f33)和19.3g/l(f84)的最终浓度,这意味着改善了最高达72%。
简略讨论使用菌株f33得到的结果
为了设计拟无枝酸菌属atcc39116菌株的新陈代谢以在发酵过程中实现更高产率,生产编码上述香草醛脱氢酶的vdh无标记基因缺失突变体,从而阻断目标产物的分解代谢。为此,建立自杀质粒p6suigusa,其在拟无枝酸菌属atcc39116中不复制,并因此必须整合进入基因组以进一步增殖。由于在放线菌中经常出现的异常重组,仅有1%的分析突变体在基因组的想要位点处表现出正确同源重组。由于缺少gusa基因的基础载体p6sui的整合非常少,推测该基因与拟无枝酸菌属atcc39116基因组发生重组。
尽管如此,发现该系统特别有利于异质体和均质体的表型鉴定,其显著简化了现有技术已知的用于其他放线菌的潜在突变体的筛选。
最后,该系统是一种产生定向基因敲除而不引入异源dna的有力工具。到目前为止这类基因缺失系统仍未知并且可用于向拟无枝酸菌属atcc39116引入敲除。
基因缺失可通过诊断pce以及基因位点测序验证(图1)。基于我们以前的研究,我们检测了与目前仍在工业中应用的野生型相比。新突变体菌株拟无枝酸菌属atcc39116δvdh(菌株f33)的香草醛的积累和随后分解代谢的性能。
发酵实验确定了我们在之前的生物转化实验中所观察到的。香草醛在突变体f33中的分解代谢减少了将近90%。拟无枝酸菌属atcc39116,由于香草醛的连续降解,积累香草酸最高达浓度3.2g/l,而vdh‐缺失突变体仅表现出残余的分解代谢,导致0.34g/l的香草酸浓度(图2c)。进一步的代谢调节甚至可以改善这一效果,因为醛脱氢酶或氧化酶活性甚至存在于拟无枝酸菌属atcc39116。事实上,敲除vdh被认为是一致的代谢调节以停止或至少减少所需产物降解的关键步骤。
根据这些观察f33以提高的速率直接积累香草醛(图2b)。香草酸是拟无枝酸菌属atcc39116中阿魏酸分解代谢的第一种溢出产物,而vdh缺失突变体f33直接积累香草醛而不会初始合成香草酸。野生型中初始香草酸合成和随后香草醛积累之间的关系已经由其他研究已知,其中在发酵实验中使用菌株或其中香草酸在瓶装实验中表现出提高香草醛的合成。
观察到减少的分解代谢导致香草醛显著更高的最终浓度以及更高的摩尔产率。f33以1.4g/l/h的最大速率积累香草醛(野生型:最大1.15g/l/h)。强化的合成导致最终浓度为17.5g/l(表2),其与野生型细胞相比提高了多于3g/l(表2:14.1g/l)。此外,菌株f33将阿魏酸以91.0%的摩尔产率代谢为香草醛,其相对于现有技术表现出多于16个百分比的增强。
简略讨论使用菌株f84得到的结果
为了避免底物的分解代谢并且优化拟无枝酸菌属atcc39116δvdh的代谢网络,在组成型启动子perme*的控制下,将基因ech和fcs的额外副本整合至基因组进一步改善新菌株f33。基因编码香草醛合成代谢相关的阿魏酰辅酶a合成酶和烯酰辅酶a水合酶/醛缩酶。
采用上述使用p6suigusa的相同策略将过表达载体整合在前vdh位点。尽管会发生自杀质粒的一些异常整合,最后产生基因组中整合perme*::ech::fcs片段的突变体。再次使用诊断pcr(图1)和产生的扩增物的测序验证基因组织。
随后在与f33和野生型相同的发酵条件下测试新突变体拟无枝酸菌属atcc39116δvdh::perme*::echfcs(菌株f84)。与另两种菌株不同,f84的阿魏酸分解代谢被显著强化,这显然是由于培养基中显著更低的底物浓度,尽管在整个发酵过程中以更高的速率供给阿魏酸(图2a)。因此在发酵最后阿魏酸分解代谢停止之前,可以在更短时间内加入更大量的底物。
此外,ech和fcs的组成型表达导致显著强化的香草醛合成,尤其在生物转化阶段的最初8小时。先前观察到的野生型的适应阶段被完全克服,因为在加入阿魏酸后立即以1.6g/l/h合成香草醛(图2b)。
由此所需产物的总产率也可以提升至19.3g/l,相对于加入底物的总摩尔香草醛产率为92.0%。如果在发酵最后培养基中仍有0.91g/l的阿魏酸,菌株以96.2%的摩尔产率代谢使用的底物,该摩尔产率基本接近理论可能值。
事实上,与f33相比f84表现出甚至略有减少的香草酸积累,尽管两种菌株由于相应基因的缺失都不表达vdhatcc39116(图2c)。由于缺失突变体中香草酸的形成似乎与香草醛的量不相关,但是却以某种方式与阿魏酸分解代谢相关,可以认为香草酸通过不依赖香草醛的路径合成。在依赖辅酶a的β‐氧化脱乙酰途径中,首先通过fcs活化阿魏酸,随后进一步通过ech副本处理。与在拟无枝酸菌属atcc39116中产生香草醛的非β‐氧化途径相反,常见中间体4‐羟基‐3‐甲氧基苯基‐β‐羟基丙酰基‐辅酶a进一步通过香草基‐辅酶a转化为香草酸,而不形成香草醛。
在f84中强化表达ech被认为导致直接形成香草醛,而较少的4‐羟基‐3‐甲氧基苯基‐β‐羟基丙酰基‐辅酶a可以转化为香草酸。这一推断也通过以下观察结果支持:当阿魏酸的分解代谢在发酵最后减少时,香草酸的形成受阻。另一方面在野生型中,由于香草醛脱氢酶的固有活性由香草醛持续形成香草酸。
给出的发酵数据构成了所有示出菌株相对于最终香草醛浓度、摩尔产物产率以及总发酵的运行时间的最佳总体生产力。通过提高进料速率,可以实现较短的运行时间,得到可比的最终浓度,但是更低的摩尔产物产率,基于加入的阿魏酸的量。另一方面使用菌株f84可以实现高达96.4%的更高摩尔产率,如果想要的最终香草醛浓度以及供给的阿魏酸量更低。此外,如果以大于5.5g/h的速率供给阿魏酸,可以以22.3g/l生产香草醛(图5)。
总之,通过根据本发明的发酵方法合成香草醛,尽管通过改变一些过程参数,以更低的总产率为代价可以实现更高的最终浓度,却似乎受限于产物的抗微生物性能。香草醛毒性实验清楚示出香草醛对细胞的抗菌作用(图4)。尽管细胞在发酵过程中似乎能耐受高于10g/l的香草醛浓度,阿魏酸的分解代谢在培养最后停止并且发酵参数清楚说明细胞代谢崩溃。由于阿魏酸的活化依赖于atp和辅酶a,进一步的降解可能被限于辅助因子的可用性。这也通过以下事实支持:为了提供额外的碳源和能量源,添加含葡萄糖进料溶液导致更高的最终浓度以及分解代谢提高的速率。
然而,酚类化合物,如香草醛,已经表现出影响或甚至抑制dna、rna和蛋白质合成、吸收葡萄糖、ph动态平衡、以及离子梯度,因为它们由于其疏水性质最可能靶向细胞质膜。此外,已经示出醛基甚至提高了这种毒性作用,由于其表现出高反应性并且可以与dna和蛋白质形成共价键。因此进一步提高的香草醛形成被认为受限于细胞生存力。尽管酶在发酵最后可能仍然有活性,由于辅助因子不可用,进一步分解代谢会被阻碍。为了保持非毒性浓度而提取香草醛表现出提高了制备方法的总产率并且可以被用于克服本问题。
得到突变体f33和f84的方法
本发明的另外两个实施方案涉及用于得到拟无枝酸菌突变体f33和f84的方法。
步骤1
根据本发明的两种方法的相同第一步涉及生产自杀质粒用于产生拟无枝酸菌
属atcc39116的无标记缺失突变体,包括以下步骤:
●用naei/sacii消化大肠杆菌‐拟无枝酸菌穿梭载体p6apra以删除拟无枝酸菌复制起点的主要部分。
●得到含有用于在大肠杆菌中复制的oriv、安普霉素抗性基因和一些用于克隆的独特限制位点的片段(3575bp)。
●重新接合片段,并用得到的质粒p6sui转化大肠杆菌。
●随后,由psetgus分离葡萄糖醛酸苷酶基因gusa作为spei/ecorv片段并克隆至nhei/drai‐线性化自杀质粒p6sui,得到p6suigusa。
步骤2
第二步涉及突变体结构f33(包括删除香草醛脱氢酶基因vdh)包括以下步骤:
●通过同源重组实现vdh基因的精确删除。使用以下核苷酸扩增拟无枝酸菌属atcc39116基因组中vdh的上游和下游侧翼区:
上游区域:
vdhlf_for3(5’‐tcgtacttcgcggtgatctcgtgg‐3’)
和
vdhlf_rev_rf(5’‐gcgcatcaggagtgacacgcgcggcgggggcg‐3’);
下游区域:
vdhrf_for_lf(5’‐cgcccccgccgcgcgtgtcactcctgatgcgc‐3’)和
vdhrf_rev3(5’‐ttgcagtcctttgtagagcgacacg‐3’)。
●纯化得到的扩增物并在随后的使用引物vdhlf_for3和vdhrf_rev3的融合pcr中结合;
●将得到的包括vdh上游和下游区域的片段δvdh克隆至自杀质粒p6suigusa的ecorv‐位点;
●由大肠杆菌移除得到的质粒p6suigusa::δvdh后,将载体转运至拟无枝酸菌属atcc39116;
●在含有50μg/ml安普霉素的固态caso培养基上选择拟无枝酸菌属atcc39116的转化细胞。此外,用前述x‐gluc覆盖得到的菌落以检测由自杀质粒编码的β‐葡萄糖醛酸苷酶活性(myronovskyietal.2011)。
●首先筛选表现出所需表型的突变体用于使用引物vdhlf_for3和vdhrf_rev3的菌落‐pcr的自杀质粒p6suigusa::δvdh的正确基因组整合,所述引物分别与重组区域的上游和下游结合。
●随后,在液态caso培养基中培养表现出正确基因类型的突变体从而得到第二重组子。
●为此无抗生素培养细胞至少10代(passages),随后稀释并置于caso琼脂板。将使用x‐gluc覆盖的不表现出葡萄糖醛酸苷酶活性的单菌落置于含有和不含安普霉素的琼脂板。
●通过诊断pcr与表现出正确vdh缺失的不同引物组合分析已经失去安普霉素抗性的突变体。此外,得到的pcr‐片段通过消化和测序分析以检验拟无枝酸菌属atcc39116δvdh的基因型。
因此,得到突变体f33的方法可以总结如下:
(a1)用naei/sacii消化大肠杆菌‐拟无枝酸菌穿梭载体p6apra以删除拟无枝酸菌复制起点的主要部分,得到含有用于在大肠杆菌中复制的oriv、安普霉素抗性基因和一些用于克隆的独特限制位点的片段(3575bp);
(a2)重新接合片段,并用得到的质粒p6sui转化大肠杆菌;
(a3)由psetgus分离葡萄糖醛酸苷酶基因gusa作为spei/ecorv片段并克隆至nhei/drai‐线性化自杀质粒p6sui,得到p6suigusa;
(a4)通过同源重组删除vdh基因,其中拟无枝酸菌属atcc39116基因组中vdh的上游和下游侧翼区使用以下核苷酸扩增:
上游区域:
vdhlf_for3(5’‐tcgtacttcgcggtgatctcgtgg‐3’)
和
vdhlf_rev_rf(5’‐gcgcatcaggagtgacacgcgcggcgggggcg‐3’);
下游区域:
vdhrf_for_lf(5’‐cgcccccgccgcgcgtgtcactcctgatgcgc‐3’)和
vdhrf_rev3(5’‐ttgcagtcctttgtagagcgacacg‐3’);
(a5)纯化得到的扩增物并在随后的使用引物vdhlf_for3和vdhrf_rev3的融合pcr中结合;
(a6)将得到的包括vdh上游和下游区域的δvdh片段克隆至自杀质粒p6suigusa的ecorv‐位点;
(a7)由大肠杆菌移除得到的质粒p6suigusa::δvdh并将载体转运至拟无枝酸菌属atcc39116;
(a8)根据正确基因型选择拟无枝酸菌属atcc39116的转化体并培养得到第二重组子。
步骤c
●步骤c覆盖突变体结构f84(ech和fcs的额外基因副本在前vdh位点上整合)并包括以下步骤:
●基于拟无枝酸菌属atcc39116δvdh构建另一个突变体菌株,其在强组成型启动子perme*控制下带有ech和fcs基因的额外副本;
●使用低聚核苷酸由拟无枝酸菌属atcc39116的基因组dna一起扩增ech和fcs基因
○ech_for_saci(5’‐aaaagagctctaaggaggtgacaactgctggccgcgctcg‐3’)and
○fcs_rev_xbai(5’‐aaaatctagaccacagagtagccgcagcggg‐3’).
●使用saci和xbai消化pcr产物,并随后克隆至saci/xbai‐线性化pbluescriptsk‐。将得到的质粒psk::echfcs转运至大肠杆菌mach‐1t1并在阿魏酸生物转化实验中观察酶活性。
●随后,使用ecoicri和hindiii消化psk::echfcs,用与perme*共线的fspai/hindiii‐线性化表达载体p6perme连接得到的含有两个基因的片段(fleigeandsteinbüchel2014);
●为了整合进入拟无枝酸菌属atcc39116δvdh的基因组,使用低聚核苷酸由得到的质粒p6perme::echfcs扩增含有启动子perme*以及ech和fcs基因的片段
○p6apramcs_for(5’‐gcgagcaccggaggcagg‐3’)和
○p6apramcs_rev(5’‐cattcatccggggtcagcac‐3’).
●将平端pcr产物克隆至hincii‐线性化自杀质粒p6suigusa::δvdh,实现在δvdh‐位点的整合;
●得到的质粒被称为p6suiδvdh::perme*::echfcs并转运至拟无枝酸菌属atcc39116δvdh;
●如上所述筛选转化体,产生第二重组子并验证过表达载体在δvdh‐位点的正确整合。
因此得到突变体f84的方法可以总结如下:
(b1)用naei/sacii消化大肠杆菌‐拟无枝酸菌穿梭载体p6apra以删除拟无枝酸菌复制起点的主要部分,得到含有用于在大肠杆菌中复制的oriv、安普霉素抗性基因和一些用于克隆的独特限制位点的片段(3575bp);
(b2)重新接合片段并用得到的质粒p6sui转化大肠杆菌;
(b3)由psetgus分离葡萄糖醛酸苷酶基因gusa作为spei/ecorv片段并克隆至nhei/drai‐线性化自杀质粒p6sui,得到p6suigusa;
(b4)使用低聚核苷酸由拟无枝酸菌属atcc39116基因组dna一起扩增ech和fcs基因
ech_for_saci(5’‐aaaagagctctaaggaggtgacaactgctggccgcgctcg‐3’)
和
fcs_rev_xbai(5’‐aaaatctagaccacagagtagccgcagcggg‐3’);
(b5)用saci和xbai消化pcr产物并随后将其克隆至saci/xbai‐线性化pbluescriptsk‐,
(b6)将得到的质粒psk::echfcs转运至大肠杆菌mach‐1t;
(b7)用ecoicri和hindiii消化psk::echfcs,并用与perme*共线的fspai/hindiii‐线性化表达载体p6perme连接得到的含有两个基因的片段;
(b8)使用低聚核苷酸由得到的质粒p6perme::echfcs扩增含有启动子perme*以及ech和fcs基因的片段
p6apramcs_for(5’‐gcgagcaccggaggcagg‐3’)和
p6apramcs_rev(5’‐cattcatccggggtcagcac‐3’)
(b9)将平端pcr产物克隆至hincii‐线性化自杀质粒p6suigusa::δvdh,实现在δvdh‐位点的整合;
(b10)将得到的质粒p6suiδvdh::perme*::echfcs转运至拟无枝酸菌属atcc39116δvdh;
(b11)根据正确基因型选择拟无枝酸菌属atcc39116的转化体并培养得到第二重组子。
附图说明
图1
验证拟无枝酸菌属atcc39116δvdh(f33)和拟无枝酸菌属atcc39116δvdh::perme*::echfcs(f84)的基因置换
为了验证vdh正确的基因缺失,以及随后整合perme*::echfcs,使用染色体dna作为模板进行诊断pcr。得到的片段在1%的琼脂糖凝胶中分析。分子量标记物(m):“generulertm1kbdnaladde”(fermentasgmbh,st.leon‐rot,germany)。通道1、2和3代表使用低聚核苷酸vdhlf_for_di和vdhrf_rev2的扩增物,其示出了野生型(1,3.7kb)、突变体f33(2,2.4kb)和突变体f84(3,5.4kb)之间的区别。纯化标记的片段,并通过测序分析扩增子以最后验证正确的同源重组。
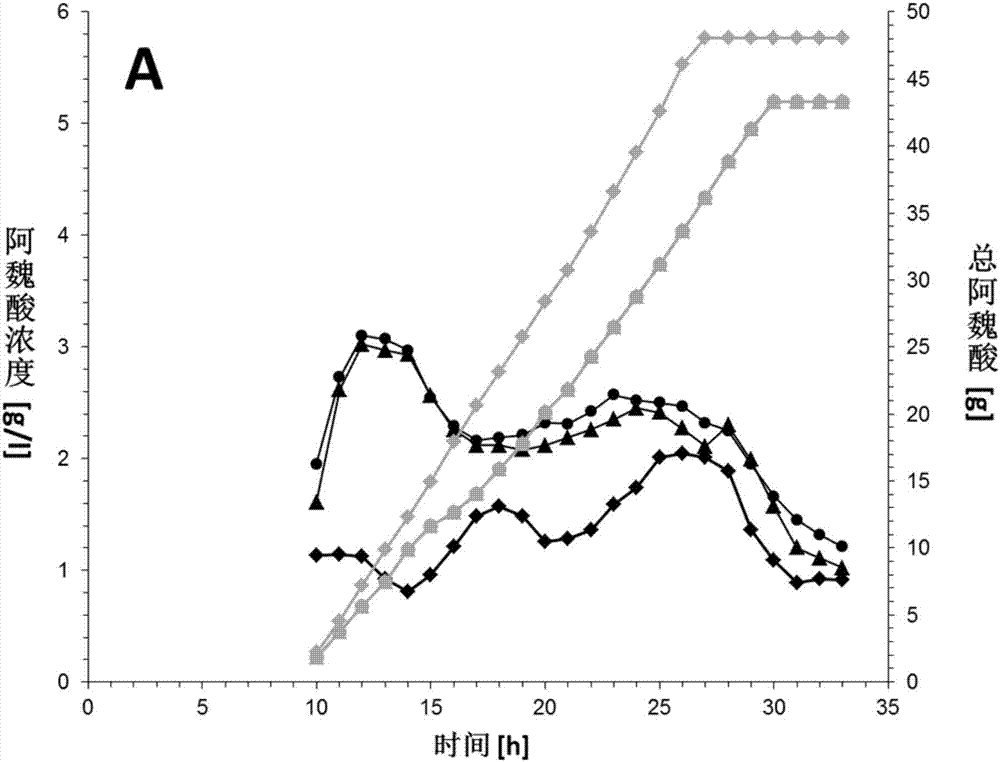
图2a、2b、2c
通过拟无枝酸菌属atcc39116和相关突变体f33和f84的阿魏酸的生物转化
在2l‐生物反应器中细胞在1000ml发酵液中于45℃下生长直至它们达到稳定生长期。随后开始加入阿魏酸溶液。在给定时间使用在材料和方法中所述的hplc分析法观察阿魏酸(a)向香草醛(b)和香草酸(c)的生物转化。在图a中灰线代表加入的阿魏酸的总量。黑线代表发酵液中中间体的浓度。(▲,
图3
ech表达的rt‐pcr分析
通过rt‐pcr分析在发酵不同部分中ech和fcs的表达。在加入阿魏酸之前以及在生物转化后7小时取样。根据材料和方法中记载的进行rna分离,cdna制备和最终pcr。在2%琼脂糖凝胶中分析得到的片段。分子量标记物m1:“generulertm1kbdnaladder”;m2:“generulertmlowrangednaladder”(fermentasgmbh,st.leon‐rot,germany)
通道1‐9代表使用低聚核苷酸ech_rt_for和fcsrt_rev以及不同模板的扩增子:1:cdna,进料前由f33样本制备;2:用于样本1的rna对比样;3:cdna,进料过程中由f33样本制备;4:用于样本3的rna对比样;5:cdna,进料前由(f84)样本制备;6:用于样本5的rna对比样;7:cdna,进料过程中由f84样本制备;8:用于样本7的rna对比样;9:用于pcr的dna‐对比样。箭头指出ech和fcs副本的预期内部624bp‐片段。
图4
香草醛毒性测试
在300mlcaso培养基中于45℃下培养拟无枝酸菌属atcc39116的细胞至培养物达到稳定生长期。提取样本,用无菌盐水(0.9%nacl)稀释并一式三份置于固态caso培养基中以测定初始细胞密度(100%)。制备45ml的六等份。将浓度为2(●)、4(▲)、7(■)和10g/l(◆)香草醛(溶于dmso)加入。作为对比实验,向其中一个培养物加入等量的dmso(+),而其他培养物没有任何添加(x)。将培养物继续在45℃下培育并在1、3和6小时后再次取样、稀释并放置于琼脂板。再次计算菌落形成单位并相对于初始细胞密度计算百分比。
图5
f84中阿魏酸的生物转化
在2l‐生物反应器中细胞在1000ml发酵液中于45℃下生长直至它们达到稳定生长期。随后开始加入阿魏酸溶液(5.5g/l/h阿魏酸)。在给定时间使用在材料和方法中所述的hplc分析法观察阿魏酸(▲)向香草醛(◆)和香草酸(●)的生物转化。曲线代表中间体在发酵液中的浓度。
实施例
材料和方法
细菌菌株、质粒和培养条件
在本研究中使用的所有细菌菌株和质粒列于下表1。
表1
本研究中使用的细菌菌株和质粒
(1)appl.environ.microbiol77:5370‐5283(2014)(2)ibid.77:5370‐5283(2011)
大肠杆菌细胞在ln(lysogenybro)培养基中于37℃下生长。拟无枝酸菌属atcc39116细胞在caso培养基中于45℃下生长(merck,darmstadt,德国)。为了选择含质粒的菌株(plasmidharboringstrains),将抗生素加入以下培养基:氨苄青霉素:100μg/ml用于大肠杆菌;安普霉素:50μg/ml用于大肠杆菌和拟无枝酸菌。通过测定400nm(拟无枝酸菌属atcc39116)或600nm(大肠杆菌)处光密度测量细胞生长。
dna分离和修饰
使用“peqgoldplasmidminiprepkiti”(peqlabgmbh,erlangen,德国)由大肠杆菌分离质粒dna。在生产商给出的条件下通过限制性内切酶(thermofisherscientificgmbh,schwerte,德国)消化dna。根据生产商说明使用phusionhot‐startiihigh‐fidelitydna聚合酶、t4dna连接酶、t4dna聚合酶和revertaid逆转录酶(thermofisherscientificgmbh,schwerte,德国)。由eurofinsmwgsynthesisgmbh(ebersberg,德国)购得低聚核苷酸。使用“peqgoldgelextractionkit”(peqlabgmbh,erlangen,德国)由琼脂糖凝胶或反应混合物移除dna片段。
dna转运
通过使用cacl2‐方法将质粒转运至大肠杆菌,而通过前述直接菌丝体转型将质粒转运至拟无枝酸菌属atcc39116。
质粒和突变体构建
为了产生拟无枝酸菌属atcc39116的无标记基因缺失突变体,构建自杀质粒。使用naei/sacii消化前述大肠杆菌‐拟无枝酸菌穿梭载体p6apra,以删除拟无枝酸菌复制起点的主要部分。得到的片段(3575bp)仍然含有用于在大肠杆菌中复制的oriv,安普霉素抗性基因和一些用于克隆的独特限制位点。重新接合片段并用得到的质粒p6sui转化大肠杆。随后由psetgus(21)分离葡萄糖醛酸苷酶基因gusa作为spei/ecorv片段并克隆至nhei/drai‐线性化自杀质粒p6sui,得到p6suigusa。
通过同源重组进行vdh基因的精确删除。使用以下低聚核苷酸扩增基因组中vdh的上游和下游侧翼区:
上游区域:
vdhlf_for3(5’‐tcgtacttcgcggtgatctcgtgg‐3’)和
vdhlf_rev_rf(5’‐gcgcatcaggagtgacacgcgcggcgggggcg‐3’);
下游区域:
vdhrf_for_lf(5’‐cgcccccgccgcgcgtgtcactcctgatgcgc‐3’)和
vdhrf_rev3(5’‐ttgcagtcctttgtagagcgacacg‐3’).
纯化得到的扩增物并在随后的使用引物vdhlf_for3和vdhrf_rev3的融合pcr中结合。得到的δvdh片段结合vdh上游和下游区域并克隆至自杀质粒p6suigusa的ecorv‐位点。由大肠杆菌移除得到的质粒p6suigusa::δvdh并将载体转运至拟无枝酸菌属atcc39116。在含有50μg/ml安普霉素的固态caso培养基上选择拟无枝酸菌属atcc39116的转化体。此外,如前所述使用x‐glu覆盖得到的菌落,以检测由自杀质粒编码的β‐葡萄糖醛酸苷酶活性(参见:appl.environ.microbiol.77:5370‐5283(2011))。
首先筛选表现出所需表型的突变体用于通过使用引物vdhlf_for3和vdhrf_rev3的菌落‐pcr的自杀质粒p6suigusa::δvdh的正确基因组整合,所述引物分别与重组区域的上游和下游结合。随后,在液态caso培养基中培养表现出正确基因型的突变体从而得到第二重组子。为此无抗生素培养细胞至少10代,随后稀释并置于caso琼脂板。将使用x‐gluc覆盖后不表现出葡萄糖醛酸苷酶活性的单菌落置于含有和不含安普霉素的琼脂板。最后通过表现出正确vdh缺失的不同引物组合的诊断pcr分析已经失去安普霉素抗性的突变体。此外,得到的pcr‐片段通过消化和测序分析以检验拟无枝酸菌属atcc39116δvdh的基因型。
基于拟无枝酸菌属atcc39116δvdh构建另一个突变体菌株,其带有在强组成型启动子perme*(18)控制下的ech和fcs基因的额外副本。使用低聚核苷酸由拟无枝酸菌属atcc39116的基因组dna一起扩增ech和fcs基因:
(i)ech_for_saci(5’‐aaaagagctctaaggaggtgacaactgctggccgcgctcg‐3’)和
(ii)fcs_rev_xbai(5’‐aaaatctagaccacagagtagccgcagcggg‐3’).
使用saci和xbai消化pcr产物,并随后克隆至saci/xbai‐线性化pbluescriptsk‐。将得到的质粒psk::echfcs转运至大肠杆菌mach‐1t1并在阿魏酸生物转化实验中观察酶活性。随后,使用ecoicri和hindiii消化psk::echfcs,与perme*共线的fspai/hindiii‐线性化表达载体p6perme连接得到的含有两个基因的片段。为了整合进入拟无枝酸菌属atcc39116δvdh的基因组,使用低聚核苷酸p6apramcs_for(5’‐gcgagcaccggaggcagg‐3’)和p6apramcs_rev(5’‐cattcatccggggtcagcac‐3’)由得到的质粒p6perme::echfcs扩增含有启动子perme*以及ech和fcs基因的片段。将平端pcr产物克隆至hincii‐线性化自杀质粒p6suigusa::δvdh以实现在δvdh‐位点的整合。
得到的质粒被称为p6suiδvdh::perme*::echfcs并转运至拟无枝酸菌属atcc39116δvdh。如上所述筛选转化体,产生第二重组子并验证过表达载体在δvdh‐位点的正确整合。
葡萄糖醛酸苷酶测定
为了检测β‐葡萄糖醛酸苷酶在琼脂板上的活性,使用1ml的1mm5‐溴‐4‐氯‐3‐吲哚基‐β‐d‐葡萄苷酸环己氨盐(x‐gluc)溶液覆盖菌落。经过短暂的潜伏期,由于形成5,5'‐二溴‐4,4'‐二氯靛青,含有水解gus活性的菌落变蓝。
2l规模的补料分批发酵
补料分批发酵实验在biostatbplus(sartorius,
测定代谢中间产物
无需之前提取通过使用“ultimate3000hplcsys”(dionex,idstein,德国)的高效液相色谱分析阿魏酸代谢的排出中间体。离心后得到培养物上清液(5分钟,16000xg)并且如果必要使用双蒸水稀释1:10至1:100。通过使用
香草醛毒性测试
在300mlcaso培养基中于45℃下培养拟无枝酸菌属atcc39116的细胞直到培养物达到稳定生长期。提取样本,用无菌盐水(0.9%nacl)稀释并一式三份置于固态caso培养基中以计算菌落形成单位用于确定初始细胞密度(100%)。随后将培养物分为45ml的六等份。加入溶度为2、4、7和10g/l的香草醛(溶解于dmso)。作为对比实验向其中一个培养物加入等量的dmso,而另一培养物无添加。将培养物继续在45℃下培育并在1、3和6小时后再次取样、稀释并放置于琼脂板。再次计算菌落形成单位并相对于初始细胞密度计算百分比。
rna分离和rt‐pcr分析
为了提取总rna提取1ml发酵液样本并通过离心(13000xg、20分钟、4℃)收获野生型拟无枝酸菌属atcc39116或突变体菌株的细胞。将得到的细胞小球再次悬浮于800μlte缓冲液(ph8.0),并如前所述分离rna。通过使用dnasei(roche,mannheim,德国)移除剩余的dna污染,随后如前所述纯化rna。使用revertaid逆转录酶(thermofisherscientificgmbh,schwerte,德国)和用于ech内部片段的低聚核苷酸ech_rt_for(5’‐gaaccccaccctgaacg‐3’)和ech_rt_rev(5’‐gagcttcagcgccacctc‐3’)进行cdna的制备。
为了重叠ech和fcs的片段使用低聚核苷酸echfcs_rt_for(5’‐gtactacatcatgaccggtgagc‐3’)和echfcs_rt_rev(5’‐cagcgcgtggtccagttc‐3’)。使用phusionhot‐startiihigh‐fidelitydna聚合酶进行最终pcr。在对比实验中通过加入rna而不进行在前逆转录步骤将dna污染由分离的rna排除。
结果
拟无枝酸菌属atcc39116的vdh‐缺失突变体的构建和表征
香草醛脱氢酶vdhatcc39116对于香草醛在拟无枝酸菌属atcc39116中的分解代谢起到实质性作用,并因此是一个用于调节菌株代谢以达到高效积累的有前途目标。用卡那霉素抗性盒替代vdh编码基因后,产生无标记缺失突变体作为新的生产菌株。为此,构建可以引入适当缺失结构以及筛选得到的异质体的自杀质粒。穿梭载体p6apra的拟无枝酸菌复制起点通过消化删除,产生非复制质粒p6sui。自杀质粒仍含有大肠杆菌复制起点、安普霉素抗性基因和一些用于克隆缺失结构的独特限制位点。为了得到筛分均质体的表型能力,将psetgus的gusa基因插入p6sui,产生p6suigusa。gusa编码β‐葡萄糖醛酸苷酶,由于使用x‐gluc覆盖后菌落强烈的蓝色,其活性可在琼脂板上简单确定。失去载体骨架的均质体没有颜色。
为了删除vdh扩增拟无枝酸菌属atcc39116基因组中位于上游和下游的侧翼区,并随后在融合pcr中结合。将得到的δvdh片段克隆至p6suigusa,并将自杀质粒p6suigusa::δvdh转运至拟无枝酸菌属atcc39116。转运后,可以观察到安普霉素抗性克隆表现出葡萄糖醛酸苷酶活性。不幸的是,对比实验中在不含缺失结构的p6suigusa转运后我们也得到了转化体。然而,当p6suigusa::δvdh转化观察到显著更多的重组。由于这些异常重组首先通过诊断pcr分析p6suigusa::δvdh在正确基因位点的整合。为此,扩增vdh位点,包括自杀质粒的克隆体通过11.5kb片段识别。随后两个正确的异质体在不加安普霉素和放置于caso琼脂板的至少10代后样本的液态caso培养基中培养。将x‐gluc重叠后未变蓝的克隆体置于含有和不含安普霉素的固态caso培养基。失去安普霉素抗性的克隆体在pcr反应中进一步分析,以确定vdh缺失。为此,使用低聚核苷酸扩增vdh位点的片段,其分别进一步与使用的侧翼区的上游和下游结合。通过2.42‐kb片段扩增确定正确的删除,其由于vdh缺失(1.461bp,图1)与野生型尺寸不同。以外,通过使用内部引物的pcr确定vdh缺失,从而证明基因没有整合在基因组的不同位点。最后通过2.42‐kb片段测序验证缺失突变体拟无枝酸菌属atcc39116δvdh(菌株f33);得到得的序列示出了vdh整个编码序列的正确删除。
在确定vdh缺失对于拟无枝酸菌属atcc39116在生物转化实验中积累香草醛的有利效果后,在发酵过程中分析拟无枝酸菌属atcc39116δvdh的新的无标记突变体。因此,将f33与野生型对比以分析vdhatcc39116对所需产物的分解代谢的影响。在2升生物反应器中进行发酵。发酵剂培养物的初始浓度是1000ml。与之前的研究相反,使用非常简单的由4g/l葡萄糖和4g/l酵母提取物构成的发酵液。由于在之前的实验中没有观察到麦芽膏的显著效果,将其由基础培养基中移除以降低成本并简化过程。分两步进行发酵。首先,细胞生长至它们达到稳定生长期,其由于po2的显著增多而明显。随后通过连续加入底物,阿魏酸开始生物转化为香草醛。根据阿魏酸在培养基中的浓度,其每间隔1h通过hplc分析,调节进料速率。由于酚类化合物的毒性,需要将底物浓度保持在低于5g/l,但不低于1g/l以确保为细胞提供适当的补给。将葡萄糖和酵母提取物加入进料溶液,因为之前的测试发酵示出补给有利地影响生物转化。
阿魏酸的分解代谢以及香草醛的积累分别在野生型和f33初始适应阶段后开始(图2b)。在第一个3小时中阿魏酸浓度由于连续供给而提高。当细胞适应生物转化条件后将两个发酵中的底物浓度减小直到浓度通过调节进料速率被恒定保持在2至3g/l之间。28h后停止进料,野生型和突变体发酵过程中,底物浓度降低至约1g/l。阿魏酸分解代谢不受vdh缺失的影响,因为两个过程以可比较的方式进行(图2a)。
然而,对于所需产物香草醛和意外副产物香草酸的积累观察到显著区别。菌株f33的香草醛合成显著增多并在23h的生物转化后达到17.5g/l的最终浓度。与之相比,野生型拟无枝酸菌属atcc39116在相同时间内仅形成至多14.1g/l的香草醛。在两种过程中都没有观察到香草醛浓度的进一步增大,尽管细胞中仍有阿魏酸(图2a)。因此,33h后结束发酵。
对于香草醛的分解代谢,f33表现出香草酸形成的显著减少(0.34g/l的最终浓度),而野生型持续分解香草醛,使发酵最后的香草酸为3.2g/l。仅检测到痕量其他分解代谢中间体如愈创木酚或原儿茶酸以及其他副产物如香草醇。
向两个发酵中加入总量43.3g的阿魏酸。对于最终香草醛浓度菌株f33以91.0%的总摩尔产率积累所需产物,而野生型仅以74.8%的产率形成香草醛。结果示于表2。
表2
通过代谢工程改善菌株:2升规模的发酵方法的结果
ech表达的逆转录酶(rt‐)‐pcr分析
发酵实验示出,在生物转化阶段的第一小时内细胞适应阿魏酸进料前,拟无枝酸菌属atcc39116和突变体f33低效降解阿魏酸以及积累香草醛(图2)。进行rt‐pcr分析以判定,诱导效应是否发生在基因表达层面。为此,(i)在加入底物前生长阶段的最后以及(ii)在生物转化7小时后,香草醛合成速率已经给达到其最大值,分别由f33和野生型实验提取发酵液样本。根据材料和方法中的记载通过离心收集细胞并分离rna。阿魏酰辅酶a合成酶(fcs)和烯酰辅酶a水合酶/醛缩酶(ech),其负责拟无枝酸菌属atcc39116中阿魏酸分解代谢为香草醛,分别由ech和fcs基因编码。两个基因的开放阅读框相邻地位于拟无枝酸菌属atcc39116的基因组,其中fcs的起始密码子(gtg)与ech的停止密码子(tga)重叠。除了编码基因的功能外,该遗传组织示出两个基因在相同启动子的控制下共转录。为了提供证据证明供给阿魏酸之后引发了基因表达,通过rt‐pcr分析ech的转录。因此,内部529bp片段在制备cdna后通过逆转录扩增。
rt‐pcr分析揭示了供给阿魏酸后ech‐表达的强烈诱导作用。对于加入底物前提取的样品,仅可以检测到模糊条带(图s1,通道1),对于在发酵的生物转化过程中提取的样品观察到强烈的扩增子(图s1,通道3)。在使用rna作为模板而不进行先前逆转录反应的对比pcr实验中可以排除分离rna的dna污染(图s1,通道2和4)。
额外的rt‐pcr分析揭示了ech和fcs作为操纵子在单一mrna中转录,因为可以使用结合ech的正向引物和结合fcs的反向引物扩增两个基因的重叠624bp片段(图3,通道3、5、7)。诱导效果在该实验中同样明显,因为对于在发酵的生物转化过程中提取的样品观察到强烈的扩增子(图3,通道3)。
拟无枝酸菌属atcc39116δvdh过表达突变体的构建和应用
为了进一步改善阿魏酸向香草醛的转化,产生了ech和fcs过表达突变体。为此,在强组成型启动子perme*的控制下将两种基因的额外副本整合至f33的基因组。为了整合,选择前vdh‐位点,因为vdh缺失和用卡那霉素抗性盒替换的基因都不会导致负极性效果。此外,自杀质粒p6suigusa::δvdh可用于通过同源重组进行定向重组。
首先,由拟无枝酸菌属atcc39116基因组一起扩增两个基因并克隆至与perme*共线的拟无枝酸菌表达载体p6perme。在第二步中用启动子一起扩增基因,得到的片段平端克隆至p6suigusa::δvdh的vdh缺失结构。得到的p6suiδvdh::perme*::echfcs质粒用于转化菌株f33。尽管得到了安普霉素抗性克隆,我们同样面对如上所述自杀质粒异常整合的问题。因此,首先通过基因组区域的扩增分析自杀质粒在vdh‐位点的整合。可以确定含有vdh侧翼区和自杀质粒的15.1‐kb片段的正确整合。在验证了想要的同源重组后,进一步培养克隆体以实现如对于vdh缺失所述的第二重组子。在一个突变体里可以实现过表达载体的精确整合并通过诊断pcr以及pcr产物测序确定(图1)。新突变体拟无枝酸菌属atcc39116δvdh::perme*::echfcs(菌株f84)应当结合了vdh敲除与提高的阿魏酸分解代谢速率的有利影响。
为了验证新的想要的表型,在2升规模的发酵实验中培养菌株f84。为了分析ech和fcs过表达对阿魏酸分解代谢和香草醛积累的影响,在如上所述的相同条件下的平行实验中培养野生型拟无枝酸菌属atcc39116和产生的突变体f33和f84。通过加入阿魏酸溶液开始生物转化后ech和fcs增强表达的效果很明显(图2a)。在阿魏酸的分解代谢增多并达到其最大值之前,野生型和菌种f33表现出约3h的延迟期,f84中阿魏酸向香草醛的转化在补充改善性能后立即开始。尽管与野生型和f33(最高至2.6g/h)相比,以更大的速率(最高至3.5g/h)加入阿魏酸,在发酵的整个时间段阿魏酸的浓度不断降低。由于更高的转化率调整进料策略,可以在仅18小时的更短时间阶段内加入总共4.9g的更多底物(图2a)。
对于由f84合成的香草醛积累也表现出增强的效果,其中在第一小时内没有之前观察到的适应阶段(图2b)。这导致在6小时的生物转化阶段后1.6g/l/h的最大产率以及8.1g/l的香草醛浓度(野生型:7.8g/l,10h)。如所希望的,提高的阿魏酸代谢为香草醛导致19.3g/l的更高最终浓度,以及相对于所加底物的92.0%的总摩尔香草醛产率(图2)。对于消耗的阿魏酸,如果最后培养基中仍有0.91g/l底物,当不再合成香草醛时,则摩尔产率甚至更高(96.2%)。
考虑到香草醛的分解代谢,新突变体f84表现出与f33可比的性能。这可能是由于两种菌株中vdh缺失。有意思的是,f84表现出甚至略有降低的香草酸的形成,尽管在培养该突变体时产生更多香草醛(图2c)。
香草醛毒性测试
发酵实验揭示了香草醛产生的未知限制,尽管细胞中仍有阿魏酸(图2c)。已知香草醛和其他酚类化合物表现出抗微生物性能。尽管已知拟无枝酸菌属atcc39116能够比其他测试有机体耐受更高浓度的香草醛,记录的发酵参数清楚示出,在这些细菌中通过提高所需产物的浓度也可以影响细胞代谢。当过程接近最后阶段溶解氧气的消耗以及细胞密度显著降低。
为了说明香草醛对拟无枝酸菌属atcc39116生存力的影响,在摇瓶中培养细胞直到它们达到稳定生长期,也可以在发酵实验中进行。随后由该培养物制备6等份,并加入2g/l至10g/l不同浓度的香草醛。在给定时间点由培养物提取样品,稀释并置于琼脂板以计算菌落形成单位。结果是四次独立重复的均值。
实验清楚示出香草醛的抗微生物效果(图4)。细胞在活细胞浓度初始降低后耐受最高至4g/l的浓度,7g/l在6小时内会导致细胞全部死亡。此外,10g/l的香草醛浓度在1小时内会导致90%细胞死亡;在培育3小时后检测不到活细胞。由于在发酵实验中检测到更高的香草醛浓度,也检测了细胞对产物的适应性是否导致更好的耐受性。
为此,将2g/l香草醛加入培养物,因为在初始实验中示出细胞能够耐受(图4)。随后,在培养1或3小时后,将浓度提高至10g/l。然而,该实验的结果示出细胞的适应性无法更高的耐受香草醛。再次暴露于10g/l香草醛导致随后几小时高达90%的细胞死亡。
- 还没有人留言评论。精彩留言会获得点赞!