毛细管法制备左旋奥拉西坦晶型I的方法与流程
本发明涉及左旋奥拉西坦,具体涉及一种左旋奥拉西坦晶型i的制备方法。
背景技术:
左旋奥拉西坦(oxiracetam),化学名为(s)-4-羟基-2-氧代-1-吡咯烷乙酰胺,是新一代脑代谢改善药,吡咯烷酮类(环gabob)衍生物,吡拉西坦类似物,可促进磷酰胆碱和邻酰乙醇胺合成,促进脑代谢,通过血脑屏障对特异性中枢神经通路有刺激作用,改善智力和记忆。对脑血管病、脑损伤、脑瘤(术后)、颅内感染、痴呆、脑变性疾病等具有良好疗效。适用于轻中度血管性痴呆、老年性痴呆以及脑外伤等症引起的记忆与智能障碍。(s)-4-羟基-2-氧代-1-吡咯烷乙酰胺由意大利史克比切姆公司于1974年首次合成,1987年上市,对记忆尤其是思维的集中比吡拉西坦更好,且毒性较小,研究表明其左旋体的疗效更好,(s)-4-羟基-2-氧代-1-吡咯烷乙酰胺结构如下:
为有效将(s)-4-羟基-2氧代-1-吡咯烷乙酰胺开发成药品,需要一种具有易于制造并且可接受的化学和物理稳定性的固态形式,以促进其加工与流通储存。对于增强化合物的纯度和稳定性而言,结晶固体形态一般优于非晶型形态。目前公开的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型有i、ii、iii三种晶型,其中晶型i具有较好的稳定性。cn102249975a公开了一种(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i及其制备方法,(s)-4-羟基-2氧代-1-吡咯烷乙酰胺经过水与丙酮二次结晶后制得晶型i,该晶型在12.500,13.940,15.000,16.540,17.400,19.320,20.520,20.840,21.980,23.340,25.120,25.840,26.240,27.660,28.100,30.040,30.660,31.040,31.780,34.300,35.180,37.060,38.020和42.240度的2θ角下有特征峰,依照该专利的制备方法得到的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺i,晶型纯度(化学纯度)99.3%,但是其光学纯度在98-99%左右。为了满足医药工业的需要,需要开发一种制备更高光学纯度(s)-4-羟基-2氧代-1-吡咯烷乙酰胺i的方法。
技术实现要素:
本发明的目的是提供一种(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的制备方法,该方法制备工艺简单,制得的产品手性纯度高。
本发明所涉及的技术方案如下:
一种(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的制备方法,其特征在于,采用以下步骤:
用有机溶剂将(s)-4-羟基-2氧代-1-吡咯烷乙酰胺溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为35~60℃,湿度为rh(%)为30%~50%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i;所述有机溶剂为乙酸乙酯、异丙醇、环己烷或正丁醇。
本发明制备得到的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i在衍射角度2θ为12.500,13.940,15.000,16.540,17.400,19.320,20.520,20.840,21.980,23.340,25.120,25.840,26.240,27.660,28.100,30.040,30.660,31.040,31.780,34.300,35.180,37.060,38.020和42.240度处有衍射峰,与cn102249975a披露的晶型一致。
本发明开辟了一条全新的培养(s)-4-羟基-2氧代-1-吡咯烷乙酰胺单晶的路线,以毛细管法在特定的溶剂与温湿度的配合下,从而成功的制得了(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i,大大推动了(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型的科学研究及应用。
为了进一步提高本发明的收率与纯度,优选空气中挥发的温度为40~50℃,湿度为rh(%)为30%~40%。
为了进一步提高本发明的纯度,本发明原料(s)-4-羟基-2氧代-1-吡咯烷乙酰胺的纯度大于99.5%。
为了保证原料的纯度,上述(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的制备方法,其特征在于,先制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺,然后通过毛细管法培养(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i,反应路线为:
采用以下步骤:
1)、先用甲醇将s-4-氯-3-羟基丁酸乙酯溶解,然后在40~60℃下加入叠氮化钠反应4~6小时得到中间体i,其中s-4-氯-3-羟基丁酸乙酯与叠氮化钠摩尔比为1:1~2;
2)用dmso将中间体i溶解,以金属钯为催化剂与氢气进行还原反应得到中间体ii,反应温度为40~50℃,反应时间为6~8小时;
3)用dmso将中间体ii溶解,碳酸钾为催化剂,与溴乙酸叔丁酯反应6~8小时,反应温度为40~60℃;所述中间体ii与溴乙酸叔丁酯的摩尔比为:1:1~2,中间体ii与所述碳酸钾的摩尔比为:1:2~3;
4)用叔丁醇将中间体ⅲ溶解,在80~100℃条件下进行关环反应得到中间体iv,反应时间为7~9小时;
5)将中间体iv与氨水在20~30℃下反应10~15小时,得到(s)-4-羟基-2氧代-1-吡咯烷乙酰胺粗品,所述中间体iv:氨的摩尔比为中间体iv:氨=1:12~15,以氨气甲醇溶液中的氨计,所述氨水的浓度为25-28%;
6)将(s)-4-羟基-2氧代-1-吡咯烷乙酰胺粗品加热溶解于水中,活性炭脱色,过滤除去活性炭,减压浓缩除水,当剩余水量为加入产物重量2~3倍时停止浓缩,0~5℃低温冷却结晶,获得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺;所述浓氨水的浓度为25-28%;
7)用有机溶剂将(s)-4-羟基-2氧代-1-吡咯烷乙酰胺溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为40~50℃,湿度为rh(%)为30%~40%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i;所述有机溶剂为乙酸乙酯、异丙醇、环己烷或正丁醇。
本发明采用s-4-氯-3-羟基丁酸酯和叠氮化钠为起始原料制备(s)-4-羟基-2氧代-1-吡咯烷乙酰胺,线路简单,中间体及产物均不需要柱层析,制备过程中不产生难以去除的杂质
附图说明
图1是(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的粉末衍射图;
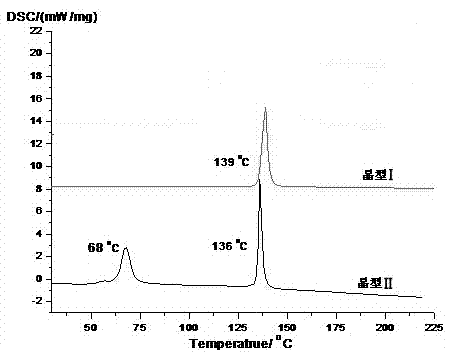
图2是(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i与晶型ii的差示扫描热分析图(dsc);
图3是(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的拉曼光谱图;
图4是(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的热重分析图;
图5是(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的圆二色谱图。
具体实施方式
下面通过实施例对本发明进行具体的描述,有必要在此指出的是以下实施例只用于对本发明进行进一步说明,不能理解为对本发明保护范围的限制,该领域的技术人员可以根据上述本发明内容对本发明作出一些非本质的改进和调整。
实施例1
用乙酸乙酯(2ml)将纯度大于99.5%的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺(20mg)溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为40℃,湿度为rh(%)为30%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型。
将制得的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型经过x射线粉末衍射试验,结果如图1,衍射峰解析如下表:
各种晶型的粉末衍射峰
得到的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型在衍射角度2θ为12.500,13.940,15.000,16.540,17.400,19.320,20.520,20.840,21.980,23.340,25.120,25.840,26.240,27.660,28.100,30.040,30.660,31.040,31.780,34.300,35.180,37.060,38.020和42.240度处有衍射峰,与cn102249975a披露的晶型i一致。
得到的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i产生的红外光谱在以下波数显示出吸收峰:
羟基νo-h:(3403cm-1)、酰胺νn-h(3355cm-1、3184cm-1)、亚甲基νc-h(2926cm-1、2881cm-1)、羰基νc=o(1672cm-1)、δch2(剪式)(1489cm-1)、羟基δo-h(面内)(1399cm-1)、伯酰胺δn-h(1307cm-1)、δc-o(1082cm-1)、伯酰胺δn-h(面外)(672cm-1)。
将实施例1制得的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i进行光学纯度测定:
取(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i,精密称取数量(相当于含(s)-4-羟基-2氧代-1-吡咯烷乙酰胺120mg)置100ml量瓶,加流动相超声溶解并定容制成每1ml中含(s)-4-羟基-2氧代-1-吡咯烷乙酰胺1.2mg的溶液,作为供试品溶液。
精密量取该溶液20ul,注入液相色谱仪,记录色谱图,按面积归一化法计算未知杂质的含量。
测定所用的色谱条件为:
仪器:岛津lc-2010aht高效液相色谱仪;
工作站名称:lc-solutio;
色谱柱:agop(4.6×100mm,5μm);
流动相:乙腈:磷酸缓冲液(ph6.0)=15:85;
检测波长:210nm;
流速:1ml/min;
柱温35℃;
计算公式如下:
公式中,ai为主药活性成分(s)-4-羟基-2氧代-1-吡咯烷乙酰胺的峰面积;
σa为s-构型和r-异构体4-羟基-2氧代-1-吡咯烷乙酰胺的峰面积之和。
经三次测量,取平均值,得实施例1的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的光学纯度为99.96%。
对于使用毛细管法进行(s)-4-羟基-2氧代-1-吡咯烷乙酰胺的晶型培养而言,不同的溶剂,挥发温度以及空气的湿度等条件均可能导致不同的结果。如下面的对比例1-3,空气湿度过大则无法得到单一晶型,湿度较小为(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i,挥发温度与湿度不匹配可能会得到晶型i和晶型ii的混合物。
对比例1
用乙酸乙酯(2ml)将纯度大于99.5%的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺(20mg)溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为40℃,湿度为rh(%)为90%的空气中进行挥发,结果无法制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺单一晶型。
对比例2
用乙酸乙酯(2ml)将纯度大于99.5%的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺(20mg)溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为40℃,湿度为rh(%)为70%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型,该晶型在衍射角度2θ为10.669、13.25、13.847、14.198、16.729、17.934、18.746、18.816、20.273、20.413、21.431、21.617、21.663、23.38、24.324、24.415、26.069、26.107、27.901、28.621、28.925、29.449、29.484、31.702、36.516、37.685、39.721度处有衍射峰,与cn102558013a披露的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型ii一致。
对比例3
用乙酸乙酯(2ml)将纯度大于99.5%的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺(20mg)溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为80℃,湿度为rh(%)为70%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型,经过结构鉴定,为(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i与晶型ii的混合物。
实施例2
(l)中间体i的制备:
取原料s-4-氯-3-羟基丁酸乙酯5g,加入一单颈瓶中,加入甲醇10ml,加入叠氮化钠5g搅拌,保持温度50℃左右反应4小时,停止反应得黄色溶液。加入水20ml,用乙酸乙酯20ml萃取,浓缩除去乙酸乙酯,得到黄色油状物中间体i。经核磁检测,中间体i为:1h-nmr(300mhz,cdcl3):δ1.42-1.73(m,2h)2.76-2.67(absystem,m,2h,),3.31-3.23(absystem,m,2h),3.75(s,3h),4.40(m,1h),3.70(s,1h).
中间体i为:
(2)中间体ii的制备
将步骤(1)获得的中间体i溶解于50ml的dmso中,加入10%钯碳催化剂1.2g,通入氢气在45℃左右搅拌6小时,点板见原料反应完全,停止反应,浓缩除去溶剂得到淡黄色油状物中间体ii。经核磁检测,中间体ii:1h-nmr(300mhz,d2o):1.30(m,3h),2.76-2.67(absystem,m,2h,),3.31-3.23(absystem,m,2h),4.12(m,2h),4.40(m,1h),4.70(bs,3h)。
中间体ii为
(3)中间体ⅲ的制备
将步骤(2)获得的中间体ii溶解于50ml的dmso中,在40℃左右,加入碳酸钾17.3g(3eq),有大量固体生成,搅拌五分钟,开始滴加溴乙酸叔丁酯9ml(2eq),滴加过程有放热现象,滴加完毕后继续搅拌6小时,点板见原料反应完全,停止反应,加入ea(乙酸乙酯)50ml,水30ml,固体完全溶解,将水层用氯化钠固体饱和,分出有机层,水层用ea20ml萃取两次,合并有机层,有机层用2m的盐酸20ml洗三次,合并盐酸水相,有机相弃去,水相用碳酸氢钠调节ph至8,固体氯化钠饱和,ea30ml萃取三次,合并有机相,无水硫酸镁干燥,浓缩除去溶剂得到淡黄色油状物,低温下固化得到中间体ⅲ。经核磁检测,中间体ⅲ:1h-nmr(300mhz,d2o):1.28-1.4(m,12h),2.28-2.53(m,2h),2.58-2.83(m,2h)3.51(s,2h),4.09-4.12(m,3h)。
中间体ⅲ为
(4)中间体iv的制备
将步骤(2)获得的中间体ⅲ用50ml叔丁醇溶解,升温至85℃反应8小时,得到一红褐色溶液,点板见原料反应完全。停止反应,浓缩除去甲苯,加入ea(乙酸乙酯)溶解,过滤除去盐,活性炭脱色,浓缩除去得黄色油状物得到中间体iv。经核磁检测,中间体iv为:1h-nmr(300mhz,cdcl3)1.402(m,9h),2.38(dd,1h),2.69(dd,1h),3.34(dd,1h),3.77(dd,lh),3.93(d,lh),4.18(d,1h),4.30(bs,1h),4.50(m,1h)。
中间体iv:
(5)(s)-4-羟基-2氧代-1-吡咯烷乙酰胺的制备
将步骤(4)获得的中间体iv加入浓氨水(浓度为26%)20ml,室温搅拌14小时,点板见原料反应完全,停止反应,浓缩去除水和氨气,得到黄色油状物,加入丙酮溶解油状物,加入少量晶种搅拌,析出固体,少量丙酮冲洗瓶壁,-10℃结晶5小时,过滤得到类白色粗品23.5g,化学纯度99.0%。
(6)将该粗品溶解于100ml的水中,加热使其溶解,活性炭脱色半小时,过滤除去活性炭,冷却结晶,5℃放置过夜,次日过滤得白色固体21.8g,化学纯度99.6%,产物中不含有
(s)-4-羟基-2氧代-1-吡咯烷乙酰胺结构式如下:
(7)(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型i的制备:
用环己烷(5ml)将步骤(6)制得的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺(70mg)溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为40℃,湿度为rh(%)为45%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型,经过x粉末衍射验证,与实施例1制得的晶型i相同,且光学纯度为99.90%。
参照实施例1制得实施例3-5,部分参数按照以下运行:
实施例3-5制得的(s)-奥拉西坦化学纯度高,其中不含有
实施例6
用乙酸乙酯(5ml)将纯度大于99.6%的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺(100mg)溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为45℃,湿度为rh(%)为35%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型,用x粉末衍射法验证,制得的晶型与实施例1的晶型i相同,且光学纯度为99.93%。
实施例7
用异丙醇(10ml)将纯度为99.5%的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺(300mg)加热溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为35℃,湿度为rh(%)为30%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型,用x粉末衍射法验证,制得的晶型与实施例1的晶型i相同,且光学纯度为99.95%。
实施例8
用环己烷(10ml)将99.5%的(s)-4-羟基-2氧代-1-吡咯烷乙酰胺(80mg)溶解,然后通过虹吸吸入毛细管当中,用蜡封住毛细管的一端,放置于温度为60℃,湿度为rh(%)为40%的空气中进行挥发,制得(s)-4-羟基-2氧代-1-吡咯烷乙酰胺晶型,用x粉末衍射法验证,制得的晶型与实施例1的晶型i相同,且光学纯度为99.90%。
- 还没有人留言评论。精彩留言会获得点赞!