依鲁替尼药物杂质的制备方法与流程
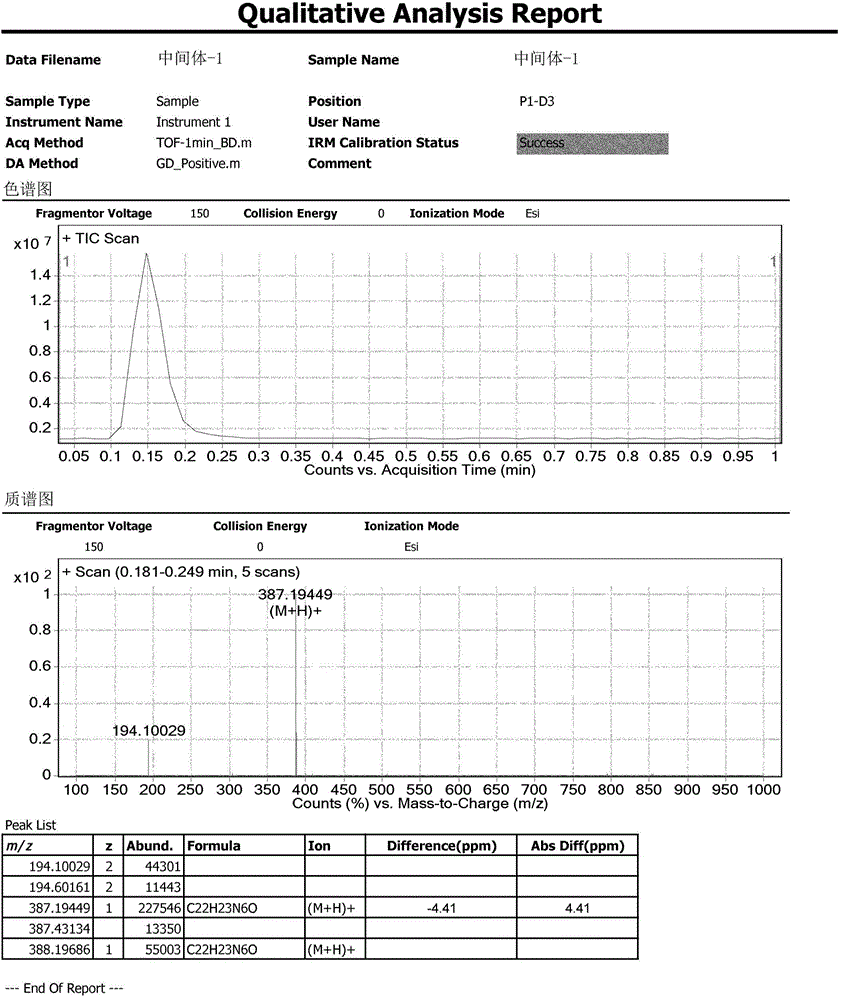
本发明属于药物合成领域,涉及原料药依鲁替尼生产过程中工艺杂质及其制备。特别涉及依鲁替尼工艺杂质及其制备方法。
背景技术:
:Ibrutinib由美国CeleraGenomics公司于2007年首先研制,后转让给加州的Pharmacyclics公司开发,2011年强生公司的子公司杨森制药(Jassen)参与合作开发。目前尚无标准的中文译名,因此本申请人在此将其音译为“依鲁替尼”。该药已于2013年11月获FDA批准,用于既往接受过至少一次来那度胺或其他药物治疗的套细胞淋巴瘤(MCL)患者的治疗。依鲁替尼是首个每日一次、单一制剂、口服布鲁顿酪氨酸激酶(BTK)抑制剂。2014年2月12日依鲁替尼被FDA批准增加适应症,用于治疗慢性淋巴细胞白血病(CLL)。2014年7月28日,FDA再次宣布,扩大批准依鲁替尼适应症,将其用于染色体17P缺失的慢性淋巴细胞白血病(CLL)患者。2015年1月29日,依鲁替尼的适应症扩展至治疗Waldenstrom巨球蛋白血症,在突破性治疗中,该药获得扩展批准,这是首例唯一一例经FDA批准的Waldenstrom巨球蛋白血症治疗。2014年10月,依鲁替尼被欧洲药物管理局(EMA)批准上市,用于治疗套细胞淋巴瘤(MCL)以及慢性淋巴细胞白血病(CLL)。依鲁替尼是布鲁顿酪氨酸激酶(BTK)小分子抑制剂,与BTK活性位点的半胱氨酸残基形成共价键,导致BTK酶活性抑制。BTK是B-细胞抗原受体(BCR)和细胞因子受体通路的信号分子。BTK的作用是通过对B-细胞表面受体发出信号指令,从而激活B-细胞运输,趋化,和黏附所需通路。非临床研究显示依鲁替尼抑制恶性B-细胞增殖和体内生存,同时也抑制体外细胞迁移和底物黏附。复发性B-细胞淋巴瘤患者服用依鲁替尼,剂量≥2.5mg/kg/day(以人均体重70公斤计,人均日剂量服用依鲁替尼大于175 mg/day),24小时后观察到外周血液单核细胞BTK活性位点超过90%被占领。依鲁替尼的化学名称为:1-[(3R)-3-[4-氨基-3-(4-苯氧基苯基)-1H吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮,结构式为:现有技术披露了七种工艺杂质或降解杂质,它们在合成依鲁替尼的过程中获得,然而FDA审批报告只报道了手性异构体的杂质结构和体内代谢物(PCI-45227)的结构,对其它杂质的结构均未被描述。只有一篇文献(专利WO2013/184572,第90-91页)中提到了依鲁替尼A晶型纯度检测的方法,该方法用以0.1%三氟乙酸水溶液为缓冲液(A),以0.1%三氟乙酸的乙腈为有机溶剂(B),色谱柱为Gemini-NXC18,4.6×150mm,3μm,梯度洗脱的HPLC方法,该方法并没有对制备工艺中特定杂质进行分离。依鲁替尼的公斤级合成路线在中国专利申请号201510165869.6中报道,该路线的策略为:以已经商品化、工艺路线成熟的3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-4-胺(SM1)和(S)-1-叔丁氧羰基-3-羟基哌啶(SM2)为起始原料,在三苯基磷和偶氮二甲酸二异丙酯的作用下,在四氢呋喃溶液体系中进行Mitsunobu反应,反应结束,直接在浓盐酸的作用下脱保护,再经氢氧化钠中和析晶,得到中间体-1:(R)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1H-吡唑并[3,4-d]嘧啶-4-胺。该中间体在二氯甲烷溶剂中,以N,N-二异丙基乙胺作碱,与丙烯酸酐缩合进行酰胺化反应,经过乙醇/水体系重结晶制得成品依鲁替尼。上述工艺路线与其他路线相比具有如下优点:(1)两步反应的总收率高于其他报道的收率。(2)中间体-1及依鲁替尼的提纯方法均为重结晶,使依鲁替尼的工业化生产成为可能;(3)在Mitsunobu反应中使用廉价的三苯基磷代替聚合物连接的三苯基磷,降低了工业化生产成本;(4)在中间体-1的生产中采用“一锅法”,将Mitsunobu反应与脱保护反应合并,简化了工艺操作。(5)将酰胺化试剂由丙烯酰氯改为丙烯酸酐,避免了难于通过重结晶去除的杂质生成。该路线是具有工业化生产价值的合成策略。目前,尚无对这条生产路线中产生的工艺杂质的报道。技术实现要素:杂质研究是药品研发的重要内容,贯穿于药品研发的始终,直接影响药品的安全性、有效性以及质量可控性。为了给依鲁替尼的质量研究提供有关物质对照品,提高依鲁替尼的质量标准,为依鲁替尼的安全用药提供重要的指导,本发明通过对依鲁替尼工艺研究过程取样进行HPLC和LC-MS分析,研究、合成并确证了依鲁替尼生产路线中的工艺杂质。中国专利申请号201510165869.6报道的依鲁替尼的合成方法以及市售的依鲁替尼胶囊(Imbruvica购自PharmacyclicsJanssen公司,140mg, 批号:L0404951)中,会涉及一系列的工艺杂质,经发明人确认其化学名称和结构式见下表以上杂质的产生途径如下:杂质A:本杂质为市售胶囊剂中杂质,应该为原研工艺杂质,在本工艺多批次中从未检出。杂质B:在中国专利申请号201510165869.6报道的依鲁替尼的合成方法第二步酰胺化反应中,酰胺化发生在依鲁替尼中间体-1(表示为YLTN-1)的两个位置,哌啶环中的氮以及嘧啶环上的氮,产生双酰胺化产物杂质B-1,后者由于热力学作用,发生分子内关环,生成更加稳定的双嘧啶环杂质B。杂质C:在中国专利申请号201510165869.6报道的依鲁替尼的合成方法第二步酰胺化反应中,起始原料丙烯酸酐中含有少量乙酸酐杂质,与YLTN-1发生酰胺化反应生成杂质C。杂质D:在中国专利申请号201510165869.6报道的依鲁替尼的合成方法第二步酰胺化反应中,依鲁替尼(YLTN)与副产物丙烯酸在碱的催化下(可能是N,N-二异丙基乙胺、也可能为依鲁替尼自身的含氮杂环)发生Baylis-Hillman反应而产生杂质D。杂质E、F、G为依鲁替尼活性成分或包含依鲁替尼的药物制剂的强制降解样品中的主要降解杂质,其中杂质E为依鲁替尼或依鲁替尼制剂制备过程中氧化降解的主要产物,杂质F、G为依鲁替尼或依鲁替尼制剂制备过程中的降解产物,也是依鲁替尼碱性条件下强制降解或水解的主要降解产物。杂质H:在中国专利申请号201510165869.6报道的依鲁替尼的合成方法第二步酰胺化反应中,起始原料丙烯酸酐含有少量丙酸酐杂质,会与中间体-1发生反应生成杂质H:由此,在一个方面,本发明涉及如下化合物:化合物A,化学名称为:(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-基)-3-氯丙基-1-酮,并具有以下结构:化合物B,化学名称为:(R)-8-(1-丙烯酰基哌啶-3-基)-10-(4-苯氧基苯基)-3,4-二氢吡唑并[4,3-e]嘧啶并[1,2-c]嘧啶-2(8H)-酮,并具有以下结构:化合物C,化学名称为:(R)-1-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-基)-乙酮,并具有以下结构:化合物D,化学名称为:(R)-4-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-羰基)戊烯-4-酸,并具有以下结构:化合物E,化学名称为:1-((R)-1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-氧化物,并具有以下结构:化合物F,化学名称为:1,3-二((R)-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)丙烷-1-酮,并具有以下结构:化合物G,化学名称为:1-((R)-3-(4-((3-((R)-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)-3-氧代丙基)氨)-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)丙基-2-烯-1-酮,并具有以下结构:化合物H,化学名称为:(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-基)-丙基-1-酮,并具有以下结构:在具体的实施方案中,上述化合物A-H为分离形式,优选为基本纯的形式,更优选具有高于约95%的纯度。另一方面,本发明涉及上述化合物A-H的一种或多种在检测依鲁替尼或含有依鲁替尼的药物制剂的样品纯度中作为参比标准或对照品的应用,其中依鲁替尼的化学名称为1-[(3R)-3-[4-氨基-3-(4-苯氧基苯基)-1H吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮,结构式为:附图说明图1:中间体-1:(R)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1H-吡唑并[3,4-d]嘧啶-4-胺的1H-NMR;图2:中间体-1:(R)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1H-吡唑并[3,4-d]嘧啶-4-胺的高分辨质谱;图3:依鲁替尼:1-[(3R)-3-[4-氨基-3-(4-苯氧基苯基)-1H吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮的1H-NMR;图4:依鲁替尼:1-[(3R)-3-[4-氨基-3-(4-苯氧基苯基)-1H吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮的高分辨质谱。图5:杂质A:(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧 啶-1-基]哌啶-1-基)-3-氯丙基-1-酮的1H-NMR;图6:杂质A:(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-基)-3-氯丙基-1-酮的高分辨质谱;图7:杂质B:(R)-8-(1-丙烯酰基哌啶-3-基)-10-(4-苯氧基苯基)-3,4-二氢吡唑并[4,3-e]嘧啶并[1,2-c]嘧啶-2(8H)-酮的1H-NMR;图8:杂质B:(R)-8-(1-丙烯酰基哌啶-3-基)-10-(4-苯氧基苯基)-3,4-二氢吡唑并[4,3-e]嘧啶并[1,2-c]嘧啶-2(8M)-酮的高分辨质谱;图9:杂质C:(R)-1-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-基)-乙酮的1H-NMR;图10:杂质C:(R)-1-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-基)-乙酮的高分辨质谱。图11:杂质D:(R)-4-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-羰基)戊烯-4-酸的1H-NMR;图12:杂质D:(R)-4-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-羰基)戊烯-4-酸的高分辨质谱;图13:杂质H:((R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-基)-丙基-1-酮的1H-NMR;图14:杂质H:(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-基)-丙基-1-酮的高分辨质谱;图15:杂质E:1-((R)-1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-氧化物的1H-NMR。图16:杂质E:1-((R)-1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-氧化物的高分辨质谱。图17:杂质F:1,3-二((R)-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)丙烷-1-酮的1H-NMR。图18:杂质F:1,3-二((R)-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)丙烷-1-酮的高分辨质谱。图19:杂质G:1-((R)-3-(4-((3-((R)-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)-3-氧代丙基)氨)-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)丙基-2-烯-1-酮的1H-NMR。图20:杂质G:1-((R)-3-(4-((3-((R)-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)-3-氧代丙基)氨)-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)丙基-2-烯-1-酮的高分辨质谱;具体实施方式为了进一步对本发明进行说明,下面将通过具体实施例进行说明,但是以下的实施例对本发明的保护范围不构成任何限制。实施例1:中间体-1:(R)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1H-吡唑并[3,4-d]嘧啶-4-胺(YLTN-1)的合成向500mL的反应瓶中加入155mL四氢呋喃,搅拌下依次加入3-(4-苯氧基苯基)-1H-吡唑[3,4-D]嘧啶-4-胺(SM1)(5g,1eq)、(S)-1-叔丁氧羰基-3-羟基哌啶(SM2)(4.97g,1.5eq)、三苯基膦(13g,3.0eq)。控温25℃条件下,在30分钟内,滴加偶氮二甲酸二异丙酯的四氢呋喃溶液(将10g,3.0eq偶氮二甲酸二异丙酯溶于10mL的四氢呋喃中)。滴加完成后,控温25℃,继续反应5小时(TLC监测:乙酸乙酯∶甲醇=10∶1)。搅拌下减压蒸馏。控温15℃,向剩余物中滴加30mL浓盐酸,滴加时间30分钟,滴加完成后,控温25℃,继续反应2小时。用二氯甲烷萃取水相三次(每次75mL),保留水相。用50mL乙酸乙酯萃取一次。剩余水相充分搅拌,控温15℃,滴加约45g20%的氢氧化钠水溶液,使用pH试纸监测反应液,pH=5~6(滴加时间约30分钟)得浅黄色固体。将上述固体通过乙醇重结晶两次,得到类白色粉末2.87g,收率:45%。1H-NMR(400Mz,DMSO-d6)δ:8.226(s,1H),7.656~7.634(m,2H),7.435~7.395(m,2H),7.185~7.094(m,5H),4.689~4.635(m,1H),3.081~3.043(m,1H),2.957~2.930(m,1H),2.901~2.873(m,1H),2.490~2.457(m,1H),2.125~2.015(m,2H),1.750~1.717(m,1H),1.589~1.518(m,1H)(详见图1);ESI-HRMS谱图显示分子离子峰m/z=387.19449[MW+H]+,所对应的分子量与提供的结构式理论计算值(387.19279)相符。绝对误差为4.41ppm,在高分辨质谱误差范围之内。(详见图2)实施例2:依鲁替尼的制备氮气保护下,向100mL三口瓶中依次加入50mL二氯甲烷,中间体-1(5g,1eq),N,N-二异丙基乙胺(2g,1.2eq)。控温-10℃条件下,开始滴加丙烯酸酐(1.96g,1.2eq)的二氯甲烷溶液,滴加时间30分钟,滴加完成后,溶液由浑浊变澄清,保温-10℃,充分搅拌至原料反应完全(TLC检测,甲醇∶乙酸乙酯∶三乙胺=1∶5∶0.05)。反应液用200mL5%柠檬酸水溶液洗涤,去除水相,浓缩蒸除二氯甲烷。得依鲁替尼粗品:3.63g。1H-NMR(400Mz,DMSO-d6)δ:8.259(s,1H),7.654~7.674(m,2H),7.405~7.444(m,2H),7.109~7.195(m,5H),6.676~6.743(m,1H),6.046~6.152(m,1H),5.570~5.713(m,1H),4.690~4.716(m,1H),4.554~4.583(m,0.5H),4.208(m,1H),4.052~4.085(m,0.5H),3.674~3.731(m,0.5H),3.184~3.214(m,1H),2.972~3.027(m,0.5H),2.245~2.282(m,1H),2.128(m,1H),1.903~1.937(m,1H),1.577~1.605(m,1H)(详见图3);ESI-HRMS谱图显示分子离子峰:441.20366[MW+H]+,依鲁替尼分子离子峰的理论计算值为:441.20335[M+H]+,绝对误差为0.71ppm,符合高分辨质谱误差范围,实测值与理论值相符。(详见图4)。实施例3:杂质A:(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基1哌啶-1-基)-3-氯丙基-1-酮的合成。氮气保护下,向100mL的三口瓶中加入50ml二氯甲烷,搅拌下依次加入中间体-1:(R)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1H-吡唑[3,4-d]嘧啶-4-胺(YLTN-1)(1.00g,1eq),N,N-二异丙基乙胺(0.40g,1.2eq),降温至-20~-10℃,开始滴加3-氯丙酰氯(1.46g,1eq),滴加完毕,溶液由浑浊变澄清,继续搅拌20~30分钟,LC-MS检测,原料消失,减压蒸馏,蒸除二氯甲烷至无馏分蒸出,得到粗品用柱层析方法提纯,洗脱比例为:甲醇∶乙酸乙酯=1∶10,收集洗脱液共400mL,减压蒸馏,蒸除溶剂至无馏分蒸出。得类白色粉末830mg为(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-基)-3-氯丙基-1-酮。1H-NMR(400Mz,CDCl3)δ:8.382(d,J=13.2Hz,1H),7.660~7.638(m,2H),7.419~7.380(m,2H),7.200~7.083(m,5H),5.751(m,2H),4.891~4.853(m,1H),4.842~4.812 (m,0.5H),4.558(m,0.5H),4.066~4.033(m,0.5H),3.844~3.833(m,1H),3.773~3.746(m,0.5H),3.355(m,0.5H),3.191(m,0.5H),2.877~2.843(m,2H),2.812~2.795(m,1H),2.434~2.270(m,2H),2.048~1.955(m,1H),1.756~1.689(m,1H)。(详见图5)ESI-HRMS谱图显示分子离子峰m/z=477.17930[MW+H]+,所对应的分子量与提供的结构式理论计算值(476.17275)相符。绝对误差为1.52ppm,在高分辨质谱误差范围之内。(详见图6)。杂质A在HPLC谱图上相对主成分依鲁替尼的相对保留时间(RRT)约为1.06。实施例4:杂质B:(R)-8-(1-丙烯酰基哌啶-3-基)-10-(4-苯氧基苯基)-3,4-二氢吡唑并[4,3-e]嘧啶并[1,2-c]嘧啶-2(8H)-酮的合成。氮气保护,向250mL的三口瓶中加入100ml二氯甲烷,搅拌下依次加入中间体-1:(R)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1H-吡唑[3,4-d]嘧啶-4-胺(YLTN-1)(2.00g,1eq),N,N-二异丙基乙胺(1.30g,2.0eq)降温至-20℃~10℃。开始滴加丙烯酸酐(1.33g,2eq),滴加过程控温-10℃以下;滴毕,继续搅拌20~30分钟,LC-MS检测,有目标产物生成,控温30~40℃,真空度:-0.08MPa,减压蒸馏,蒸除二氯甲烷至无馏分蒸出;得到粗品用柱层析方法提纯,洗脱比例为:甲醇∶乙酸乙酯=1∶5,收集洗脱液,控温30~40℃,真空度:-0.08MPa,减压蒸馏,蒸除溶剂至无馏分蒸出。得类白色粉末531mg为(R)-8-(1-丙烯酰基哌啶-3-基)-10-(4-苯氧基苯基)-3,4-二氢吡唑并[4,3-e]嘧啶并[1,2-c]嘧啶-2(8H)-酮。1H-NMR(400Mz,CDCl3)δ:8.352(m,2H),7.874(m,2H),7.367~7.328(m,2H),7.144~7.050(m,5H),6.591~6.558(m,1H),6.335~6.293(m,1H),5.747~5.658(m,1H),4.803(m,1H),4.649~4.622(m,0.5H),4.300(m,0.5H),4.333~4.300(m,2H),4.153(m,0.5H),4.051~4.019(m,0.5H),3.751~3.680(m,0.5H),3.354~3.217(m,1H),2.902(m,0.5H),2.789~2.770(m,2H),2.397~2.223(m,2H),2.194~2.022(m,1H),2.005~1.718(m,1H)(详见图7)。ESI-HRMS谱图显示分子离子峰m/z=495.21580[MW+H]+,所对应的分子量与提供的结构式理论计算值(494.20664)相符。绝对误差为3.82ppm,在高分辨质谱误差范围之内(详见图8)。杂质B在HPLC谱图上相对主成分依鲁替尼的相对保 留时间(RRT)约为0.97。实施例5:杂质C:(R)-1-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-基)-乙酮的合成将中间体-1:(R)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1H-吡唑[3,4-d]嘧啶-4-胺(YLTN-1)(2.00g,1eq)溶解于二氯甲烷(50ml)中,加入N,N-二异丙基乙基胺(0.80g,2eq),降温至-10℃,氮气保护,滴加乙酸酐(0.53g,1eq),加毕,反应30分钟,LC-MS监控,反应完成,将体系减压浓缩至干,剩余物通过硅胶柱层析,洗脱液:乙酸乙酯∶甲醇=10∶1,得类白色粉末1.38g为(R)-1-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基]哌啶-1-基)-乙酮。1H-NMR(400Mz,CDCl3)δ:8.352(d,J=16Hz,1H),7.657~7.623(m,2H),7.402~7.364(m,2H),7.187~7.069(m,5H),5.971(m,2H),4.849~4.841(m,0.5H),4.841~4.821(m,1H),4.574~4.541(m,0.5H),4.045~4.005(m,0.5H),3.864~3.831(m,0.5H),3.744~3.685(m,0.5H),3.318~3.253(m,0.5H),3.201~3.141(m,0.5H),2.802~2.745(m,0.5H),2.403~2.249(m,2H),1.962~1.697(m,2H)(详见图9);ESI-HRMS谱图显示分子离子峰m/z=429.20416[MW+H]+,所对应的分子量与提供的结构式理论计算值(428.19607)相符。绝对误差为1.89ppm,在高分辨质谱误差范围之内(详见图10)。杂质C在HPLC谱图上相对主成分依鲁替尼的相对保留时间(RRT)约为0.96。实施例6:杂质D:(R)-4-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-羰基)戊烯-4-酸的合成氮气保护下,将依鲁替尼1-[(3R)-3-[4-氨基-3-(4-苯氧基苯基)-1H吡唑并[3,4-d]嘧啶-1-基]-1-哌啶基]-2-丙烯-1-酮(2g,1eq)溶解于二氯甲烷(50ml)中,依次加入N,N-二异丙基乙基胺(0.70g,2eq),丙烯酸(0.98g,3eq),反应升温至35℃,搅拌36小时,将体系减压浓缩至干,剩余物通过硅胶柱层析,洗脱液:乙酸乙酯∶甲醇=8∶1,得类白色粉末103mg为(R)-4-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-羰基)戊烯-4-酸,1H-NMR(400Mz,DMSO-d6)δ:12.309(br,1H),8.263(S, 1H),7.687~7.666(m,2H),7.422~7.461(m,2H),7.122~7.211(m,5H),6.009~6.072(m,0.5H),5.570~5.660(m,0.5H),4.647~4.772(m,1H),4.516~4.547(m,0.5H),4.200-4.232(m,0.5H),4.069-4.101(m,0.5H),3.913~3.945(m,0.5H),3.629-3.655(m,0.5H),3.129~3.157(m,1H),2.878(m,0.5H),2.445-2.513(m,4H),2.206~2.264(m,1H),2.085~2.125(m,1H),1.861~1.936(m,1H),1.522~1.666(m,1H)(详见图11);ESI-HRMS谱图显示分子离子峰m/z=513.22606[MW+H]+,所对应的分子量与提供的结构式理论计算值(512.21720)相符。绝对误差为3.09ppm,在高分辨质谱误差范围之内(详见图12)。杂质D在HPLC谱图上相对主成分依鲁替尼的相对保留时间(RRT)约为1.02。实施例7:杂质H:(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-基)-丙基-1-酮的合成氮气保护下,将中间体-1:(R)-3-(4-苯氧基苯基)-1-(哌啶-3-基)-1H-吡唑[3,4-d]嘧啶-4-胺(YLTN-1)(2.00g,1eq)溶解二氯甲烷(50mL)中,加入N,N-二异丙基乙基胺(0.80g,2eq),降温至-10℃,氮气保护,滴加丙酸酐(0.67g,1eq),加毕,反应30分钟,LC-MS监控,反应完成,将体系减压浓缩至干,剩余物通过硅胶柱层析,洗脱液:乙酸乙酯∶甲醇=10∶1,得浅粉色粉末1.72g为(R)-1-(3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑并[3,4-d]嘧啶-1-基)-哌啶-1-基)-丙基-1-酮。1H-NMR(400Mz,CDCl3)δ:8.338~8.302(m,2H),7.658~7.627(m,2H),7.440~7.400(m,2H),7.224~7.097(m,5H),4.881~4.634(m,1H),4.881~4.826(m,1H),4.108~3.094(m,1H),2.762(m,1H),3.310~3.167(m,1H),2.452~2.315(m,4H),1.213~1.157(m,3H),2.069~1.691(m,2H)(详见图13);ESI-HRMS谱图显示分子离子峰m/z=443.25380[MW+H]+,所对应的分子量与提供的结构式理论计算值(442.21172)相符。绝对误差为3.66ppm,在高分辨质谱误差范围之内(详见图14)。杂质H在HPLC谱图上相对主成分依鲁替尼的相对保留时间(RRT)约为1.04。实施例8:杂质E:1-((R)-1-丙烯酰基哌啶-3-基)-4-氨基-3-(4-苯氧基苯 基)-1H-吡唑并[3,4-d]嘧啶-1-氧化物的制备(1)杂质E的制备取实施例2制备的依鲁替尼约5.17g,置500ml反应瓶中,加150ml80%乙腈-水溶解,再加30%双氧水30ml,室温搅拌过夜,反应结束,向反应液中加入硫代硫酸钠,至无气泡产生,35℃减压浓缩至无液体流出,再加3倍水相体积的乙酸乙酯,萃取,收集有机相,合并,35℃减压浓缩至干,得依鲁替尼杂质E粗品2.65g。LC-MS检测粗品纯度为51.26%,液相色谱上相对于依鲁替尼峰的相对保留时间为0.98,质荷比[MW+H]+为457.2。杂质E的1H-NMR图参见图15,高分辨质谱参见图16。(2)杂质E粗品的提纯将2.95g上述制备的杂质E粗品用DMSO溶解成浓度约为150mg/ml的溶液,采用以下制备色谱法进一步纯化:色谱柱(购自艾杰尔科技):采用粒径10μm的C18填料,用异丙醇匀浆后装柱(50mm×250mm);流动相:水和乙腈流速:120ml/min;检测波长:254nm;进样量:10ml梯度洗脱,洗脱程序为:时间(min)流动相A(%)流动相B(%)0653510653530455550455550.01208055208055.016535606535收集保留时间为30~37min的峰所对应的馏分;LC-MS或HPLC检测杂质E的纯度。35℃减压浓缩所收集的馏分至无液体流出,加入3倍体积的乙酸乙酯萃取,收集有机相,合并,加无水硫酸钠干燥,过滤,滤液30℃减压浓缩至干,得到2.06g浅粉色固体,经HPLC测定,纯度为98.97%,总收率为36.6%。(3)杂质E的结构鉴定杂质E的高分辨质谱(ESI源)显示其质荷比[M+H]+为457.19930,所对应的分子量与提供的结构式理论计算值456.19099的绝对误差为2.27ppm,符合高分辨质谱的误差范围。参考式(2)依鲁替尼的结构,结合杂质E的1H-NMR、13C-NMR谱鉴定其结构式如下式E所示。杂质E高分辨质谱显示其分子量与所提供的分子式是完全相符的;表1杂质E的核磁共振1H-NMR、13C-NMR解析结果杂质E的核磁共振氢谱、碳谱所显示的信息能够对所提供的式E化合物分子结构式上氢、碳全部归属。因此,依鲁替尼杂质E与所提供的式E分子结构式是相符的。实施例9:杂质F:1,3-二((R)-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)丙烷-1-酮和杂质G:1-((R)-3-(4-((3-((R)-3-(4-氨基-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)-3-氧代丙基)氮)-3-(4-苯氧基苯基)-1H-吡唑[3,4-d]嘧啶-1-基)哌啶-1-基)丙基-2-烯-1-酮的制备。(1)杂质F和杂质G的制备在1000ml反应瓶中加入约20g实施例2制备的依鲁替尼,加入250ml乙腈和200ml5M氢氧化钠水溶液,80℃水浴加热反应40min,冷却至室温,加5MHCl中和至中性,35℃减压浓缩至无液体留出,加入5倍水相体积的乙酸乙酯萃取,收集上层有机相,合并,35℃减压浓缩至干,得到依鲁替尼杂质F、G粗品18.72g,LC-MS检测粗品纯度,根据对应MS离子 峰,液相色谱上相对依鲁替尼峰的相对保留时间约为0.94(杂质F)和1.15(杂质G)的峰为目标物,其粗品中的含量分别为16.9%(杂质F)和18.4%(杂质G)。杂质F的1H-NMR图参见图17,高分辨质谱参见图18。杂质G的1H-NMR图参见图19,高分辨质谱参见图20。(2)杂质F和杂质G粗品的提纯将上述制备的18.72g杂质F、G的粗品加DMSO溶解成10ml的溶液,上样;中压制备柱(购自Agela公司):AgelaXBPC18填料,480g,填料粒径:20~35μm流动相:水(0.05%TFA)和纯乙腈流速:40ml/min检测波长:254nm上样量:~5g梯度洗脱,洗脱程序为:时间(min)流动相A(%)流动相B(%)075255752530653540653550505055505055.017525607525分别收集所有峰的馏分,LC-MS或HPLC确认目标产物。收集30~33min的馏分(杂质F)和40~44min的馏分(杂质G),合并相同馏分,分别减压浓缩,至无液体流出,水相中加入3倍体积的乙酸乙酯萃取,收集有机相,加无水硫酸钠干燥,过滤,收集滤液。减压浓缩至干,分别得依鲁替尼杂质F纯品2.21g和依鲁替尼杂质G二次分离粗品2.57g。经 LC-MS或HPLC检测分析,依鲁替尼杂质F的纯度为98.15%,依鲁替尼杂质G二次分离粗品的纯度为85.32%。(3)杂质G的二次分离上述制备的2.57g依鲁替尼杂质G二次分离粗品加10mlDMSO溶解依鲁替尼杂质G二次分离所采用的条件为:色谱柱:WatersXBridgeC830mm×100mm,5μm流动相:水和乙腈检测波长:254nm流速:25ml/min梯度洗脱,洗脱程序如下:时间(min)流动相A(%)流动相B(%)075255752515356525356525.017525307525进样体积:每次2ml/次;收集保留时间26min~30min的馏分,并用LC-MS确认目标物纯度。合并相同馏分、35℃减压浓缩至无乙腈流出,加3倍体积的乙酸乙酯萃取,合并有机相,加无水硫酸钠干燥、过滤,减压浓缩至干,得到依鲁替尼杂质G纯品1.64g,类白色固体粉末,LC-MS检测纯度为95.13%。(4)杂质F的结构鉴定杂质F的高分辨质谱(ESI源)显示其质荷比[M+H]+为827.39065,所对应的分子量与提供的结构式理论计算值826.34520的绝对误差为2.16ppm,符合高分辨质谱的误差范围。样品的高分辨质谱显示其分子量与所提供的分子式是完全相符的。参考依鲁替尼的结构,结合本品的1H-NMR、13C-NMR、H-HCOSY、HMBC、HSQC、DEPT谱鉴定其结构式如下式F所示。表2为杂质F1H-NMR、H-HCOSY、及HMBC解析数据表3杂质F13C-NMR、DEPT、HSQC、HMBC数据解析由样品的1H-NMR、H-HCOSY、及HMBC谱可知,其结构中氢质子数目与式F化合物的结构相符。由样品的13C-NMR、DEPT、HSQC及HMBC谱可知,其结构中的碳数目与式F化合物的结构相符。核磁氢谱、碳谱以及二维谱数据能对结构式上的氢和碳进行合理归属和关联。(5)杂质G的结构鉴定杂质G的高分辨质谱(ESI源)显示其质荷比[M+H]+为881.40140,所对应的分子量与提供的结构式理论计算值880.39215的绝对误差为 1.43ppm,符合高分辨质谱的误差范围。样品的高分辨质谱显示其分子量与所提供的分子式是完全相符的。结合依鲁替尼的结构,结合本品1H-NMR、13C-NMR、DEPT、H-HCOSY、HMBC及HSQC谱鉴定其结构如下式G所示。表4为杂质G的1H-NMR、H-HCOSY及HMBC数据及氢归属表5为杂质G的13C-NMR、DEPT、HSQC及HMBC数据及碳归属根据样品的1H-NMR谱、结合H-HCOSY、HMBC二维谱可知,其结构中氢质子数目与式G化合物的结构相符。由样品的13C-NMR、DEPT、 HSQC及HMBC谱可知,其结构中的碳数目与式G化合物的结构相符。实施例10:杂质A-H在依鲁替尼杂质检测中作为对照品的应用(1)杂质A-H对照溶液的配制:取上述制备的杂质A-H适量,精密称定,分别置100ml容量瓶中,加适量乙腈(其中杂质F和G加2~4mlDMSO)超声使溶解,再加乙腈定容至刻度,摇匀,分别制成每1ml含杂质A-H各约10μg的溶液,再精密量取该溶液1.0ml,置10ml容量瓶中,加80%乙腈-水稀释至刻度,摇匀,既得分别含依鲁替尼杂质A-H的对照品溶液。(2)供试品溶液的配制:上述制备的依鲁替尼粗品供试品溶液的配制:取上述实施例制备的依鲁替尼样品粗品约10mg,精密称定,置10ml容量瓶中,加80%乙腈-水溶解并定容至刻度,摇匀,制成每1ml含依鲁替尼约1mg的溶液,作为依鲁替尼原料药供试品溶液。依鲁替尼胶囊供试品溶液的配制:取依鲁替尼胶囊(Imbruvica购自PharmacyclicsJanssen公司,140mg,批号:L0404951)内容物细粉适量(含依鲁替尼约10mg),精密称定,置10m1容量瓶中,加80%乙腈-水溶解并定容至刻度,摇匀,过滤,弃去初滤液2ml,取续滤液作为依鲁替尼胶囊供试品溶液。(3)色谱条件如下:以辛基硅烷键合硅胶C8柱(4.6×250mm,5μm)作为色谱柱(购自艾杰尔科技),流动相为:水(0.05%TFA)和乙腈,流速为1.2ml/min,检测波长254nm,柱温35℃,梯度洗脱,洗脱程序如下:时间(min)流动相A(%)流动相B(%)080203802015208022208022.018020308020(4)试验步骤:取上述杂质对照溶液和供试品溶液10μl,分别注入液相色谱仪(购自美国Agilent公司,型号1260)或液质联用仪(美国Agilent公司,Agilent6120),记录色谱图,根据各杂质相对主成分依鲁替尼的相对保留时间进行定性,按照外标法(中国药典2010年版,二部,附录VD)计算杂质A-H的含量。(4)试验结果:①市售的依鲁替尼胶囊中杂质谱分析:序号含量(%)[MW+H]+杂质归属相对保留时间检测到的杂质10.01429.2C0.96检测到的杂质20.05495.2B0.97检测到的杂质30.05443.2H1.04检测到的杂质40.12477.2A1.06检测到的杂质50.13414.2/827.4F0.94检测到的杂质60.04441.3/881.3未知杂质1.03检测到的杂质70.08441.3/881.6G1.15②上述制备的依鲁替尼粗品的杂质谱分析序号含量(%)[MW+H]+杂质归属相对保留时间检测到的杂质10.01429.2C0.96检测到的杂质20.41495.2B0.97检测到的杂质30.70443.2H1.04检测到的杂质40.06513.2D1.02检测到的杂质50.14414.2/827.4F0.94检测到的杂质60.07441.3/881.3未知杂质1.03检测到的杂质70.03441.3/881.6G1.15检测到的杂质80.07457.2E0.98需要说明的是在本发明中提及的所有文献在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,以上所述的是本发明的具体实施列及所运用的技术原理,在阅读了本发明的内容后,本 领域技术人员可以对本发明做各种改动或修改而不背离本发明的精神与范围,这些等价形式同样落在本发明的范围内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1