阻燃材料及包含其的装置的制作方法
相关申请的交叉引用
本申请要求2014年12月19日提交的美国专利申请案号14/577,706的优先权,美国专利申请案号14/577,706要求2014年8月29日提交的美国临时专利申请序列号62/043,851的权益,其各自对于其所有教导整体以引用的方式并入本文中。
关于联邦资助的研究的声明
本发明是在政府支持下以国家科学基金会(nsfepscorring-trueiii)授予的拨款号0447416;国家科学基金会小型企业创新研究规划授予的拨款号oii-0610753;国家科学基金会小型企业技术转让研究i期规划授予的拨款号iip-0740289;和nasagrc授予的合同号nnx10cd25p进行。该政府在本发明中拥有一定权利。
本公开涉及阻燃材料及包含阻燃材料的装置。在一些情况下,本文提供的阻燃材料可包含聚(吡啶鎓)盐部分和氧化膦部分。
背景技术:
聚合物由于其可调谐的机械性质和加工容易性而常用于各种产品中。阻燃聚合物为对高温降解具有抵抗性的聚合物。在包括构造诸如摩天大楼、轮船和飞机机舱的小封闭空间的许多领域中需要阻燃聚合物。在紧密空间中,如果发生火灾,逃生的能力则被减损,增加了火灾危险。防火安全聚合物还在航空航天材料、电子仪器的绝缘材料及诸如帆布帐篷的军队材料中作为胶粘剂应用。然而,常见的聚合物可燃性高且在燃烧期间可生成毒性气体和烟气。使聚合物对燃烧更具抵抗性的一种常见方法是使滞燃添加剂包含在聚合物中。然而,诸如多氯联苯和溴化滞燃剂的滞燃添加剂可造成某些健康问题。
已经合成、表征并评价了六种新型耐高温的含氧化膦的聚(4,4'-(对亚苯基)-双(2,6-二苯基吡啶鎓))聚合物p-1、p-2、p-3、p-4、p-5和p-6。它们以高产率和高纯度合成。高玻璃化转变温度(tg>240℃)和高残碳率(charyield)(在700℃下,>50%)分别通过差示扫描量热法(dsc)和热解重量分析(tga)测定。这些离子聚合物展示出优良的可加工性、成薄膜性、耐高温性、防火性和阻燃性、涂覆性、粘附性、机械和拉伸强度及n型(电子传输)性质。在聚合物主链中并入氧化膦和双(苯基吡啶鎓)部分产生高玻璃化转变温度和阻燃性质。使用有机平衡离子使得这些离子聚合物可自许多常见的有机溶剂中容易地加工。多种这样的聚合物可通过利用含双吡喃鎓盐、含氧化膦的二胺和以组合式样的平衡离子的结构变体来合成。这些结构使得它们对于许多应用具有很大的吸引力,这些应用包括作为用于汽车、飞行器、动力和推进系统的涂料和结构组件材料;消防员服装;印刷电路板;用于电子和电气组件的箱体和外壳;建筑材料;床垫;地毯;室内装饰品和家具;及用于保护重要书面文件的极薄涂层。
本文提供的阻燃聚合物可包含吡啶鎓盐部分、氧化膦部分及其组合。在一些情况下,本文提供的阻燃聚合物可包含各自包含至少一个吡啶鎓盐和/或至少一个氧化膦部分的重复单元。在一些情况下,本文提供的阻燃聚合物可为吡啶鎓盐部分和氧化膦部分的无规共聚物。在一些情况下,本文提供的阻燃聚合物基本不含卤素。
一个或多个实施方式的详细描述在以下描述中阐述。其他特点和优势将从以下描述、附图和权利要求书中显而易见。
技术实现要素:
本发明涉及包含至少一个吡啶鎓盐部分和至少一个氧化膦部分的阻燃聚合物及由其制造的装置。
通过考虑发明详述和附图将显而易见本发明的其他方面。
附图说明
提供以下附图以便于理解本文提供的描述和实施例。
图1描绘本文提供的阻燃聚合物的各种例示性实施方式的化学结构;
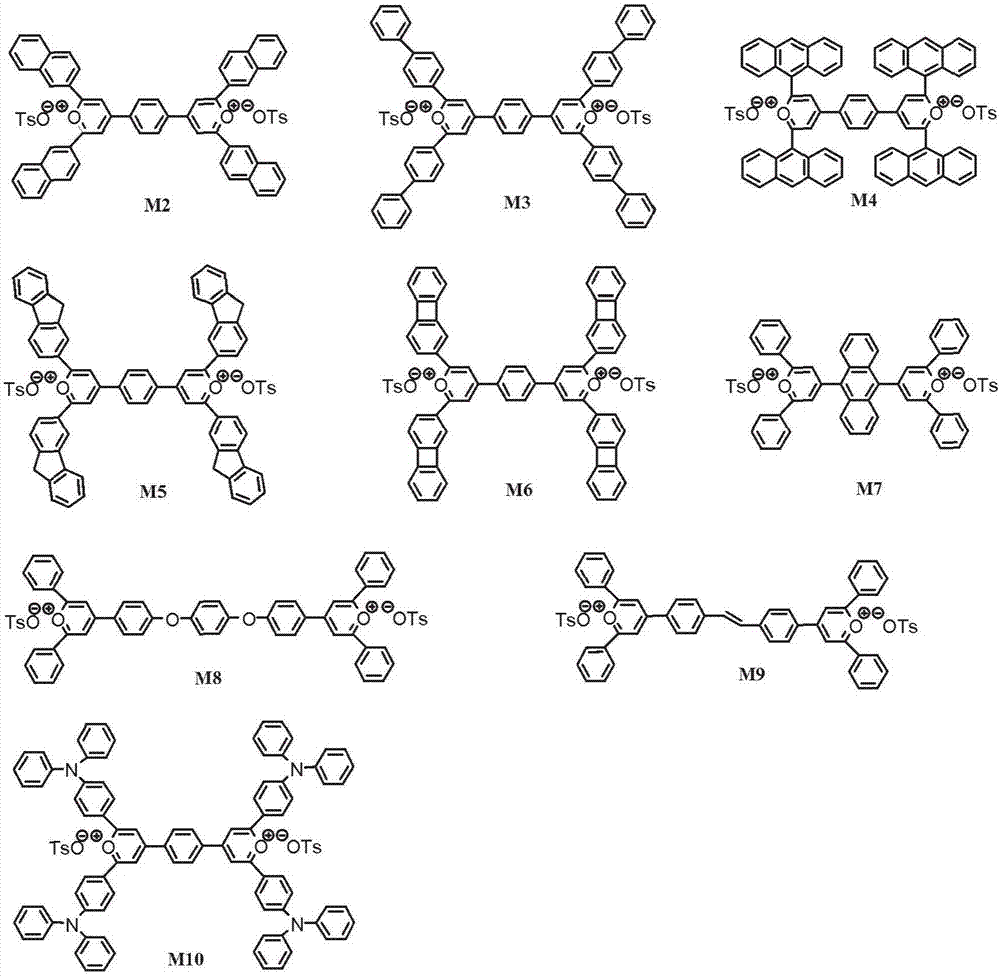
图2描绘可包含在本文提供的阻燃聚合物中的单体组分m2~m10的例示性化学结构变体;
图3描绘可包含在本文提供的阻燃聚合物中的平衡离子组分y1~y15的例示性化学结构变体;
图4描绘可包含在本文提供的阻燃聚合物中的芳族氧化膦组分ar1~ar9的例示性化学结构变体;
图5(a)和图5(b)分别显示本文提供的例示性阻燃聚合物的1hnmr光谱和13cnmr光谱;
图6(a)和图6(b)分别显示本文提供的另一例示性阻燃聚合物的1hnmr光谱和13cnmr光谱;
图7(a)~7(c)描绘本文提供的三种例示性阻燃聚合物的dsc温谱图;
图8(a)和图8(b)显示本文提供的两种代表性阻燃聚合物的tga曲线;
图9显示本文提供的例示性阻燃聚合物的光学吸收谱;
图10显示本文提供的例示性阻燃聚合物的光学吸收对浓度的曲线;
图11显示本文提供的例示性阻燃聚合物的荧光光谱;
图12(a)和图12(b)分别显示在玻璃基材上的例示性阻燃聚合物的薄膜的吸收光谱和荧光光谱;
图13描绘在玻璃基材上的例示性阻燃聚合物的薄膜的光学显微照片;
图14描绘在锡基材上的例示性阻燃聚合物的薄膜的弯曲的照片;
图15描绘在高热处理之前和之后在玻璃基材上的例示性阻燃聚合物的薄膜的光学照片;
图16描绘在高热处理之前和之后在玻璃基材上的例示性阻燃聚合物的轻敲模式(tapping-mode)afm高度、3d和横截面图像及数据;
图17描绘在参考聚合物(rp;顶行)和例示性阻燃聚合物(底部三行)的直接火焰/燃烧期间采集的一系列照片;
图18描绘六种例示性阻燃聚合物随时间而变的放热率(w/g)。
具体实施方式
聚合物由于其可调谐的性质和加工容易性而在我们的日常生活中广泛使用。这些品质使得聚合物可用于从胶粘剂和润滑剂到用于飞行器的结构组件和窗的应用。然而,常见的聚合物可燃性也高且在燃烧期间生成毒性气体和烟气。因此,需要研发具有阻燃性质的聚合物,特别是对于在建筑、运输和服装中使用的材料的改善的耐火性能而言。同时,一些含有溴或氯部分的耐火材料由于其在燃烧期间对健康和环境的不利影响而正被逐步停止使用。因此,研发可安全地且以环境自觉的方式制造并利用的有效耐火聚合物具有重大意义。
含氮聚合物已经在具有合乎需要的电致发光(el)、导电和液晶(lc)性质的离子聚合物的设计和合成方面受到关注,这使它们在许多工艺应用中成为有吸引力的材料。在含氮的el聚合物中,聚(吡啶鎓)盐已经在用于显示和照明、日光采集的电子和光电装置、传感器、光子装置、汽车和飞行器部件的应用及在电子工业中的封装方面收到相当大的重要性和关注度。这些离子el聚合物展示出包括氧化还原行为、导电率、电致变色、光致变色、热致变色和lc性质的许多重要性质。所述离子聚合物为用于通过连续的逐层沉积技术经由静电相互作用构造功能性多层组合件的适当聚合物。离子el聚合物(也称作电活性聚合物(eap))具有诱发比在用刚性且脆性的电活性陶瓷的情况下可能的移动大高达两个数量级的应变的能力。当与形状记忆合金相比较时,eap材料还具有较高的响应速度、较低的密度和改善的回弹性。已知通过环变换聚合反应合成光活性且电活性的离子聚合物及其性质的表征。这些离子聚合物通常具有高玻璃化转变温度tg,且为热稳定且热氧化稳定的。它们高度结晶且具有优良的成膜性质。尽管常规聚(吡啶鎓)盐具有非常吸引人的性质且热稳定,但是它们缺乏离子el聚合物材料的稳健、高温、高效、高机械强度、低成本和轻质性。
经过近数十年,诸如金属、木材、玻璃和陶瓷的常规材料日益被合成聚合物替换,这归因于合成聚合物的通用性、低密度、机械性质及其可塑模的容易性。然而,与金属相比较,这些优势被其可燃性和在高温下的低稳定性所遮蔽。相当大的注意力已经集中在制备滞燃聚合物上,并且,尤其广泛使用含磷的聚合物。已经将磷部分并入包括环氧树脂、聚(酰胺酸)、聚碳酸酯、聚(氯乙烯)、聚酯、聚酰亚胺和聚(甲基丙烯酸甲酯)的不同聚合主链中。在具有含磷部分的聚合物之中,具有氧化膦部分的聚合物具有滞燃性质、热氧化稳定性、可溶于有机溶剂中、可混性和对其他化合物的粘附性。尽管在聚合物主链中并入氧化膦部分产生改善的性质,然而,这些聚合物的生产成本因为其难以加工而过高。当前,没有可成本有效地生产并加工且其性质可使用直接了当的合成路线调谐好的实用的高温、高性能且轻质的含氧化膦的电活性且光活性的离子聚合物。因此,需要研发具有耐高温性和光致发光效率的可容易加工的含氧化膦的离子el聚合物。离子聚合材料与以大分子构造的耐高温氧化膦基团的组合具有传递所述新颖性的潜能。
在详细解释本发明的任何实施方式之前,应理解本发明在适用于以下说明书中所阐述或在图中所说明的构造的详述和组件的配置方面不受限制。本发明能够存在其他实施方式且能够以多种方式实践或进行。同样,应理解本文使用的措辞和术语是出于描述的目的且不应认为是限制。本文使用的“包括”、“包含”或“具有”及其变体意在包涵其后所列项和其等价物以及补充项目。
还应该理解本文叙述的任何数值范围包括自下限值到上限值的所有值。例如,如果将浓度范围陈述为1%~50%,意图在于诸如2%~40%、10%~30%或1%~3%等值明确列举在本说明书中。这些仅仅是被视为在本申请中明确陈述的在所列举的最低值与最高值之间且包括最低值和最高值的数值的具体预期且所有可能的组合的实例。
应当理解的是如本文使用的术语“约”与术语“大约”的意义是一样的。作为说明,使用术语“约”指示值包括略超出所提到的值的值。变化可归因于诸如实验误差、制造公差、在平衡条件下的变化等的条件。在一些实施方式中,术语“约”包括所提到的值加或减10%。在所有情况下,其中已经将术语“约”用于描述值,应该了解本公开还支持精确的值。
贯穿本说明书提到“一个实施方式”或类似语言是指关于该实施方式描述的特定特点、结构或特征包括在本文提供的本发明的至少一个实施方式中。因此,贯穿本说明书的短语“在一个实施方式中”和类似语言的出现可(但未必一定)全部指同一实施方式。
此外,本文提供的方法和装置的所述特点、结构或特征可在一个或多个实施方式中以任何合适的方式组合。在下文描述中,提供许多细节以提供实施方式的全面理解。然而,相关领域的技术人员应该认识到这些实施方式可在不存在这些细节中的一个或多个的情况下或在具有其他方法、组分、材料等的情况下实践。在其他情况下,没有详细显示或描述众所周知的结构、材料或操作以避免模糊实施方式的方面。
如本文使用的术语“烷基”为1~24个碳原子的支链或无支链的饱和烃基,诸如甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、戊基、己基、庚基、辛基、癸基、十四烷基、十六烷基、二十烷基和二十四烷基。所述烷基可为环状的,诸如环己基或环戊基。“低碳烷基”为含有1~6个碳原子的烷基。所述烷基可未被取代或被包括但不限于烷基、烯基、炔基、芳基、芳氧基、卤化物、卤烷基、硝基、氨基、酯、酮、醛、羟基、羧酸或烷氧基的一个或多个基团取代。
术语“烯基”在本文中定义为具备至少一个c=c双键的c2-c20烷基。术语“炔基”在本文中定义为具备至少一个c-c三键的c2-c20烷基。所述烯基或炔基可未被取代或被包括但不限于烷基、烯基、炔基、芳基、芳氧基、卤化物、卤烷基、硝基、氨基、酯、酮、醛、羟基、羧酸或烷氧基的一个或多个基团取代。
术语“氨基”在本文中定义为具有式-nr’r”的具备至少一个氮原子的基团,其中r'和r"各自独立地选自氢、烷基、芳基和杂芳基,或r'和r"连同其所连接的氮可形成环结构。基团r'和r"可任选被例如一个或多个取代基取代。所述氨基可为伯氨基(即,rnh2)、仲氨基(即,rrnh)、叔氨基(即,rrrn)或季铵基团(即,rrrrn+)。氨基的实例包括但不限于-nh2;烷基氨基,诸如-nhch3、-nhch2ch3和-nhch(ch3)2;二烷基氨基,诸如-n(ch3)2和-n(ch2ch3)2;和芳基氨基,诸如-nhph。
如本文使用的术语“芳族物”为含有至少一个共轭环的化合物,且包括芳基和杂芳基。
如本文使用的术语“芳基”为含有至少一个芳族环的单环、双环或三环烃环系,其中能够取代的任何环原子都可被取代(例如,被一个或多个取代基取代)。芳基部分的实例包括但不限于苯基、萘基、蒽基、芴基和亚联苯基。所述芳基可未被取代或被包括但不限于烷基、烯基、炔基、芳基、芳氧基、卤化物、卤烷基、硝基、氨基、酯、酮、醛、羟基、羧酸或烷氧基的一个或多个基团取代。被取代的芳基的实例包括联苯基、苯氧基苯基、氨基苯氧基苯基和三氟甲基-氨基苯氧基苯基。
如本文使用的术语“杂芳基”为具有1~3个杂原子(如果是单环)、1~6个杂原子(如果是双环)或1~9个杂原子(如果是三环)的芳族3~8元单环、8~12元双环或11~14元三环环系,其中至少一个杂原子并入芳族基团的环内。杂原子的实例包括但不限于氮、氧、硫和磷。所述杂芳基可未被取代或被包括但不限于烷基、烯基、炔基、芳基、芳氧基、卤化物、卤烷基、硝基、氨基、酯、酮、醛、羟基、羧酸或烷氧基的一个或多个基团取代。杂芳基的实例包括吡啶基、吡嗪基、呋喃基、噁唑基、噻吩基、嘧啶基和苯并环化环系如苯并咪唑和喹啉。
本文描述含有至少一个吡啶鎓部分和至少一个氧化膦部分的新阻燃离子聚合物的设计、合成、表征和评价及包含这一聚合物的装置。例示性阻燃离子聚合物示于图1中。
本文提供的阻燃聚合物可内在地阻燃。在某些实施方式中,本文提供的阻燃聚合物可由至少两种通用组分构成:含有至少一个吡啶鎓部分的单体组分(m)和含有至少一个氧化膦部分的芳族组分(ar)。所述聚合物可具有在式1中示出的通用结构:
其中m+为阳离子单体组分,y-为阴离子平衡离子,ar为芳族组分,且n为2~约100,000的数值。在某些实施方式中,m+为吡啶鎓部分,y-为阴离子平衡离子,且ar为芳基氧化膦。
本文提供的阻燃聚合物可包含作为m+的任何合适吡啶鎓盐部分,包括衍生自在图2中示出的单体的那些。在某些实施方式中,在本文提供的阻燃聚合物中的吡啶鎓盐部分可以包含一个或多个芳族基团。在一些实施方式中,所述吡啶鎓盐部分包含一个或多个芳基或杂芳基。在一些实施方式中,在本文提供的阻燃聚合物中的吡啶鎓盐部分可包含一个或多个苯基。在一些实施方式中,在本文提供的阻燃聚合物中的吡啶鎓盐部分可包含至少两个吡啶鎓基团。
在一些实施方式中,在本文提供的阻燃聚合物中的吡啶鎓盐部分可包含经由一个或多个诸如芳基或杂芳基的芳族基团键结在一起的至少两个吡啶鎓基团。在一些实施方式中,在本文提供的阻燃聚合物中的吡啶鎓盐部分可包含经由一个或多个苯基键结在一起的至少两个吡啶鎓基团。在一些实施方式中,在本文提供的阻燃聚合物中的吡啶鎓盐部分可包含至少两个芳族基团,诸如至少两个苯基。所述吡啶鎓盐部分可包含经由氧、苯基、o-苯氧基(即,-o-苯基-o-)、烯基、萘基或蒽基彼此键结或连接的至少两个芳族基团。其他合适的吡啶鎓盐部分在下文论述。
在本文提供的阻燃聚合物中的吡啶鎓盐部分可包含任何合适的负电性离子(y-)。在一些情况下,在本文提供的阻燃聚合物中使用的吡啶鎓盐部分可选自由如在图1和图3中所示的平衡离子构成的基团。合适的平衡离子的实例包括芳基和杂芳基磺酸盐、芳基和杂芳基硼酸盐及二卤烷基磺酰胺,诸如甲苯磺酸盐和三氟甲磺酰亚胺。
本文提供的阻燃聚合物可包含任何合适的芳族氧化膦部分(ar)。在本文提供的阻燃聚合物中的氧化膦部分在一些情况下可包含至少一个芳基或杂芳基,诸如至少一个苯基。在一些实施方式中,所述氧化膦部分可包含至少两个芳基或杂芳基,诸如至少两个苯基。在一些实施方式中,所述氧化膦部分可包含至少三个芳基或杂芳基,诸如至少三个苯基。在一些实施方式中,所述氧化膦部分可包含至少一个胺取代的苯基。在一些实施方式中,所述氧化膦部分包含至少一个三氟甲基取代的苯基。在一些实施方式中,所述氧化膦部分可包括三苯基膦。在某些实施方式中,所述氧化膦部分可包含苯氧基苯基、氨基苯氧基苯基或二苯氧基苯基,其中苯基可被卤基、烷基、卤烷基或氨基取代基取代。其他合适的氧化膦部分在下文论述。
在一些实施方式中,本文提供的式1的通用结构的阻燃聚合物具有在式2中所示的结构:
其中r1和r6各自为n,其中r2和r7各自为带负电的平衡离子,其中r3、r4、r8、r9、r11、r13、r14、r15和r16各自为h或包含一个或多个碳原子的基团,且其中r5、r10和r12各自为包含一个或多个碳原子的基团,且其中n为在2至约100,000之间的数值。
在一个实施方式中,r1和r6各自为n,且r2和r7各自单独地选自甲苯磺酸盐和三氟甲磺酰亚胺。在某些实施方式中,r3、r4、r8、r9、r11、r13、r14、r15和r16各自单独地选自h、烷基、芳基和杂芳基。在一些实施方式中,r5、r10和r12各自单独地选自烷基、烯基、炔基、芳基和杂芳基。在一个实施方式中,n为在2至约100,000之间的数值。在某些实施方式中,r1和r6各自为n;r2和r7各自单独地选自甲苯磺酸盐和三氟甲磺酰亚胺;r3、r4、r8、r9、r11、r13、r14、r15和r16各自单独地选自h、烷基、芳基和杂芳基;r5、r10和r12各自单独地选自烷基、烯基、炔基、芳基和杂芳基;且n为在2至约100,000之间的数值。
在一些实施方式中,r3、r4、r8、r9、r13、r14、r15和r16中的至少一个包括芳基或杂芳基,诸如苯基。在一些实施方式中,r10、r11和r12中的至少一个包括芳基或杂芳基,诸如苯基。在一些实施方式中,r3、r4、r5、r8、r9、r10、r11和r12各自包括苯基。在一些实施方式中,r3、r4、r5、r8、r9、r10、r11和r12各自由苯基组成。在一些实施方式中,n在2至约70,000之间,或在2至约65,000之间。在某些实施方式中,n不超过约100,000,为至少约10,000或至少约30,000。在一个实施方式中,n在约30,000至约65,000之间。
本文提供的阻燃聚合物可包含吡啶鎓盐部分和氧化膦部分。图1描绘本文提供的包含吡啶鎓盐部分的阻燃聚合物的各种实施方式的化学结构。如在图1中所示,例示性聚合物p-1包含含有吡啶鎓盐部分和氧化膦部分的重复单元,所述吡啶鎓盐部分包括双(2,6-二苯基吡啶鎓)二甲苯磺酸盐,所述氧化膦部分包括三苯基氧化膦。如所示,p-1包括甲苯磺酸盐平衡离子(ots-)。p-2与p1类似,但包括三氟甲基取代的三苯基氧化膦部分。p-3与p-1类似,但包括在各个双(2,6-二苯基吡啶鎓)二甲苯磺酸盐和各个三苯基氧化膦之间的氧-苯基。p-4与p-1类似,但包括n(tf)2-平衡离子,而不是甲苯磺酸盐平衡离子。p-5与p-2类似,但包括n(tf)2-平衡离子,而不是甲苯磺酸盐平衡离子。p-6与p-3类似,但包括n(tf)2-平衡离子,而不是甲苯磺酸盐平衡离子。
图2描绘可用于形成聚合物的吡喃鎓盐化学结构变体m2~m10的单体,包括在图1中描绘的聚合物p-1和p-4的双(2,6-二苯基吡啶鎓)二甲苯磺酸盐部分。m2~m10为具有变化的化学构造的例示性吡喃鎓盐,在与含胺的部分聚合时,由其制成聚(吡喃鎓)盐,聚(吡喃鎓)盐可具有与在图2中的m2~m10中所示相同的结构,但具有被氮原子置换的一个或多个吡喃鎓氧原子。m2~m10各自可用以形成本文提供的聚合物。
图3描绘可作为在本文提供的聚合物中的平衡离子使用的例示性化学结构变体y1~y15。例如,y1~y15可被在图1中描绘的聚合物中的甲苯磺酸盐或三氟甲磺酰亚胺平衡离子取代。在一些情况下,如所示,本文提供的平衡离子可为脂族的。在一些情况下,如所示,本文提供的平衡离子可为芳族的。在一些情况下,如所示,本文提供的平衡离子可具有单一负电荷。在一些情况下,如所示,本文提供的平衡离子可为带双倍负电。
图4描绘可作为在本文提供的聚合物中的芳族氧化膦部分使用的例示性化学结构变体ar1~ar9。ar1~ar9为可使用环变换聚合反应与双吡喃鎓化合物反应以形成本发明聚合物的芳族二胺氧化膦化合物的实例。
新型含氧化膦的聚(4,4’-(对亚苯基)-双(2,6-二苯基吡啶鎓))离子聚合物p-1、p-2、p-3、p-4、p-5和p-6的合成使用环变换聚合反应。已经研究这些离子聚合物的热稳定性、耐火性、残碳率、机械强度、涂覆和成膜、光学吸收、光致发光、电化学和形态学性质以将它们确定为潜在的高温且高性能的材料。合成方法为通用的且服从低成本大规模生产。该通用性源自大批这样的聚合物可通过利用含双吡啶鎓盐、含氧化磷的二胺和以组合式样的平衡离子的结构变体来合成的事实。因此,化学结构改性允许精细调谐其阻燃性质。
本文提供的阻燃聚合物可用于任何合适的产品中。在一些情况下,本文提供的阻燃聚合物可用于建筑物(例如,摩天大楼、公寓大楼、帐篷等)或车辆(例如,汽车、军用运输机、飞机、航天器等)中。在一些情况下,本文提供的阻燃聚合物可作为用于电子设备的绝缘材料使用。在一些情况下,本文提供的阻燃聚合物可作为膜使用。在一些情况下,本文所述的带不同电荷的聚合物的多个纳米层可用于制造膜。
本文提供的阻燃聚合物可具有以下优势中的一个或多个。在一些情况下,本文提供的阻燃聚合物可相对廉价以便制造和/或加工。在一些情况下,本文提供的阻燃聚合物可大量制造。在一些情况下,本文提供的阻燃聚合物可用常见的有机溶剂加工。在一些情况下,本文提供的阻燃聚合物可具有良好的成膜性质。在一些情况下,本文提供的阻燃聚合物可具有高残碳率和/或低易燃性。
使用在主链中的苯代双吡啶鎓二甲苯磺酸盐和苯基氧化膦部分两者的一个目的在于研发具有高热稳定性、高玻璃化转变温度和增强的光物理、电化学、热和机械性质的先进聚合结构材料。
4,4'-(1,4-亚苯基)-双(2,6-二苯基吡喃鎓)二甲苯磺酸盐单体(化合物m)和含氧化膦的二胺、双(3-氨基苯基)苯基氧化膦(m-dappo)、双(3-氨基苯基)-3,5-双(三氟甲基)苯基氧化膦(batfpo)和双(4-氨基苯氧基-4-苯基)苯基氧化膦(p-bappo)的化学结构显示如下:
双吡喃鎓二甲苯磺酸盐单体(在上文显示的化合物中标记为化合物m)为用于设计并研发具有液晶(lc)性质和发光性质两者的离子聚合物的优良构筑嵌段。化合物m为可在本文公开的聚合物的合成中使用的单体的实例。其含有增加热稳定性的在芳族环中稳定的苯基和杂原子。因为这些离子聚合物为阳离子的,所以它们具有通过连续静电沉积技术与阴离子聚合物一起构造多层组合件的重大潜力。选择含苯基氧化膦的芳族二胺m-dappo、batfpo和p-bappo,这归因于它们的高热氧化稳定性、高玻璃化转变温度和对于含磷聚合物的阻燃行为的潜力。预计将所述聚合物初始暴露于原子氧在表面上产生钝化磷酸盐透明层,进一步防止在苛刻热环境中由原子氧引起的侵蚀/腐蚀。
本文提供的阻燃聚合物可用于任何合适的产品中。在一些情况下,本文提供的聚合物可为结构组分、胶粘剂和/或润滑剂。在一些情况下,可将本文提供的聚合物施用到诸如结构组件的物件上,以改善物件的耐火性质。包含本文提供的聚合物的合适结构组件包括建筑物和/或车辆的结构组件,诸如窗、木梁和干墙。在一些情况下,本文提供的聚合物可包含在医疗装置中。在一些情况下,本文提供的聚合物可用于使电气组件(例如,接线)或设备绝缘。本文提供的聚合物可具有阻燃性质。在一些情况下,本文提供的聚合物在燃烧时不制造毒性气体。本文提供的聚合物可基本不含溴、氯和/或卤素,其如果存在于燃烧的聚合物中,则可制造有害气体。
在一些情况下,本文提供的聚合物可使用逐层沉积(lbl)技术形成。lbl技术为用于由两种带相反电荷的聚合物产生多层的均匀纳米构造的高度受控且广泛范围的方法。聚(吡啶鎓)盐为带正电的聚合物,它们为构造纳米层的适当配偶体,其可简单地通过改变带负电的聚合物的性质而在精确控制下构造。存在许多带负电的聚合物:合成型(人造),例如,聚苯乙烯磺酸盐;和天然生物聚合物,例如dna。在lbl沉积期间,平衡离子可从带正电聚合物和带负电聚合物中洗掉。厚度、表面电荷和组成可经由改变阴离子聚合物的性质、浸泡/洗涤周期和反应条件来控制。另外,各种形态可通过改变下伏基材模板的构造来实现。在一些情况下,本文提供的lbl沉积的聚合物可作为膜使用。在一些情况下,本文提供的lbl聚合物膜可用于药物传送、基因传染、神经干细胞的电刺激或组织工程化。在一些情况下,生物传感器可包括本文提供的lbl聚合物膜。
本公开的例示性实施方式在以下实施例中提供。提出以下实施例以说明本文公开的发明且帮助普通技术人员完成和使用这些实施方式。这些是实施例,而不是想要以任何方式限制本文公开的本发明的范围。
单体的合成和表征。化合物4,4’-(1,4-亚苯基)-双(2,6-二苯基吡喃鎓)二甲苯磺酸盐单体(化合物m)根据已知程序以两个步骤合成(方案1)。在第一步骤中,使对苯二甲醛与苯乙酮缩合以提供所要的四甲酮i。随后将产物自甲苯重结晶以提供38.0g(产率89%)的化合物i的灰白色晶体。i的化学结构和纯度通过元素、1hnmr和差示扫描量热法(dsc)(在206℃下熔融吸热)分析来确认。
在第二步骤(方案1)中,接着通过用作为氢化物受体的三苯甲基甲苯磺酸盐处理使四甲酮i与单体m环化脱水。该氢化物受体自三苯基甲醇和对甲苯磺酸原位产生。产物通过过滤收集,用(ch3co)2o和乙醇仔细洗涤,随后自乙酸重结晶,且获得所要4,4’-(1,4-亚苯基)-双-(2,6-二苯基吡喃鎓)二甲苯磺酸盐单体的黄色晶体,产率为75%。化合物m的化学结构和纯度通过1h和13cnmr和元素分析确认。nmr峰值位置和在芳族质子与脂族质子之间的整合比与化合物的计算值十分吻合。根据dsc分析的若干吸热线在161℃(tm)、195℃和304℃(ti)下观察到,其匹配先前报道的值。
方案1.合成4,4'-(1,4-亚苯基)-双(2,6-二苯基吡喃鎓)二甲苯磺酸盐单体(化合物m)。
双(3-氨基苯基)苯基氧化膦(m-dappo)通过如在方案2中所示的两步法合成。在第一步骤中,使三苯基氧化膦(tpo)ii与90%发烟hno3和96%h2so4反应以提供双(3-硝基苯基)苯基氧化膦(m-dnppo)。将粗产物在真空中干燥并自乙醇重结晶两次且获得m-dnppo(产率65%)。元素和1hnmr分析证实m-dnppo的化学结构和高纯度。
在第二步骤中,双(3-氨基苯基)苯基氧化膦(m-dappo)通过使用氯化亚锡还原m-dnppo来制备。该方法通常用于合成该二胺,因为在氢化振荡器中在pd/c上用氢气还原生成低产率的产物(28%)。将固体粗产物过滤,用水洗涤至中性,且自氯仿重结晶以给出m-dappo,产率84%。m-dappo的化学结构和高纯度也通过元素、1h和13cnmr分析确认。m-dappo的dsc温谱图在氮气中在10℃/min的加热和冷却速率下获得。在第一加热周期中,熔融吸热的峰最大值为208℃,这完全吻合所报道的熔点值(mp=203℃)。
方案2.合成双(3-氨基苯基)苯基氧化膦(m-dappo)。
方案3概述双(3-氨基苯基)-3,5-双(三氟甲基)苯基氧化膦(batfpo)的合成。该二胺经由由3,5-双(三氟甲基)溴苯与二苯基次膦酰氯准备的格利雅反应,接着是硝化和还原反应来合成。将固体粗产物过滤且用大量的水洗涤直至中性,以提供粗产物batfpo,将其风干。随后通过升华进一步纯化,以提供纯batfpo,产率86%。1h和13cnmr光谱和元素分析证实其化学结构和纯度。显示在dsc温谱图的第一加热周期中在δh=9.0kcal/mol下在228℃下的tm(lit.mp=226~227℃)。
方案3.双(3-氨基苯基)-3,5-双(三氟甲基)苯基氧化膦(batfpo)的合成。
第三种二胺化合物双(4-氨基苯氧基-4-苯基)苯基氧化膦(p-bappo)在如在方案4中所示的两步法中合成。首先,在四氢呋喃(thf)中在镁存在下使对溴氟苯与苯基膦酰二氯反应制备为灰白色固体的双(4-氟苯基)-苯基氧化膦(p-fppo),产率89%且mp=128~130℃。
在第二步骤中,使p-fppo与4-氨基苯酚在k2co3存在下在n,n-二甲基乙酰胺(dmac)中反应以提供p-bappo。粗产物通过柱色谱法经硅胶以2%甲醇-乙酸乙酯溶离来纯化以供给为灰白色固体粉末的p-bappo,产率82%。化学结构通过1h和13cnmr及dsc分析(mp=98~100℃)确认。
方案4.双(4-氨基苯氧基-4-苯基)苯基氧化膦(p-bappo)的合成。
离子聚合物的合成和表征。含苯基氧化膦的聚(4,4'-(对亚苯基)-双(2,6-二苯基吡啶鎓))离子聚合物(p-1)通过如在方案5中所示的环变换聚合反应制备。该聚合反应实质上是在4,4'-(1,4-亚苯基)-双(2,6-二苯基吡喃鎓)二甲苯磺酸盐单体(化合物m)和双(3-氨基苯基)苯基氧化膦(m-dappo)之间的缩聚反应,释放作为缩合产物的水。
方案5.使用环变换聚合反应合成聚(4,4′-(对亚苯基)-双(2,6-二苯基吡啶鎓))聚合物p-1、p-2和p-3。
化合物m在加热下在非挥发性的无毒的与水混溶的溶剂二甲亚砜(dmso)中在130~140℃下与m-dappo聚合24小时。在该聚合期间产生的缩合产物水自反应介质中作为甲苯/水共沸物蒸馏出来。浅黄色固体离子聚合物p-1通过用蒸馏水沉淀和在甲醇中溶解的两个周期分离。以高纯度的77~80%的产率在纯化之后实现。应注意到所述反应介质贯穿整个聚合反应时间维持作为均质溶液,因此容许生成高分子量聚合物。该缩聚反应与其他缩聚反应形成对比,其中聚合物通常自溶液过早地沉淀出来,因此限制聚合物的最大分子量。聚合物p-1通过元素分析、1h和13cnmr及凝胶渗透层析色谱(gpc)分析全面表征。
图5(a)和图5(b)显示离子聚合物p-1在d6-dmso中在室温下的1h和13cnmr光谱。其1hnmr光谱显示对于双吡啶鎓盐的芳族部分的质子在δ=8.93和8.70ppm下的独特共振和对于在甲苯磺酸盐平衡离子中的芳族部分和甲基的质子在δ=7.44和7.05及2.25ppm下的一组共振。所有芳族质子(49h)和脂族质子(6h)的相对整合比完美吻合自在该聚合物的重复单元中的那些获得的计算值。正如所料,其13cnmr光谱含有在适当化学位移下的脂族碳信号和芳族碳信号两者。
使用类似的环变换聚合反应(如在方案5中所示)和纯化方法,离子聚合物p-2和p-3通过使化合物m与相应的二胺(batfpo和p-bappo)在dmso中在130~140℃下聚合24小时获得,产率分别为77%(黄色固体)和79%(深褐色固体)。离子聚合物p-2和p-3两者的化学结构也通过元素、1h和13cnmr光谱分析确认。图6(a)和图6(b)显示在室温下取得的在d6-dmso中的聚合物p-2的1h和13cnmr光谱。像聚合物p-1的1hnmr光谱的分析(参见上文)一样,其芳族质子(47h)和脂族质子(6h)的相对整合比完美吻合从在该聚合物的重复单元中的那些获得的计算值。正如所料,其13cnmr光谱也含有在适当化学位移下的脂族碳信号和芳族碳信号两者。
另外的离子聚合物通过相应初级离子聚合物在所要离子存在下的简单易位反应来制备,如在方案6中所示。该反应引起原始甲苯磺酸盐离子与新引入的离子交换。使用该易位反应,离子聚合物p-4、p-5和p-6通过使甲苯磺酸盐离子与三氟甲磺酰亚胺离子交换而由相应离子聚合物p-1、p-2和p-3以高产率(95%)和纯度制备。在此应注意,尽管具有三氟甲磺酰亚胺离子的离子聚合物可由三氟甲磺酰亚胺改性的单体m直接制造,但将需要的酸性很大的双三氟甲烷磺酰基酰亚胺平衡离子的处理则非常不便。
方案6.使离子聚合物p-1转化成p-4的易位反应
p-4、p-5和p-6离子聚合物的化学结构和纯度通过元素、1h和13cnmr光谱分析确认。容易的环变换聚合消除了对于单体或成品聚合物的最终纯化的需要。这类聚合物的纯化通过在包括水的良性溶剂中简单溶解和沉淀来方便地执行。此外,该聚合不需要玻璃仪器的特殊件、不需要特殊的催化剂且不需要严格地排除水分,这将能够扩大这类离子聚合物的合成的规模。因此,这些耐高温的离子聚合物代表降低其对环境和污染的影响的吸引人的供选例。该易位反应提供低成本大规模生产具有可调谐性质的各种离子聚合物的良好潜力。
分子量和溶解度.36~65kda范围内的离子聚合物的数均分子量(mn)汇总于表1中。重要地,与具有相当宽的分子量分布的其他市售聚合物相比较,多分散指数(pi)低(<1.73)。该性质提供具有可为各种应用定制的非常特殊的性质的聚合物。基于离子聚合物的分子量数据,环变换聚合反应对于合成高分子量的含磷离子聚合物似乎相当有效。
表1.离子聚合物p-1~p-6的热性质和gpc数据
atd为聚合物的质量减少其原始质量的5重量%时的分解温度。
聚合物的分子量可通过利用双吡喃鎓单体和二胺在聚合反应中的非化学计量比来调节。关于溶解度,发现p-1、p-2和p-3离子聚合物可容易地溶解于甲醇、乙醇和乙腈中。另一方面,聚合物p-4、p-5和p-6可溶于丙酮和乙腈中。然而,它们在水、丙醇、甲苯、氯仿、四氢呋喃和二氯甲烷中不溶解或溶解度很差,这提示着这些聚合物对水、水蒸气和有机气体具有高耐受性,正如将其作为涂料或结构组件材料所需要的。
热性质.为了测定离子聚合物的热性质,差示扫描量热法(dsc)和热解重量分析(tga)分别地在10℃/min的加热和冷却速率下和在氮气中在10℃/min的加热速率下执行。离子聚合物p-1、p-2和p-3的dsc温谱图示于图7(a)、7(b)和7(c)中。由于溶剂损失而引起的在112℃下的广泛吸热在p-1的dsc温谱图(图7a)的第一加热周期中观察到,而在第二加热周期中没有观察到这样的吸热。p-1的高玻璃化转变温度tg=276℃在其dsc温谱图中显而易见。然而,在进一步加热的情况下,由于低温结晶在320℃下的tg之后观察到放热。用于p-2,观察到275℃的玻璃化转变温度(图7b),然而,在高达350℃的较高温度下没有观察到低温结晶吸热。对于p-3,观察到tg=243℃的玻璃化转变温度(图7c)。对于含有三氟甲磺酰亚胺平衡离子的离子聚合物p-4、p-5和p-6,通过dsc分析观察到在230~253℃范围内的tg。根据聚合物的化学结构,tg为231~275℃(表1)。这些结果指示这些离子聚合物具有高tg值。
图8(a)和8(b)显示在氮气中在10℃/min的加热速率下获得的两种代表性离子聚合物p-2和p-4的tga曲线。离子聚合物的分解温度(td)也汇集在表1中。发现,对于含甲苯磺酸盐平衡离子的离子聚合物(p-1、p-2和p-3)而言,td值在343~352℃的温度范围内,且对于含三氟甲磺酰亚胺平衡离子的聚合物(p-4、p-5和p-6)而言,td值在418~439℃的温度范围内,所有六种离子聚合物在td下仅出现5%的重量损失。这些分解温度比不含氧化膦部分的聚(对亚苯基-二苯基吡啶鎓)离子聚合物的分解温度(245℃)高超过100℃。一般而言,由于氟代阴离子的高热稳定性,与含有三氟甲磺酰亚胺平衡离子的聚合物相比较,含有甲苯磺酸盐平衡离子的聚合物具有较高tg,但具有较低td。tga分析还展示所有六种离子聚合物(p-1~p-6)在700℃下都具有高残碳率(p-1,52%;p-2,53%;p-3,49%;p-4,54%;p-5,47%;和p-6,45%)。这些结果提示这些离子聚合物具有高td值和残碳率。
加工、耐水分和蒸气、光学/荧光性质、薄膜、粘附、机械和拉伸强度.发现离子聚合物p-1、p-2和p-3可容易地溶解于甲醇、乙醇和乙腈中,而p-4、p-5和p-6由于不同的平衡离子三氟甲磺酰亚胺离子而可溶于丙酮和乙腈中。然而,所有这些离子聚合物在水、丙醇、甲苯、氯仿、四氢呋喃和二氯甲烷中都不溶解或溶解性很差。这些离子聚合物的溶解度概况汇总于表2中。这些结果指示含氧化膦的离子聚合物对水分、水蒸气和有机气体具有高度抵抗性,这是用于涂料和结构组件材料的首要标准。
表2.离子聚合物p-1~p-6在不同溶剂中的溶解度概况
+=可溶;+–=略溶,–=不溶
这些离子聚合物的光学吸收和荧光性质分别使用shimadzuuv-2401pcuv-vis分光光度计和ptiquantamastertmqm-4/2005型分光荧光计检验。图9~11显示p-1在甲醇中的吸收和荧光光谱。p-1聚合物在uv-可见区域(200~400nm)中吸收,最大吸收值(λmax)在342nm下,这归因于π-π*跃迁。p-1具有高摩尔吸光系数(~105m-1cm-1)且显示与浓度的线性相关性。在甲醇溶液中,p-1聚合物发射具有在458nm下的峰最大值的蓝光。对于离子聚合物p-2~p-6,也观察到了在甲醇溶液中的类似光学吸收和荧光性质。发现这些聚合物吸收在200~400nm波长范围内的光并发射在uv-可见区域(350~650nm)内的光。吸收强度和发射强度两者随着在0.1μm和5μm之间的增加的聚合物浓度而线性增加。
这些离子聚合物具有优良的薄膜形成性质。图12(a)和图12(b)显示通过旋涂涂布在玻璃基材上的p-1聚合物薄膜的代表性吸收和荧光光谱。p-1~p-6的薄膜也在200~400nm区域内吸收,峰值在约245nm下。正如所料,它们展示出弱荧光。如在图13中所示,如通过光学显微法所观察,离子聚合物的薄膜形成对准结构。
离子聚合物的薄膜的对准结构的形成通过原子力显微镜(afm)分析(下文论述)进一步检验。由于该对准结构,可将这些离子聚合物用于增强光电装置的性能。
为了理解离子聚合物的粘附、剥离、机械和拉伸强度,离子聚合物在金属基材上的大面积薄膜通过喷涂制备。图14显示通过自甲醇溶液中喷涂制备的在金属锡基材(金属基材没有经过任何表面处理)上的离子聚合物p-1的大面积薄膜的照片。p-1薄膜在锡基材上的大致表面积为约15cm×约7cm。在锡基材上的p-1薄膜的大致厚度为约2~5mμ。
该等薄膜即使在以不同角度弯曲和矫直金属基材之后也保留其完整性。它们经过至少约4~5月仍保留其完整性。如在图14中所示,在弯曲-矫直金属基材时,没有观察到薄膜的剥离和斑纹。对于离子聚合物p-2~p-6,也观察到了类似性质。这些结果提示它们显示出对金属基材的优良粘附性、高机械和拉伸强度,且具有在涂料中和/或作为用于汽车、飞行器、发动机和动力/推进系统的结构组件材料应用的优良潜力。
薄膜的高温热处理和形态.离子聚合物的热稳定性还通过在500℃下在空气中热处理其薄膜并探测其在热处理之前(上部照片)和在热处理之后(下部照片)的形态来确定,如在图15中所示。在500℃下在空气中热处理之后,观察到薄膜从黄色到棕色的颜色改变,这可归因于由氧化膦基团形成玻璃状磷酸盐层。然而,没有观察到离子聚合物的薄膜的结构对准的显著改变。
对于形态研究,将离子聚合物的薄膜从其甲醇溶液溶剂流延到抛光的硅基材(1x1cm)上并在60℃下在真空下干燥12小时。热处理过的样品通过利用加热板在550℃下加热离子聚合物薄膜来制备。这些薄膜在硅基材上的大致厚度为约1mμ。
afm图像使用具有作为纳米探针的单硅-晶体尖端的nano-scopeiii显微镜(digitalinstrumentsinc.,veecometrologygroup,santabarbara,ca)以标准轻敲模式获得。在热处理之前和之后在硅基材上的p-1离子聚合物薄膜的表面形态的轻敲模式afm高度、3d和横截面图像示于图16中。从自在热处理之前(在图16中的上部数据)和在热处理之后(在图16中的下部数据)p-1离子聚合物的薄膜获得的afm结果的分析中观察到0.08913mμ/0.61544mμ的均方根(rms)表面粗糙度、0.52786mμ/1.81704mμ的区域平均高度和1.07694mμ/3.11783mμ的区域最大高度。对于p-1薄膜而言,与在热处理之前的值相比较,在热处理之后观察到较高的rms表面粗糙度、区域平均高度和区域最大高度,这归因于因形成玻璃状磷酸盐层引起的表面形态的变形。对于在该系列中的其他离子聚合物p-2~p-6的薄膜也观察到类似的afm形态变化。
耐火性和阻燃性质.离子聚合物p-1~p-6和参考聚合物(rp)聚噻吩的耐火性和阻燃性质通过将它们用丙烷喷灯焰直接点燃/燃烧并探测其灼烧和/或火焰点燃和延迟行为来研究,如在图17中所示。离子聚合物各自的燃烧试验为基于nasa向上火焰蔓延性试验(nasaupwardflamepropagationtest)(nasa标准6001和astmd6413)的垂直式火焰试验。所有试验都在化学通风柜中利用敞开式设计装备在20%周围氧环境中进行。所有聚合物样品都用清洁的不锈钢钩牢固固定。将一张清洁的纸置于聚合物样品下面以便容易地观察任何滴流。随后将聚合物样品暴露于在其底部边缘处的点火源历时10秒至超过5分钟。将丙烷喷灯焰用作点火器。录像记录所有火焰研究,因此可确定续焰、阴燃和成焦时间。同样记录熔融和滴流的程度。
使用该一致且可重复的方法来评价包括参考聚合物的聚合物样品的阻燃性质。对于rp聚噻吩,观察对火焰点燃的迅速反应(在1秒内)和在2-3秒内的完全灼烧(图17)。在离子聚合物的情形下,尽管观察到变形和由于成焦引起的从黄色到黑色的颜色改变,但是通过用丙烷喷灯焰直接点燃历时超过5分钟没有观察到火焰、火焰蔓延、熔融和滴流。这些结果指示本文研发的含苯基氧化膦的离子聚合物(p-1~p-6)展示出优良的耐火和阻燃性质。
图18描绘聚合物p-1、p-2、p-3、p-4、p-5和p-6的样品随时间而变的释热率(hrr),以w/g为单位。锥形热量测定能够提供关于聚合物的燃烧的有用信息且有效用于聚合物的阻燃性质的实验室评价。该技术测量释热率(hrr)和峰值释热率(phrr);这两者都被视为评价材料的阻燃性质的主要参数。释放的总热量(thr)是另一相关参数,其代表直至火焰熄灭时释放的热量的总和。例如,阻燃剂bpc-聚碳酸酯聚合物和bpc-聚丙烯酸酯聚合物分别具有29和21j/gk及3.0和7.6kj/g的hrr和thr值,这两种阻燃剂皆衍生自1,1-二氯-2,2-(4-羟基苯基)乙烷(bpc)。高度滞燃的聚合物也显示出低phrr值。
电化学性质.为了理解离子聚合物p-1~p-6与在光电装置中的电荷输送过程相关的电子结构,在离子聚合物的薄膜上执行循环伏安法(cv)测量。cv测量在eg&gprincetonappliedresearch恒电位仪/稳流仪器(263a型)上在0.1m六氟磷酸四丁铵(tbapf6)在乙腈中的电解质溶液中执行。将铂(pt)丝电极用作反电极和工作电极,且将ag/ag+电极用作参比。将二茂铁/二茂铁离子(fc/fc+)氧化还原对用作内标物。将关于ag/ag+电极获得的电位值转化成饱和甘汞电极(sce)标值。
离子聚合物p-1~p-6的薄膜通过将pt丝浸泡到0.5~1重量%甲醇溶液中涂布在pt工作电极上并在真空下在80℃下干燥6小时。各薄膜在电极上的大致厚度为约1mμ。所有离子聚合物都显示相对于sce具有-1.83~-1.80v的起始还原电位的可逆还原。正式还原电位也非常类似,相对于sce,在-1.95~-1.91v范围内。估计p-1~p-6的电子亲和势(ea,lumo水平)几乎相同,在约2.57ev与约2.59ev之间。对于所有六种离子聚合物观察到不可逆的氧化。通过粗略估计,发现这些离子聚合物的电离电位(ip,homo水平)为约5.40ev。这些结果提示这些离子聚合物的电化学性质主要由双(2,6-二苯基吡啶鎓)主链结构决定。所观察到的可逆还原、高电子亲和势和不可逆氧化提示这些离子聚合物为固有的n型(电子传输)且空穴阻挡的材料。
该工作包括经由环变换聚合和易位反应合成并表征六种具有可调谐性质的新型的含氧化膦的离子聚合物。这些材料的研发旨在满足安全、易于加工、可调谐材料的需要。下列的方法能够经由简单的聚合反应利用dmso作为聚合反应的溶剂且利用甲醇-水来纯化而以高产率和高纯度生成离子聚合物。这些工作还容许通过诸如平衡离子交换的步骤直截了当地调节性质。获得了具有高玻璃化转变温度(tg>230℃)和相对较高的分解温度(大于340℃)的聚合物。发现这些聚合物即使在暴露于明火5分钟之后也避免点燃,证实了其充当高温阻燃材料的能力。这些聚合物易于抵抗水分和常见的有机溶剂,且发现其具有优良的成膜性质。还证实了这些聚合物吸收在光谱的紫外范围内的光并生成在可见光区域内的电致发光的能力。可将光活性、电活性且实用的耐高温的离子聚合物用于电子仪器、光电子学、防火防蚀涂层和用于汽车、飞行器、发动机、动力和推进系统的结构组件的领域中。另外,可将它们用于消防队员服装、印刷电路板、建筑材料和用于保护未偿付债券、债券和股票、不动产房地契和契据的极薄涂层。
实验细节
材料.对苯二甲醛、苯乙酮、三苯基甲醇、对甲苯磺酸单水合物、3,5-双(三氟甲基)溴苯、二苯基次膦酰氯、4-氨基苯酚、苯基次膦酰二氯、乙酸、乙酸酐、对溴氟苯、乙酸乙酯、三苯基氧化膦、硝酸、硫酸、盐酸、氢氧化钾、碳酸氢钠、na2so4、sncl2.2h2o、pd/c和mg屑自tci购买且以原样使用。出于合成和纯化目的,使用如自sigma-aldrich获得的试剂级溶剂(乙醇、丙醇、甲苯、苯、氯仿、乙醚、己烷、四氢呋喃、二氯甲烷、氯化乙烯、乙腈、二甲亚砜)。将自aldrich获得的光谱光度测量级溶剂用于光学吸收和荧光测量。将自aldrich获得的高纯度乙腈(纯度>99.9%)用于电化学测量。
通用表征方法.单体和聚合物的1hnmr和13cnmr光谱在variannmrj400光谱仪,400mhz在298°k下使用cdcl3和d6-dmso两者作为溶剂来记录。它们的元素分析自numegaresonancelaboratory,ca执行。为了评定聚合物的分子量,凝胶渗透层析色谱(gpc)在50℃下以1ml/min的流速运行。gpc仪器使用同时具有viscotek301型三重的检测器阵列的waters515泵。所述阵列含有激光折射仪、差示粘度计和光散射检测器,在单一仪器中具有直角激光散射(rals)和低角度激光散射(lals)两者,其具有固定的内检测器系统和可调控到高达80℃的温度控制。所述仪器用自polymerstandardservicesusa,inc获得的p-50支链淀粉标准物校准。分离使用自viscotek购买的viscogeli-mbhmw-3078柱实现。注射100-200μl的在含有0.01mlibr的dmso中的2mg/ml聚合物溶液的等分试样。dn/dc值通过注射不同体积来校正以评定趋势。所有数据分析都使用viscotektrisec软件执行。聚合物的热解重量分析用universalv3.0gtainstruments进行。使用在n2下的10℃/min的加热速率,操作从室温进行到800℃。差示扫描量热(dsc)测量在tainstrumentsq100dsc上在氮气中进行。使用在氮气中的加热-冷却-加热法,其中起始加热速率为40℃/min,冷却速率为20℃/min,且最终加热速率为10℃/min。自dsc温谱图的第二加热周期记录所有瞬变。这些离子聚合物的光学吸收和荧光性质分别使用shimadzuuv-2401pcuv-vis分光光度计和ptiquantamastertmmodelqm-4/2005型分光荧光计研究。
合成4,4'-(1,4-亚苯基)-双(2,6-二苯基吡喃鎓)二甲苯磺酸盐单体(m)。将对苯二甲醛(10.0g)和苯乙酮(54.3g)的混合物在250ml的95%乙醇中在65℃下搅拌。在起始化合物溶解之后,在剧烈搅拌下经30分钟逐滴加入koh(10.5g)在10ml水中的溶液。立即形成黄色沉淀物。随后将非均质反应混合物在回流下加热,直至其经5小时的时间变为粉红色。在此期间,将对双查尔酮再溶解且使其与另外两当量的苯乙酮反应以形成所要的四甲酮i(方案1),其也沉淀出来。将反应混合物滤出,且棕褐色固体通过过滤收集以提供41.0g粗产物。将其自甲苯重结晶以提供38.0g(产率89%)的化合物i的灰白色晶体(方案1)。i的化学结构和纯度通过元素和1hnmr分析确认。
对于i到化合物m的转化(即,在方案1中的第二步骤),将三苯基甲醇(7.8g)和对甲苯磺酸单水合物(5.8g)加到100ml乙酸酐中并在室温下搅拌3小时。随后将固体四甲酮i(7.2g)加到反应混合物中,并将混合物加热到100℃历时1小时。非均质混合物变得透明。在冷却时,出现红色晶体且其通过过滤,用乙酸酐和乙醇小心洗涤来收集。随后将产物从乙酸中重结晶并在真空中干燥以提供7.9g的m(产率75%)。d6-dmso中的m的1hnmr,δh:9.35(4h,s,芳族间o+),9.21(4h,s,1,4-亚苯基),7.58-8.93(20h,m,苯基),7.46-7.47(4h,d,j=6.7hz,甲苯磺酸盐),7.09-7.10(4h,d,j=7.7hz,甲苯磺酸盐),2.27(6h,s,ch3)。
双(3-氨基苯基)苯基氧化膦(m-dappo)。将量为11.14g的三苯基氧化膦(tpo)ii置于装备有搅拌器、氮气入口和温度计的250ml烧瓶中。加入体积为60ml的96%h2so4且只要反应物已溶解,就可将溶液在冰/盐浴中冷却到-5℃。经1小时的时间逐滴加入5.80g的90%发烟hno3在40mlh2so4中的溶液。在添加期间将样品维持在-5℃下,升高到室温并留在该温度下8小时。将所获得的浅黄色溶液倾入400ml冰水中,产生通过倾析收集的粘稠固体。随后将该固体溶解于chcl3中且用碳酸氢钠水溶液洗涤直至其为中性。有机层经na2so4干燥过夜并过滤。溶剂通过旋转蒸发除去。将粗产物在真空中干燥并从乙醇中重结晶两次以生成9.6g(26.1mmol,产率65%)的纯产物。执行元素和1hnmr分析以证实m-dnppo的化学结构。
在第二步骤中,将m-dnppo(5.0g)加到sncl2.2h2o在50ml浓hcl和100ml乙醇中。将该混合物保持在室温下搅拌2小时,且随后另外加热2小时。将反应混合物冷却到室温,并倾入60g的koh在200ml冰水中的溶液来冷却,且有力地搅拌。将固体粗产物过滤,用h2o洗涤直至中性,且从氯仿中重结晶以给出3.5g(11.4mmol,产率84%)。m-dappo的化学结构通过元素、1h和13cnmr光谱分析确认。c18h17n2op(308.32)的分析计算值:c,70.12;h,5.56;n,9.09;实验值:c,70.17;h,5.82;n,9.43。
双(3-氨基苯基)-3,5-双(三氟甲基)苯基氧化膦(batfpo).将1.05g镁屑和50ml乙醚装入装备有磁力搅拌器、冷凝器、干燥管、温度计和氮气入口的250ml三颈圆底烧瓶中。将溶液在冰浴中冷却到5℃以下,随后经1小时的时间逐滴加入9.71g的3,5-双(三氟甲基)溴苯,同时有力地搅拌。让混合物另外反应5小时。随后经1小时的时间逐滴加入7.00g的二苯基次膦酰氯。在另外反应18小时之后,获得褐色溶液。接着,将10%硫酸水溶液加到溶液中,达到ph1,接着加入250ml水和乙醚以形成水层和有机层。在滗析乙醚层之后,将水相用乙醚洗涤两次且将所有有机层合并且通过蒸发干燥,产生3,5-双(三氟甲基)苯基二苯基氧化膦(tfpo)浅褐色固体。随后将tfpo溶解于氯仿中并用10%碳酸氢钠洗涤数次且用水洗涤三次。有机层通过旋转蒸发器浓缩并在室温下储存12小时且另外在冰箱中储存12小时。tfpo的纤维质灰白色晶体通过真空过滤收集并通过在己烷中重结晶进一步纯化。
在第二步骤中,双(3-硝基苯基)-3,5-双(三氟甲基)苯基氧化膦(dntfpo)通过使用浓硫酸和浓硝酸使tfpo硝化来制备。将纯化的tfpo(10.0g)加到装备有氮气入口、温度计、干燥管和机械搅拌器的250ml三颈烧瓶中。将浓硫酸(23ml)加到烧瓶中以在室温下溶解化合物tfpo。将溶液用冰水浴冷却到0℃。经1小时的时间将硝酸(4.7ml)逐滴加入溶液中,同时有力地搅拌并维持0℃。让混合物反应8小时且随后注入650g粉碎的冰中。将所得浅黄色固体用氯仿提取,接着用碳酸氢钠水溶液洗涤,直至ph达到7。将溶剂用旋转蒸发器除去且将所得固体自纯乙醇中重结晶两次,提供10.0g的dntfpo的浅黄色晶体。
双(3-氨基苯基)-3,5-双(三氟甲基)苯基氧化膦(batfpo)随后通过用氯化亚锡还原dntfpo来制备。将量为4.0g的batfpo加到10.8gsncl2.2h2o在30ml浓hcl和60ml乙醇中的溶液中,并保持在室温下搅拌2小时且另外加热2小时。将反应混合物冷却到室温,注入40gkoh在200ml冰水中的溶液中且有力地搅拌。将固体粗产物过滤且用大量的h2o洗涤直至中性,给出粗产物batfpo,且使其风干。其随后通过升华进一步纯化以提供3.0g(产率86%)。1h和13cnmr光谱和元素分析证实其化学结构和纯度。c20h15n2of6p(444.32)的分析计算值:c,54.07;h,3.40;n,6.30;实验值:c,53.88;h,3.80;n,6.39。显示在dsc温谱图的第一加热周期中在δh=9.0kcal/mol下在228℃下的tm(lit.mp=226~227℃)。
双(4-氨基苯氧基-4-苯基)苯基氧化膦(p-bappo).首先,经3小时的时间向在冰浴中的2.2g镁在50mlthf中的浆液中逐滴加入对溴氯苯(15g)在thf(40ml)中的溶液。让其在室温下搅拌过夜。在此期间,出现灰色溶液。将混合物再次冷却到0℃且在搅拌下经3~4小时的时间逐滴加入苯基次膦酰二氯(8.35g)在20mlthf中的溶液。随后将混合物温至室温并搅拌12小时。将混合物用10%h2so4淬灭且搅拌1小时。加入乙醚(200ml)以分离有机层,且将水层用乙醚(3×75ml)提取。将合并的乙醚提取物用nahco3溶液和水洗涤,且经硫酸钠干燥。将溶剂在减压下除去以供给浅褐色油。粗产物通过自thf/己烷(1:1)中结晶来纯化以提供为灰白色固体的双(4-氟苯基)-苯基氧化膦(p-fppo)(产率89%),mp128~130℃。
在第二步骤中,将p-fppo(4g)、4-氨基苯酚(3.06g)和k2co3(5.28g)在dmac(15ml)中的磁力搅拌的混合物在氮气下加热到回流历时24小时。随后将反应混合物冷却到室温并在有力搅拌下注入冰水中。沉淀的灰白色固体产物使用真空过滤收集在布氏漏斗中,且用水充分洗涤以除去盐和未反应的4-氨基苯酚。粗产物通过柱色谱法经硅胶以2%甲醇-乙酸乙酯溶离来纯化以供给为灰白色固体粉末的p-bappo,产率82%。化学结构通过1h和13cnmr及dsc分析(mp=98~100℃)确认。
含有苯基氧化膦的聚(4,4’-(对亚苯基)-双(2,6-二苯基吡啶鎓))离子聚合物.第一含苯基氧化膦的聚(4,4’-(对亚苯基)-双(2,6-二苯基吡啶鎓))离子聚合物(p-1)通过如下的环变换聚合反应来制备:6.5873g化合物m在加热下在130~140℃下在dmso中与m-dappo(2.3000g)聚合24小时(参见方案5)。在聚合期间产生的水自反应介质中作为甲苯/水共沸物蒸馏出来。浅黄色固体p-1聚合物通过用蒸馏水沉淀和在甲醇中溶解的两个周期分离,产率80%。p-1通过元素分析及1h和13cnmr光谱分析全面表征。c72h55n2o7s2p(1155.33)的分析计算值:c,74.85;h,4.80;n,2.42;s,5.55;实验值:c,73.36;h,4.82;n,2.44;s,5.45.
使用相同的环变换聚合反应(方案5)和纯化方法,离子聚合物p-2(产率77%;黄色固体)通过使化合物m(8.4800g,6.56mmol)与batfpo(2.918g,6.56mmol)在dmso中在130~140℃下聚合24小时来获得。该聚合物通过用蒸馏水沉淀而以定量产率基本分离。其通过再溶解于甲醇中且通过在加入蒸馏水的情况下随后再沉淀来进一步纯化。类似地,离子聚合物p-3(产率79%;深褐色固体)通过使化合物m(5.3787g,6.09mmol)与p-bappo(3.0000g,6.09mmol)在dmso中在130~140℃下聚合24小时来获得。该聚合物通过用蒸馏水沉淀而以定量产率分离。其通过再溶解于甲醇中且通过在加入蒸馏水的情况下随后再沉淀来进一步纯化,以产生离子聚合物p-3。
离子聚合物p-2和p-3两者的化学结构也通过元素、1h和13cnmr光谱分析确认。p-2聚合物的元素分析:c79h53n2o7f6s2p(1291.34)的分析计算值:c,68.83;h,4.14;n,2.17;s,4.97;实验值:c,66.38;h,4.69;n,2.28;s,5.74。p-3聚合物的元素分析:c84h63n2o9s2p(1339.55)的分析计算值:c,75.32;h,4.74;n,2.09;s,4.79;实验值:c,71.87;h,5.24;n,2.05;s,5.00。
离子聚合物p-4通过离子聚合物p-1与三氟甲磺酰亚胺锂在dmso中的易位反应来制备(方案6)。将1.40g(1.21mmol)聚合物p-1溶解于50mldmso中。向聚合物p-1的dmso溶液中缓慢加入三氟甲磺酰亚胺锂(0.73g,2.54mmol)。在持续搅拌下将所得溶液保持在50℃下48小时。在通过旋转蒸发器减少dmso溶液的体积之后,将反应混合物加到蒸馏水中,提供所要离子聚合物p-4。其通过真空过滤收集,用大量的蒸馏水洗涤数次,且在真空中在100℃下干燥72小时并称重以给出1.58g(1.50mmol)的聚合物p-4(产率95%)。c62h41n4o9f12s4p(1373.24)的分析计算值:c,54.23;h,3.01;n,4.08;s,9.34;实验值:c,54.41;h,3.19;n,4.02;s,9.05。
使用类似的易位反应,离子聚合物p-5由离子聚合物p-2制备。将1.40g(1.08mmol)p-2溶解于50mldmso中。向该聚合物的dmso溶液中加入三氟甲磺酰亚胺锂(0.73g,2.28mmol)。将所得溶液保持在50℃下48小时。在通过旋转蒸发减少dmso溶液之后,将反应混合物加到蒸馏水中,提供所要离子聚合物p-5。其通过真空过滤收集,用大量的热蒸馏水洗涤数次。将该程序再重复一次且随后将收集的聚合物在真空中在100℃下干燥72小时并称重以给出1.54g(1.02mmol)的聚合物p-5。然而,该聚合物的1hnmr光谱显示一对双重峰[δ=7.09(d)和7.46ppm(d)],提示甲苯磺酸盐离子被三氟甲磺酰亚胺离子不完全交换。为了完成甲苯磺酸盐离子的交换以生成聚合物p-5,使易位反应进行第三次,产生成功的易位反应产物。
使用相同的程序,离子聚合物p-6通过p-3与三氟甲磺酰亚胺锂在dmso中的易位反应制备。离子聚合物p-5和p-6的化学结构和纯度通过元素、1h和13cnmr光谱分析确认。聚合物p-5的元素分析:c64h39n4o9f18s4p(1509.23)的分析计算值:c,50.93;h,2.60;n,3.71;s,8.50;实验值:c,50.26;h,2.71;n,3.75;s,10.25。聚合物p-6的元素分析:c74h49n4o11f12s4p(1557.43)的分析计算值:c,57.07;h,3.17;n,3.60;s,8.23;实验值:c,54.57;h,3.77;n,3.55;s,8.88。
循环伏安法测量.循环伏安法实验在eg&gprincetonappliedresearch恒电位仪/稳流器(263a型)上进行。收集数据且将其通过270型电化学分析系统软件分析。在如先前所述的所有实验中使用三电极电化学电池。将铂丝电极用作反电极和工作电极两者,且将银/银离子(在0.1magno3溶液中的ag,bioanalyticalsystem,inc.)用作参比电极。ag/ag+(agno3)参比电极在实验开始时通过对作为内标物的二茂铁/二茂铁离子运行循环伏安法来校准。借助于内标物二茂铁离子/二茂铁(fc+/fc),将参考ag/ag+电极获得的电位值转化成饱和甘汞电极(sce)标值。所有离子聚合物的薄膜通过将pt丝浸泡到在甲醇中的粘稠溶液中且随后将其在真空烘箱中在80℃下干燥8小时而涂布在pt工作电极上。在所有实验中使用0.1mtbapf6在混合水/乙腈溶剂中的电解质溶液。在各实验之前,在三电极电池中的所有溶液都用超纯氮气吹扫10~15分钟,且在实验期间使用n2覆盖层。
参考文献
下列引用各自以引用的方式整体完全地结合到本文中来。
[1](a)c.negrell-guirao,b.boutevin,g.david,a.fruchier,r.sonnier,j.-m.lopez-cuesta,polym.chem.2011,2,236;(b)c.negrell-guirao,b.boutevin,macromolecules2009,42,2446;(c)f.laoutid,l.bonnaud,m.alexandre,j.-m.lopez-cuesta,p.dubois,mater.sci.eng.r2009,63,100;(d)e.d.weil,s.v.levchik,j.firesci.2008,26,243;(e)s.v.levchik,e.d.weil,j.firesci.2006,24,345;(f)s.v.levchik,e.d.weil,polym.int.2005,54,11;(g)s.v.levchik,e.d.weil,polym.int.2004,53,1901;(h)s.-y.lu,i.hamerton,prog.polym.sci.2002,27,1661;(i)g.e.zaikov,s.m.lomakin,j.appl.polym.sci.1998,68,715;(j)c.f.cullis,m.m.hirschler,thecombustionoforganicpolymers,oxforduniversity:newyork,1980;(k)j.w.lyons,thechemistryandusesoffireretardant,wiley:newyork,1970.
[2](a)c.wang,m.kilitziraki,j.a.h.macbride,m.r.bryce,l.e.horsburgh,a.k.sheridan,a.p.monkman,i.d.w.samuel,adv.mater.2000,12,217;(b)h.hong,r.sfez,e.vaganova,s.yitzchaik,d.davidov,thinsolidfilms2000,366,260;(c)y.z.wang,a.j.epstein,acc.chem.res.1999,32,217;(d)x.zhang,a.s.shetty,s.a.jenekhe,actapolym.1998,49,52.
[3](a)s.a.jenekhe,m.m.alam,y.zhu,s.jiang,a.v.shevade,adv.mater.2007,19,536;(b)c.j.tonzola,m.m.alam,s.a.jenekhe,macromolecules2005,38,9839;(c)c.j.tonzola,m.m.alam,s.a.jenekhe,macromol.chem.phys.2005,206,1271;(d)t.w.kwon,m.m.alam,s.a.jenekhe,chem.mater.2004,16,4657.
[4](a)y.eichen,g.nakhmanovich,v.gorelik,o.epshtein,j.m.poplawski,e.ehrenfreund,j.am.chem.soc.1998,120,10463;(b)a.j.epstein,y.z.wang,s.w.jessen,j.w.blatchford,d.d.gebler,l.-blin,t.l.gustafson,t.m.swager,a.g.macdiarmid,macromol.symp.1997,116,27;(c)y.z.wang,d.d.gebler,d.k.fu,t.m.swager,a.g.macdiarmid,a.j.epstein,synth.met.1997,85,1179;(d)k.a.bunten,a.k.kakkar,macromolecules1996,29,2885.
[5](a)d.d.gebler,y.z.wang,j.w.blatchford,s.w.jessen,l.-b.lin,t.l.gustafson,h.l.wang,t.m.swager,a.g.macdiarmid,a.j.epstein,j.appl.phys.1995,78,4264;(b)t.yamamoto,t.muruyama,z.-hzhou,t.ito,t.fukuda,y.yoneda,f.begum,t.ikeda,s.sasaki,h.takezoe,a.fukuda,k.kubota,j.am.chem.soc.1994,116,4832.
[6]t.kawai,t.yamaue,m.onoda,k.yoshino,synth.met.1999,102,971.
[7](a)h.han,p.r.vantine,a.k.nedeltchev,p.k.bhowmik,j.polym.sci.parta:polym.chem.2006,44,1541;(b)p.k.bhowmik,h.han,i.v.nedeltchev,j.polym.sci.parta:polym.chem.2002,40,2015;(c)p.k.bhowmik,h.han,j.j.cebe,r.a.burchett,a.m.sarker,j.polym.sci.parta:polym.chem.2002,40,659;(d)p.k.bhowmik,a.h.molla,h.han,m.e.gangoda,r.n.bose,macromolecules1998,31,621;(e)p.k.bhowmik,s.akhter,h.han,j.polym.sci.parta:polym.chem.1995,40,1927;(f)p.k.bhowmik,h.han,j.polym.sci.parta:polym.chem.1995,40,1745;(g)h.han,p.k.bhowmik,trendspolym.sci.1995,3,199;(h)p.k.bhowmik,w.xu,h.han,j.polym.sci.parta:polym.chem.1994,32,3205.
[8]p.masson,p.gramain,d.guillon,macromol.chem.phys.1999,200,616.
[9]w.-y.zheng,r.-h.wang,k.levon,z.y.rong,t.taka,w.pan,macromol.chem.phys.1995,196,2443.
[10]y.cao,p.smith,polymer1993,34,3139.
[11](a)a.merz,s.reitmeier,angew.chem.,int.ed.engl.1989,28,807;(b)j.s.moore,s.i.stupp,macromolecules1986,19,1815;(c)h.akahoshi,s.toshima,k.itaya,j.phys.chem.1981,85,818;(d)m.s.simon,p.t.moore,j.polym.sci.,parta:polym.chem.1975,13,1.
[12](a)t.s.jo,a.k.nedeltchev,b.biswas,h.han,p.k.bhowmik,polymer2012,53,1063;(b)a.k.nedeltchev,h.han,p.k.bhowmik,l.ma,j.polym.sci.parta:polym.chem.2011,49,1907;(c)a.k.nedeltchev,h.han,p.k.bhowmik,l.ma,j.polym.sci.parta:polym.chem.2010,48,4611;(d)a.k.nedeltchev,h.han,p.k.bhowmik,l.ma,j.polym.sci.parta:polym.chem.2010,48,4408;(e)a.k.nedeltchev,h.han,p.k.bhowmik,polym.chem.2010,1,908;(f)p.k.bhowmik,h.han,a.k.nedeltchev,h.d.mandal,j.a.jimenez-hernandez,p.m.mcgannon,j.appl.polym.sci.2010,116,1197;(g)p.k.bhowmik,h.han,a.k.nedeltchev,h.d.mandal,j.a.jimenez-hernandez,p.m.mcgannon,polymer2009,50,3128;(h)p.k.bhowmik,s.kamatam,h.han,a.k.nedeltchev,polymer2008,49,1748;(i)p.k.bhowmik,h.han,a.k.nedeltchev,polymer2006,47,8281;(j)p.k.bhowmik,h.han,a.k.nedeltchev,j.polym.sci.parta:polym.chem.2006,44,1028;(k)p.k.bhowmik,h.han,j.j.cebe,i.k.nedeltchev,s.-w.kang,s.kumar,macromolecules2004,37,2688;(l)p.k.bhowmik,r.a.burchett,h.han,j.j.cebe,polymer2002,43,1953;(m)p.k.bhowmik,r.a.burchett,h.han,j.j.cebe,macromolecules2001,34,7579;(n)p.k.bhowmik,r.a.burchett,h.han,j.j.cebe,j.polym.sci.parta:polym.chem.2001,39,2710.
[13]i.k.spiliopoulos,j.a.mikroyannidis,j.polym.sci.parta:polym.chem.2001,39,2454.
[14](a)f.w.harris,k.c.chuang,s.a.x.huang,j.j.janimak,s.z.d.cheng,polymer1994,35,4940;(b)s.a.x.huang,k.c.chuang,s.z.d.cheng,f.w.harris,polymer2000,41,5001.
[15](a)g.decher,science1997,277,1232;(b)p.bertrand,a.jonas,a.laschewsky,r.legras,macromol.rapidcommun.2000,21,319;(c)x.arys,a.laschewsky,a.m.jonas,macromolecules2001,34,3318;(d)x.arys,p.fischer,a.m.jonas,m.m.koetse,a.laschewsky,r.legras,e.wischerhoff,j.am.chem.soc.2003,125,1859.
[16](a)z.tang,n.a.kotov,s.magonov,b.ozturk,nat.mater.2003,2,413;(b)a.laschewsky,f.mallwitz,j.-f.baussard,d.cochin,p.fischer,j.-l.h.jiwan,e.wischerhoff,macromol.symp.2004,211,135.
[17](a)e.holder,v.marin,a.alexeev,u.s.schubert,j.polym.sci.parta:polym.chem.2005,43,2765;(b)r.dobrawa,f.wurthner,j.polym.sci.parta:polym.chem.2005,43,4981.
[18]y.bar-cohen,“electroactivepolymersasartificialmuscles-realityandchallenges.”proceedingofthe42ndaiaastructures,structuresdynamicsandmaterialsconferences(sdm),gossamerspacecraftforum(gsf),heldinseattle,wa,april16-19,2001.
[19](a)y.w.chen-yang,c.y.yuan,c.h.li,h.c.yang,j.appl.polym.sci.2003,90,1357;(b)w.wu,c.q.yang,j.appl.polym.sci.2003,90,1885;(c)a.rosy,j.polym.sci.parta:polym.chem.1993,31,3187.
[20](a)y.-l.liu,g.-h.hsiue,c.-w.lan,y.-s.chiu,j.polym.sci.parta:polym.chem.1997,35,1769;(b)h-j.kim,j.-k.choi,b.-w.jo,j.-h.chang,r.j.farris,koreapolym.j.1998,6,84;(c)c.tian,h.wang,x.liu,z.ma,h.guo,j.xu,j.appl.polym.sci.2003,89,3137.
[21](a)h.galip,h.
[22](a)q.wu,j.lu,b.qu,polym.int.2003,52,1326;(b)y.-l.liu,g.-h.hsiue,y.-s.chiu,j.polym.sci.parta:polym.chem.1997,35,565.
[23](a)k.g.gravalos,j.polym.sci.parta:polym.chem.1992,30,2521;(b)y.l.liu,y.l.liu,r.j.jeng,y.-s.chiu,j.polym.sci.parta:polym.chem.2001,39,1716;(c)r.p.mateva,n.v.dencheva,j.appl.polym.sci.1993,47,1185.
[24](a)z.ma,w.zhao,y.liu,j.shi,j.appl.polym.sci.1997,63,1511;(b)k.faghihi,m.hajibeygi,j.appl.polym.sci.2004,92,3447.
[25]j.w.connell,k.a.watson,highperform.polym.2001,13,23.
[26](a)j.w.connell,p.m.hergenrother,j.g.smith,jr.,theusaasrepresentedbytheadministratorofthenasa,uspatentspecification5245044,1994;(b)j.w.connell,p.m.hergenrother,j.g.smith,jr.,(theusaasrepresentedbytheadministratorofthenasa)uspatentspecification5317078,1994;(c)j.w.connell,p.m.hergenrother,j.g.smith,jr.,(theusaasrepresentedbytheadministratorofthenasa)uspatentspecification5412059,1995.
[27](a)c.d.smith,h.grubbs,h.f.webster,a.
[28](a)j.w.connell,j.smith,jr.,j.hedrick,polymer1995,36,13;(b)j.lennhoff,g.harris,j.vaughn,d.edwards,j.zwiener,highperform.polym.1999,11,101.
[29](a)d.wilson,h.d.stenzenberg,p.m.hergenrother,polyimides;blackie:glasgow,1990;(b)y.-l.liu,g.-h.hsiue,y.-s.chiu,r.-j.jeng,j.appl.polym.sci.1996,61,1789.(c)p.schuler,h.b.mojazza,r.haghighat,highperform.polym.2000,12,113.
[30](a)m.f.
[31](a)a.k.agrawal,s.a.jenekhe,chem.mater.1996,8,579;(b)c.-j.yang,s.a.jenekhe,macromolecules1995,28,1180.
本发明的各种特点和优势在权利要求书中阐述。
- 还没有人留言评论。精彩留言会获得点赞!