一种枸橼酸托法替尼的合成方法与流程
本发明属于有机及药物合成技术领域,具体涉及一种枸橼酸托法替尼的合成方法。
背景技术:
枸橼酸托法替尼(tofacitinib citrate),化学名为3-[(3R,4R)-4-甲基-3-[甲基-(7H-吡咯并[2,3-d]嘧啶-4-基)氨基]哌啶-1-基]-3-氧代丙腈枸橼酸盐,是辉瑞公司研发的一种新型口服Janus激酶(JAK)抑制剂,2012年11月经美国FDA通过危险评估和减轻策略(REMS)批准,用于治疗成人活动期及对甲氨蝶呤反应不佳的中至重度类风湿性关节炎(RA)患者批准上市,商品名Xeljanz。还适用于银屑病、胰腺病、白血病、骨髓增生异常综合征、强直性脊柱炎、移植排斥的治疗。
枸橼酸托法替尼的化合物专利(WO2001042246)于2000.11.23申请,其中公布的合成路线如下:
该化合物专利路线,工艺稳定,原料及试剂廉价易得,是枸橼酸托法替尼合成工艺的基本路线,很多相关研究者对其工艺进行了各种改进,但仍存在一些缺陷,例如:原料5的脱苄基反应基本采用原研方法,该方法的反应时间长,而且氢气易燃易爆,存在安全隐患。CN105348287公布了利用Pd/C、HCOONH4脱去该苄基的方法,反应时间大大缩短,但在放大生产的时候仍存在甲酸铵易升华堵塞冷凝管的安全隐患,且甲酸铵不除去会在下一步的成酰胺反应中引入副产物。
中间体3的酰胺化反应,多采用氰基乙酸乙酯、氰基乙酸、氰基乙酸-N-羟基丁二酰亚胺酯等,但氰基乙酸-N-羟基丁二酰亚胺酯成本较高,氰基乙酸乙酯的反应存在不完全或副反应较多的缺陷,缩合剂或碱多采用DBU、HOBT、三乙胺等,普遍存在反应时间长(10h~48h)、反应不完全、产率低的问题。CN105085527公布了氰基乙酸在DCC缩合下反应,反应很彻底,基本无副反应,但DCC的副产物DCU在后处理的过程中难以被除尽,使得该方法存在一定局限性。
因此,开发新的合成枸橼酸托法替尼的工艺具有重要应用价值。
技术实现要素:
针对上述现有技术的不足,本发明的目的是提供一种新的枸橼酸托法替尼的合成方法,无氢气、甲酸铵带来的安全隐患,脱苄基反应和酰胺化反应皆干净彻底,基本无副反应,反应时间大大缩短,产率高,后处理简便。
为实现上述目的,本发明采用下述技术方案:
本发明第一方面提供一种枸橼酸托法替尼的合成方法,步骤如下:
(1)将式5所示的化合物溶于质子型溶剂中,加入Pd/C、HCOOH进行反应,反应温度50~70℃,反应时间1~3h,得式4所示的化合物;
(2)将式4所示的化合物与碳酸钾水溶液反应,经萃取、有机相减压浓缩,得式3所示的化合物;
(3)将式3所示的化合物在非质子型溶剂中与氰基乙酸反应,加入催化剂进行催化,反应温度为25~110℃,反应时间2~4h;然后与枸橼酸进行成盐反应,即得枸橼酸托法替尼。
上述方法,步骤(1)中,所述质子型溶剂选自甲醇或乙醇;质子型溶剂的用量为式5所示化合物的5-15倍重量。
上述方法,步骤(1)中,式5所示的化合物与Pd/C加入的质量比为1:0.1~0.3,Pd/C中钯的质量分数为5-20%;优选的,Pd/C中的钯的质量分数为10%(含水50%),式5所示的化合物与Pd/C加入的质量比为1:0.2。
上述方法,步骤(1)中,式5所示的化合物与甲酸加入的摩尔比为1:(1~6);优选为1:4。
上述方法,步骤(1)中,所述反应温度优选为65℃,反应时间优选为2h。
上述方法,步骤(2)中,所述碳酸钾水溶液的质量浓度为20%~50%,用量为式4所示化合物的3-6倍重量。
上述方法,步骤(2)中,所述有机相选自乙酸乙酯、二氯甲烷或正丁醇;优选为二氯甲烷。
上述方法,步骤(3)中,式3所示的化合物与氰基乙酸加入的摩尔比为1:(1.2~3);优选为1:1.5。
上述方法,步骤(3)中,所述催化剂选自1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)或EDCI、1-羟基苯并三氮唑(HOBT)、三乙胺组成的复合体系;优选的,所述催化剂为EDCI、HOBT、三乙胺组成的复合体系。
上述方法,步骤(3)中,催化剂为EDCI时,式3所示的化合物与EDCI的摩尔比为1:(2~6)。
催化剂为EDCI、HOBT、三乙胺组成的复合体系时,式3所示的化合物与EDCI、HOBT与三乙胺的摩尔比为1:(1~2):(1~2):(2~6);优选为1:1.2:1.2:3。
上述方法,步骤(3)中,所述非质子型溶剂选自四氢呋喃、二氯甲烷或甲苯;优选为四氢呋喃。用量为式3所示化合物的25-35重量倍。
上述方法,步骤(3)中,式3所示的化合物与枸橼酸的摩尔比为1:(1~1.5)(优选为1:1);成盐温度为25~50℃(优选为40℃),时间为1~4h(优选为2h)。
上述方法,步骤(3)中,所述成盐反应的具体方法为:催化反应完毕,水洗并除去溶剂,然后在丙酮中与枸橼酸成盐,经乙醇水溶液重结晶,得到白色的枸橼酸托法替尼。
丙酮的用量为式3所示化合物的10-20倍重量;优选为12倍重量。
所述乙醇水溶液为体积分数35%的乙醇水溶液。
本发明所述式5所示的化合物可以按在本发明申请日之前公开的任何现有技术制备,比如采用专利(WO2001042246)公开的方法制备。
本发明枸橼酸托法替尼具体的合成路线如下:
需要说明的是,对于一个有机化学全合成路线而言,其评价指标主要包括:起始原料是否适宜,步骤路线是否简短易行,总收率高低,反应的安全性,以及合成的选择性高低等。在有机化学合成反应中,提高收率、提高操作效率以及实现工业化生产,是进行步骤工艺优化的主要目标。与步骤工艺优化密切相关的因素有两类,一是有关体系组成,如反应物当量、溶剂、浓度等,这类参量决定了反应体系的物性,在放大时易于重现;二是有关操作参量,如温度、反应时间、反应物加入顺序等。本发明针对枸橼酸托法替尼现有的合成路线中存在的问题,对反应的原料、反应的参数条件等进行了优化,提高了合成反应的收率和操作效率,合成反应的条件温和,安全性高,有利于实现工业化生产。
本发明第二方面提供一种用于合成枸橼酸托法替尼的中间体,其结构式如下:
本发明的有益效果:
(1)本发明的合成方法中,以N-[(3R,4R)-1-苄基-4-甲基哌啶-3-基]-N-甲基-7H-吡咯并[2,3-d]嘧啶-4-胺为原料,利用Pd/C、HCOOH进行催化,得到新的中间体化合物(式4所示),然后再与碳酸钾水溶液反应,得到式3所示的化合物,实现对原料的脱苄基反应。消除了现有的脱苄基反应中,由于氢气或甲酸铵的参与而带来的安全隐患(氢气易燃易爆,甲酸铵容易堵塞冷凝管),而且反应时间由现有的8-10小时缩短为1-2小时,大大提高了生产效率;采用本发明的合成方法,对于式3所示化合物的产率达90%以上,适合用于工业化生产。
(2)本发明的合成方法中,由式3所示的化合物制备枸橼酸托法替尼,提高了反应的收率,大大缩短了反应时间,由10~48h缩短为2-4h,反应干净彻底,简化了后处理纯化过程,更适用于工业化生产。
附图说明
图1:式4所示化合物的氢谱图;
图2:式3所示化合物的氢谱图;
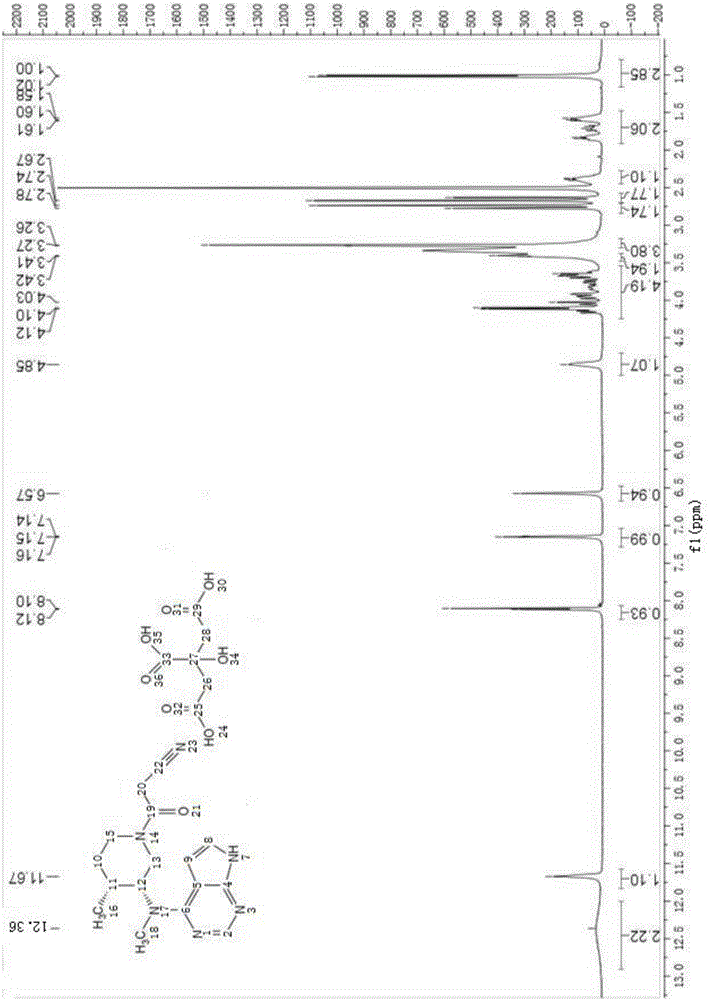
图3:枸橼酸托法替尼的氢谱图;
图4:枸橼酸托法替尼的液相图。
具体实施方式
结合实施例对本发明作进一步的说明,应该说明的是,下述实施例仅是为了解释本发明,并不对其内容进行限定。
实施例1:枸橼酸托法替尼的合成
合成路线为:
具体合成步骤如下:
(1)中间体4(式4所示的化合物)的制备
往500ml反应瓶中加入15g(45mmol)化合物5,加200ml无水甲醇,常温搅拌下分批加入3g10%Pd/C(含水50%),滴加8.3g(180mmol)甲酸,70℃加热回流2h,TLC监测反应完毕,冷却,用硅藻土滤除Pd/C,减压浓缩至干得13g白色固体4,产率100%。
(2)中间体3(式3所示的化合物)的制备
往500ml反应瓶中加入13g(45mmol)中间体4加水50ml,加37.26g碳酸钾,加100ml二氯甲烷,常温搅拌1h,水层再用二氯甲烷(100ml×3)萃取,合并有机相,减压浓缩得9.9g白色固体3,产率91%。
(3)枸橼酸托法替尼的制备
往500ml反应瓶中加入10g(41mmol)中间体3,加入250ml四氢呋喃,搅拌使溶解,加入氰基乙酸5.3g(62mmol),加入12.4g(123mmol)三乙胺,加入6.6g(49mmol)HOBT,最后加入9.4g(49mmol)EDCI,70℃加热回流2h,TLC监测反应完毕,减压浓缩,加入200ml二氯甲烷和200ml水,常温搅拌0.5h,有机相再用100ml水洗,再用100ml饱和食盐水洗,有机相减压浓缩得淡黄色托法替尼粗品。用150ml丙酮溶解,搅拌,加入7.9g(41mmol)枸橼酸,40℃反应2h,冷却。过滤,滤饼用丙酮(20ml×3)淋洗,干燥得近白色固体,经35%乙醇水溶液重结晶得19.0g白色的枸橼酸托法替尼固体粉末,产率92%,纯度99.5%。[HPLC归一化法:色谱柱Eclipse XDB-C18柱(4.6mm×250mm,5μm);流动相50mmol/L乙酸铵水溶液∶乙腈(3:1);检测波长286nm;柱温25℃;流速1.0ml/min]。
实施例2:中间体4(式4所示的化合物)的制备
往500ml反应瓶中加入15g(45mmol)化合物5,加200ml无水甲醇,常温搅拌下分批加入1.5g10%Pd/C(含水50%),滴加8.3g(180mmol)甲酸,70℃加热回流10h,TLC监测反应完毕,冷却,用硅藻土滤除Pd/C,减压浓缩至干得12.3g白色固体4,产率95%。
实施例3:中间体4(式4所示的化合物)的制备
往500ml反应瓶中加入15g(45mmol)化合物5,加200ml无水甲醇,常温搅拌下分批加入4.5g10%Pd/C(含水50%),滴加8.3g(180mmol)甲酸,70℃加热回流2h,TLC监测反应完毕,冷却,用硅藻土滤除Pd/C,减压浓缩至干得12.9g白色固体4,产率99%。
实施例4:中间体4(式4所示的化合物)的制备
往500ml反应瓶中加入15g(45mmol)化合物5,加200ml无水甲醇,常温搅拌下分批加入3g10%Pd/C(含水50%),滴加4.2g(90mmol)甲酸,70℃加热回流8h,TLC监测反应完毕,冷却,用硅藻土滤除Pd/C,减压浓缩至干得12.5g白色固体4,产率96%。
实施例5:中间体3(式3所示的化合物)的制备
往500ml反应瓶中加入13g(45mmol)中间体4加水50ml,加37.26g碳酸钾,加100ml二氯甲烷,常温搅拌1h,水层再用乙酸乙酯(100ml×3)萃取,合并有机相,减压浓缩得8.6g白色固体3,产率79%。
实施例6:中间体3(式3所示的化合物)的制备
往500ml反应瓶中加入13g(45mmol)中间体4加水50ml,加37.26g碳酸钾,加100ml二氯甲烷,常温搅拌1h,水层再用正丁醇(100ml×3)萃取,合并有机相,减压浓缩得9.2g白色固体3,产率85%。
实施例7:中间体3(式3所示的化合物)的制备
往500ml反应瓶中加入13g(45mmol)中间体4加水100ml,加37.26g碳酸钾,加100ml二氯甲烷,常温搅拌1h,水层再用二氯甲烷(100ml×3)萃取,合并有机相,减压浓缩得7.1g白色固体3,产率66%。
实施例8:枸橼酸托法替尼的制备
往500ml反应瓶中加入10g(41mmol)中间体3,加入250ml四氢呋喃,搅拌使溶解,加入氰基乙酸5.3g(62mmol),加入31.5g(164mmol)EDCI,70℃加热回流2h,TLC监测反应完毕,减压浓缩,加入200ml二氯甲烷和200ml水,常温搅拌0.5h,有机相再用100ml水洗,再用100ml饱和食盐水洗,有机相减压浓缩得淡黄色托法替尼粗品。用150ml丙酮溶解,搅拌,加入7.9g(41mmol)枸橼酸,40℃反应2h,冷却。过滤,滤饼用丙酮(20ml×3)淋洗,干燥得近白色固体,经35%乙醇水溶液重结晶得18.3g白色的枸橼酸托法替尼固体粉末,产率88.6%,纯度98.9%。[HPLC归一化法:色谱柱Eclipse XDB-C18柱(4.6mm×250mm,5μm);流动相50mmol/L乙酸铵水溶液∶乙腈(3:1);检测波长286nm;柱温25℃;流速1.0ml/min]。
实施例9:枸橼酸托法替尼的制备
往500ml反应瓶中加入10g(41mmol)中间体3,加入250ml二氯甲烷,搅拌使溶解,加入氰基乙酸5.3g(62mmol),加入12.4g(123mmol)三乙胺,加入6.6g(49mmol)HOBT,最后加入9.4g(49mmol)EDCI,70℃加热回流4h,TLC监测反应完毕,减压浓缩,加入200ml二氯甲烷和200ml水,常温搅拌0.5h,有机相再用100ml水洗,再用100ml饱和食盐水洗,有机相减压浓缩得淡黄色托法替尼粗品。用150ml丙酮溶解,搅拌,加入7.9g(41mmol)枸橼酸,40℃反应2h,冷却。过滤,滤饼用丙酮(20ml×3)淋洗,干燥得近白色固体,经35%乙醇水溶液重结晶得17.6g白色的枸橼酸托法替尼固体粉末,产率85%,纯度99.1%。[HPLC归一化法:色谱柱Eclipse XDB-C18柱(4.6mm×250mm,5μm);流动相50mmol/L乙酸铵水溶液∶乙腈(3:1);检测波长286nm;柱温25℃;流速1.0ml/min]。
实施例10:枸橼酸托法替尼的制备
往500ml反应瓶中加入10g(41mmol)中间体3,加入250ml甲苯,搅拌使溶解,加入氰基乙酸5.3g(62mmol),加入12.4g(123mmol)三乙胺,加入6.6g(49mmol)HOBT,最后加入9.4g(49mmol)EDCI,70℃加热回流4h,TLC监测反应完毕,减压浓缩,加入200ml二氯甲烷和200ml水,常温搅拌0.5h,有机相再用100ml水洗,再用100ml饱和食盐水洗,有机相减压浓缩得淡黄色托法替尼粗品。用150ml丙酮溶解,搅拌,加入7.9g(41mmol)枸橼酸,40℃反应2h,冷却。过滤,滤饼用丙酮(20ml×3)淋洗,干燥得近白色固体,经35%乙醇水溶液重结晶得18.4g白色的枸橼酸托法替尼固体粉末,产率89%,纯度98.7%。[HPLC归一化法:色谱柱Eclipse XDB-C18柱(4.6mm×250mm,5μm);流动相50mmol/L乙酸铵水溶液∶乙腈(3:1);检测波长286nm;柱温25℃;流速1.0ml/min]。
- 还没有人留言评论。精彩留言会获得点赞!